

Abstract
Astrocytes are a major cell type in the mammalian CNS. Astrocytes are now known to play a number of essential roles in processes including synapse formation and function, as well as blood-brain barrier formation and control of cerebral blood flow. However, our understanding of the molecular mechanisms underlying astrocyte development and function is still rudimentary. This lack of knowledge is at least partly due to the lack of tools currently available for astrocyte biology. ACSA-2 is a commercially available antibody originally developed for the isolation of astrocytes from young postnatal mouse brain, using magnetic cell-sorting methods, but its utility in isolating cells from adult tissue has not yet been published. Using a modified protocol, we now show that this tool can also be used to isolate ultrapure astrocytes from the adult brain. Furthermore, using a variety of techniques (including single-cell sequencing, overexpression and knockdown assays, immunoblotting, and immunohistochemistry), we identify the ACSA-2 epitope for the first time as ATP1B2 and characterize its distribution in the CNS. Finally, we show that ATP1B2 is stably expressed in multiple models of CNS injury and disease. Hence, we show that the ACSA-2 antibody possesses the potential to be an extremely valuable tool for astrocyte research, allowing the purification and characterization of astrocytes (potentially including injury and disease models) without the need for any specialized and expensive equipment. In fact, our results suggest that ACSA-2 should be a first-choice method for astrocyte isolation and characterization.
Keywords: astrocyte, cell surface protein, membrane protein, neuroscience, plasma membrane, flow cytometry, immunoblotting, immunohistochemistry, immunoisolation, single-cell RNA-seq
Introduction
Astrocytes are one of the major cell types in the central nervous system (CNS), where they are thought to perform a variety of complex functions ranging from synapse formation and modulation of synaptic transmission to formation of the blood-brain barrier and regulation of cerebral blood flow (1).
However, progress in understanding the complex roles that astrocytes play in the CNS has been hampered by a lack of tools directly targeting this cell type, such as antibodies for cell type-specific proteins, fluorescent reporter mice, and Cre-expressing lines.
One important attempt to study astrocyte function in a controlled and defined manner was the introduction of an in vitro culture preparation from neonatal rodent brains (2). Although this system has been widely used to elucidate many aspects of astrocyte function, a number of studies have questioned whether this system is fully representative of astrocytes in vivo (3, 4). These doubts have arisen due to a number of factors. First, these cultures are obtained from animals very early in postnatal development, when astrocytes are still in a proliferative phase, meaning that cells are likely to retain some “stemlike” properties (5). This is probably one of the reasons why these astrocyte cultures divide rapidly and can be passaged over many months, despite adult astrocytes in vivo showing limited proliferative ability under normal physiological conditions (3, 6, 7) (although injury, which may well be mimicked by the tissue dissociation process, has been reported to lead to cell proliferation within lesions (8)). This, in turn, is probably reflected by the flat fibroblast-like morphology taken on by these cultured cells (in comparison with the highly branched appearance of astrocytes in vivo) as well as their highly different gene expression profiles and proteomes (9, 10). Hence, there has been a recent move toward acute isolation from older postnatal animals, combined with next-generation culture systems, to better preserve the morphological and functional characteristics of recovered astrocytes (3, 11).
To date, acute isolation strategies have tended to fall into one of two types: fluorescent reporter-based methodologies (where fluorescent marker proteins are expressed under the control of an astrocyte specific promoter) or immunoisolation (using antibodies against specific cell surface antigens). Of these approaches, separation based on fluorescent protein expression has traditionally been the most widely used, principally because most of the common astrocyte markers are intracellular (11, 12). As such, astrocytes have typically been isolated from transgenic animals that stably express a fluorescent protein under the control of an astrocyte-specific promoter (e.g. Gfap (13), S100β (14), Slc1a2 (15), Slc1a3 (15), or Aldh1l1 (16)). However, the use of these animals is subject to several caveats, which mainly center on whether the fluorescent marker protein accurately recapitulates the expression pattern driven by the endogenous promoter (11). Furthermore, there is usually considerable cost involved in maintaining such lines and a need to access specialist cell-sorting equipment.
In acknowledgment of these limitations, several groups have recently revisited the selection of astrocytes using various endogenous surface antigens (such as integrin β5 and GLAST) with some success (3, 11). In fact, Miltenyi Biotec has recently commercialized a range of immuno-based purification kits known as the ACSA (astrocyte cell surface antigen) range. At present, this consists of the ACSA-1 and ACSA-2 kits, which are sold for the rapid, one-step isolation of astrocytes from a single-cell suspension, using either fluorescence-based sorting methods (FACS), or (more conveniently and inexpensively) magnetic-activated cell sorting (MACS)7 procedures.
The ACSA-1 system has been widely adopted and targets the broadly expressed astrocyte glutamate transporter GLAST (11). However, the use of a trypsin digestion step in the initial dissociation protocol (which is necessary to keep the crucial epitope on GLAST intact) is suited mainly to young postnatal tissue.
In contrast, ACSA-2 uses a papain-based digestion step, which is generally regarded as less damaging and more effective with CNS tissue (17). Hence, ACSA-2 is potentially more compatible with successful isolation (and subsequent characterization) of adult astrocytes. However, many aspects of ACSA-2 performance are unknown, including the precise identity of the epitope, its distribution and developmental profile, and the nature of expression in both normal and pathological states. The latter is of particular importance, given that reactive astrogliosis is a signature common to all neuropathologies and an essential component of any disease model (1).
Hence, in this study, we set out to fully characterize the ACSA-2 tool and show it to be a versatile and cost-effective method for the one-step isolation of astrocytes from young and adult mouse brain, including reactive astrocytes. This makes the ACSA-2 system a valuable addition to the “toolbox” for future astrocyte research (including disease modeling).
Results
A major step forward in astrocyte research would be the ability to label and characterize astrocytes from both young postnatal and adult brain (including through immunoisolation).
ACSA-2 is a commercial antibody from Miltenyi Biotec, which is marketed for the one-step isolation of astrocytes from young mice (approximately postnatal day 6). As noted in the Introduction, however, many aspects of ACSA-2 use are largely unexplored. Therefore, we decided to test the utility of ACSA-2 as a general tool for astrocyte biology (when compared with other technologies).
The ACSA-2 antibody can be used to purify astrocytes from both young and adult cortex
In a first set of experiments, we decided to investigate the yield, viability, and purity of astrocytes isolated using ACSA-2 (see Fig. 1). To ensure that our results were broadly comparable with other published techniques, we decided to use mouse cortex as a source of material.
Figure 1.

Schematic of astrocyte isolation from mouse cortex using ACSA-2. To account for the high level of myelination present in the adult CNS, the standard manufacturer's protocol was modified to include an additional myelin removal step (using Percoll PLUS and Myelin Removal Beads II, Miltenyi Biotec) immediately following tissue dissociation but before astrocyte isolation.
As expected, ACSA-2 could be used to purify astrocytes from young cortex (approximately postnatal day 6) with minimal modifications to the manufacturer's recommended protocol. Labeling and isolation were highly efficient, yielding on average 300,000 cells/cortex dissociated, with a viability of 86% (as judged by dye exclusion assays). The purity of recovered cells was assessed using two independent methods: quantitative RT-PCR for mRNA content and flow cytometry for protein expression. (Details of primers and antibodies used can be found in Tables 1 and 2, respectively). ACSA-2 routinely enriched for astrocytes and removed contaminating cells types (including neurons and neuronal precursors, oligodendrocytes and oligodendrocyte precursors, microglia, and endothelial cells) (Figs. 2 and 3).
Table 1.
Primers used in qPCR experiments
| Gene (PubMed accession number) | Primer sequence | Cell-type specificity | Primer efficiency (%) |
|---|---|---|---|
| Atp1b2 (NM_013415.5) | |||
| Forward | TGTGTTGGCAAGAGAGATGAAGA | Astrocytesa | 99.8 |
| Reverse | GGGTCACATTCAGGAACTTTACA | ||
| Slc1a3 (NM_148938.3) | |||
| Forward | CCTGGGTTTTCATTGGAGGGTTG | Astrocytes | 104.2 |
| Reverse | GTGGCAGAACTTGAGGAGGTC | ||
| Aldh1l1 (NM_027406.1) | |||
| Forward | GCAGGTACTTCTGGGTTGCT | Astrocytes | 90.67 |
| Reverse | GGAAGGCACCCAAGGTCAAA | ||
| Cx3cr1 (NM_009987) | |||
| Forward | CGTGAGACTGGGTGAGTGAC | Microglia | 93.20 |
| Reverse | GGACATGGTGAGGTCCTGAG | ||
| Mbp (NM_001025251.2) | |||
| Forward | CATCCTTGACTCCATCGGGC | Oligodendrocytes | 93.75 |
| Reverse | CAGGGTACCTTGCCAGAGC | ||
| Ocln (NM_008756.2) | |||
| Forward | CCGCCAAGGTTCGCTTATCT | Endothelia | 97.67 |
| Reverse | CGGACATGGCTGATGTCACT | ||
| Cspg4 (NM_139001.2) | |||
| Forward | AGGCTGAGGTAAATGCTGGG | Oligodendrocyte precursors | 97.67 |
| Reverse | GCAGGTGGTGAGGACAGTAG | ||
| Dcx (NM_001110222.1) | |||
| Forward | AGGTAACGACCAAGACGCAAA | Neuronal precursors | 97.00 |
| Reverse | GGGTGTAGAGATAGGAGACTGC | ||
| Syt1 (NM_001252341.1) | |||
| Forward | GGGGAAGGGAAGGAAGATGC | Neurons | 99.67 |
| Reverse | ACGGTGGCAATGGGATTTTATG | ||
| Tbp (NM_013684.3) | |||
| Forward | GCCAAGAGTGAAGAACAATCCA | Housekeeping gene | 94.33 |
| Reverse | AGAACTTAGCTGGGAAGCCC | ||
| Cyc1 (NM_025567.2) | |||
| Forward | TGCTCTTCCTGCCACAGC | Housekeeping gene | 93.67 |
| Reverse | GACCTCCACCTCCTCAGCC | ||
a As based on expression data in this paper.
Table 2.
Antibodies used in flow cytometry experiments
All antibodies used in flow cytometry were used according to the manufacturer's instructions (including recommended dilutions), except for anti-O1-eFluor 660, which was used at a 1:80 dilution.
| Antibody-fluorophore conjugate | Vendor and product code | Cell-type specificity |
|---|---|---|
| ACSA-2-PE | Miltenyi Biotec: 130102365 | Astrocytes |
| ACSA-2-APC | Miltenyi Biotec: 130102315 | Astrocytes |
| Anti-GLAST-PE | Miltenyi Biotec: 130095821 | Astrocytes |
| Anti-GLAST-APC | Miltenyi Biotec: 130098804 | Astrocytes |
| Anti-CD11B-PE | Miltenyi Biotec: 130098087 | Microglia |
| Anti-CD31-PE | Miltenyi Biotec: 130102971 | Endothelia |
| Anti-THY1.2-PE | Miltenyi Biotec: 130102960 | Neuronsa |
| Anti-NG2-PE | Miltenyi Biotec: 130097458 | Oligodendrocyte precursors |
| Anti-O4-APC | Miltenyi Biotec: 130095891 | Oligodendrocytes |
| Anti-O1-eFluor 660 | eBioscience: 506506 | Oligodendrocytes |
a Note the anti-THY1.2-PE antibody has also been reported to partially stain glia (28).
Figure 2.

Quantitative RT-PCR analysis of astrocytes isolated from young and adult mouse cortex. RNA was extracted from isolated astrocytes and reverse transcribed into cDNA. (As a positive control for relative quantification, RNA was also extracted from a single-cell suspension of whole cortex.) Quantitative PCR was then performed using a range of primers for cell type-specific markers, including Aldh1l1 (astrocytes), Cx3cr1 (microglia), doublecortin (Dcx) (neuronal precursors), myelin basic protein (Mbp) (oligodendrocytes), chondroitin sulfate proteoglycan (Cspg4) (oligodendrocyte precursors: NG2+ cells), occludin (Ocln) (endothelia), and synaptotagmin I (SytI) (neurons). Tata box-binding protein (Tbp) and cytochrome c1 (Cyc1) were used as housekeeping genes for normalization. (Full details of primer sequences used can be found in Table 1.) We routinely see a 2-fold enrichment of Aldh1l1 signal relative to control and a substantial decrease in contaminating cell types at both of the developmental ages we assayed, indicating that our astrocytes are of exceptional purity. The one exception was chondroitin sulfate proteoglycan (Cspg4), which was detectable at high levels in astrocytes isolated from P56 mice. Graphs show results from three independent cell isolation experiments (with a minimum of two technical replicates performed for each sample). qPCR runs on cells from different isolation experiments were repeated more than 3 times with the same results. The dotted line represents the level of mRNA in the positive control (whole-cortex cell suspension). a.u., arbitrary units. Graphs are plots of average ± S.D. (error bars).
Figure 3.

Flow cytometry-based analysis of astrocytes isolated from young and adult mouse cortex. Live, non-fixed astrocytes were stained using a range of fluorescently conjugated antibodies for cell type-specific markers, including ACSA-2 and anti-GLAST (astrocytes), anti-CD11B (microglia), anti-O1 and anti-O4 (oligodendrocytes), anti-NG2 (oligodendrocyte precursors: NG2+ cells), anti-CD31 (endothelia), and anti-THY1.2 (neurons). (Full details of antibodies used can be found in Table 2.) We routinely see that > 95% of all cells purified are astrocytes, indicating that the preparation is of exceptional purity (consistent with data from qPCR analysis; Fig. 2). Trace amounts of all other cell types were detected (< 2%) except in the case of NG2+ cells, where levels were higher (∼4%, although this figure could be a slight underestimation due to the sensitivity of the epitope to papain; see “Experimental procedures”). Note that THY1.2 staining was performed on astrocytes isolated from the Aldh1l1-EGFP mouse line (which expresses EGFP specifically in astrocytes), because THY1.2 was previously reported to stain both neurons and astrocytes. Graphs show results from one representative experiment. This experiment was repeated twice on separate days with identical results. In total, two biological replicates were analyzed for each condition, with at least 10,000 cells analyzed per sample (effectively 1 technical replicate per sample). Lines in each plot delineate gates; numbers represent the proportion of cells in each particular gate.
Isolation of cells from adult brain is typically seen as much more demanding, especially given the higher levels of myelin present at later ages. However, we found that additional myelin/oligodendrocyte depletion using a Miltenyi myelin removal kit solved this issue and that cell yield, viability, and purity was broadly comparable with that obtained from young brain (with the exception of a higher level of chondroitin sulfate proteoglycan (Cspg4) (which is a marker for oligodendrocyte precursors) (Figs. 2 and 3).
Finally, to determine whether ACSA-2 labels all cortical astrocytes (or just a defined subset), we prepared a suspension of adult cortical cells from the Aldh1l1-EGFP mouse line, which is widely reported to express EGFP in all astrocytes (9). This cell suspension was labeled with an ACSA-2-phycoerythrin (PE) conjugate and used for subsequent flow cytometry. All Aldh1l1-EGFP-positive astrocytes were labeled by the ACSA-2 antibody (Fig. 4).
Figure 4.

The ACSA-2 epitope is expressed on all cortical astrocytes. A suspension of cortical cells from the Aldh1l1-EGFP mouse line (which is a pan-astrocyte marker line) was generated according to the standard protocol used for magnetic isolation of astrocytes. However, although we used adult brains in the experiments, we actually omitted the use of Percoll PLUS density medium, using only HBSS with calcium and magnesium. The cell suspension was initially depleted of myelin using Myelin Removal Beads II before labeling with an ACSA-2-PE antibody for flow-cytometry experiments. All EGFP-positive astrocytes were labeled with ACSA-2 antibody, indicating that the ACSA-2 epitope is present on all cortical astrocytes. Note the small population of cells labeled with ACSA-2 alone, which could be astrocytes not labeled by Aldh1l1-EGFP or other cell types that express ACSA-2 (e.g. oligodendrocytes) that are otherwise removed during our astrocyte purification procedure. This experiment was repeated four times using independent samples (effectively 1 technical replicate per sample) on separate days. At least 20,000 cells were analyzed for each sample.
Identification of putative ACSA-2 targets
Our initial results provided strong evidence that ACSA-2 is a general astrocyte marker (with no obvious bias for a defined astrocyte subset), which can be used for the labeling and subsequent isolation of ultrapure astrocytes from both young and adult brain.
However, one major question in our minds was the identity of the ACSA-2 target. For instance, is it a commonly used astrocyte marker (such as GLT-1 or aquaporin 4) or a more “exotic” protein, which would give an additional, alternative option for astrocyte labeling and purification, depending on its distribution between brain regions and expression levels? Such a consideration is particularly relevant in disease and/or injury models, where the expression levels of some genes and proteins are known to change dramatically (4, 18).
Hence, to gain insights into the extent to which ACSA-2 may be useful as a general purpose tool for astrocyte biology, we decided to 1) identify and 2) characterize the ACSA-2 target protein under both normal and pathological conditions.
Identification of the ACSA-2 binding target: Bioinformatics analysis of single-cell sequencing data
In a first set of experiments, we decided to identify the previously unknown target of ACSA-2. To do this, we took advantage of a recently published data set produced by single-cell mRNA sequencing of CNS cells (19). To be of general utility as an astrocyte marker, we hypothesized that expression of the transcript encoding the ACSA-2 target must actually be enriched in astrocytes. Therefore, to test our hypothesis, we screened this publicly available data set for transcripts that were present in 90% plus of astrocytes but that were not expressed (or were expressed at very low levels) in other cell types. (90% was set as the cut-off because single-cell data are inherently noisy, and some transcripts in individual cells are not detected). Identified transcripts were then cross-referenced to the in situ hybridization data available in the Allen Brain Atlas to confirm CNS expression (not shown). Once this initial list was generated, we further refined it by assuming that the ACSA-2 target is a transmembrane domain-containing protein, which is present in the plasma membrane of astrocytes, criteria that are absolutely essential to successful immunoisolation of live (non-fixed) cells. The list of potential target proteins that we identified is summarized in Table 3.
Table 3.
Genes cloned for overexpression in a HEK293T system
Single cells identified as astrocytes on the basis of established marker genes (19) were analyzed to identify transcripts fulfilling the following criteria. First, transcripts had to be present in ≥90% of identified astrocytes. Second, transcripts had to encode proteins containing a predicted transmembrane region. Third, only proteins localizing to the plasma membrane were considered. Only proteins fulfilling these criteria could realistically be used in the immunoisolation of live (non-fixed) astrocytes. This table lists identified transcripts fulfilling these criteria and the corresponding PubMed accession numbers of the transcripts used to design subsequent cloning steps. Essentially, PCR primers were designed to flank ORFs of genes of interest (capital letters) as well as to incorporate unique restriction sites on both ends (lowercase letters). Sequences were then amplified by PCR before ligation into the pCAGIG plasmid. Plasmids were verified by sequencing and checked against the deposited amino acid sequence for the respective proteins. Single-nucleotide polymorphisms that did not affect the amino acid sequence were tolerated. In addition to encoding genes of interest, these constructs also encoded cytosolic GFP to act as a marker for cell transfection.
| Gene (PubMed accession number) | Protein (PubMed accession number) | Primers | Restriction sites |
|---|---|---|---|
| Slc1a2 (NM_001077514.3) | Excitatory amino acid transporter 2 (GLT-1) (NP_001070982.1) | Forward, taagtatcGAATTCATGGCATCAACAGAGGGTG; reverse, taagtatcCTCGAGTTATTTTTCACGTTTCCAAGGTTCT | EcoRI/XhoI |
| Slc1a3 (NM_148938.3) | Excitatory amino acid transporter 1 (GLAST) (NP_683740.1) | Forward, taagtatcCTCGAGATGACCAAAAGCAACGGAGAAG; reverse, taagtatcGCGGCCGCCTACATCTTGGTTTCGCTGTCT | XhoI/NotI |
| Atp1a2 (NM_178405.3) | Na+/K+-transporting ATPase subunit α-2 precursor (NP_848492.1) | Forward, taagtatcCTCGAGATGGGTCGTGGGGCAG; reverse, taagtatcGCGGCCGCTCAGTAGTACGTCTCCTTCTCC | XhoI/NotI |
| Atp1b2 (NM_013415.5) | Na+/K+-transporting ATPase subunit β-2 (NP_038201.1) | Forward, taagtatcCTCGAGATGGTCATCCAGAAAGAGAAGAAGA; reverse, taagtatcGCGGCCGCTCAGGTTTTGTTGATCCGGAGTT | XhoI/NotI |
| Ppap2b (NM_080555.2) | Lipid phosphate phosphohydrolase 3 (NP_542122.1) | Forward, taagtatcCTCGAGATGCAAAGCTACAAGTACGACAA; reverse, taagtatcGCGGCCGCCTACACCATGTTATGGTGATTGTTC | XhoI/NotI |
| Gja1 (NM_010288.3) | Gap junction α-1 protein (NP_034418.1) | Forward, taagtatcCTCGAGATGGGTGACTGGAGCGCC; reverse, taagtatcGCGGCCGCTTAAATCTCCAGGTCATCAGGCCG | XhoI/NotI |
| Tlcd1 isoform a (isoform 1) (NM_026708.2) | Calfacilitin isoform a (isoform a) (NP_080984.1) | Forward, taagtataGAATTCATGCCCCTACTGTTCCACC; reverse, taagtataGCGGCCGCTCACTCAGTCAAGAATTTGTCTTTT | EcoRI/NotI |
| Tlcd1 isoform c (isoform 3) (NM_001291236.1) | Calfacilitin isoform c (isoform c) (NP_001278165.1) | Forward, taagtatcGAATTCATGAGATGGCCGTGGGAT; reverse, taagtatcGCGGCCGCTCACTCAGTCAAGAATTTGTCTTTT | EcoRI/NotI |
ATP1B2 is the ACSA-2 target: Protein overexpression and knockdown studies
To establish whether any of the proteins listed in Table 3 was the ACSA-2 target, we used two independent but complementary techniques.
First, we decided to overexpress the identified proteins in a heterologous (HEK293T cell-based) expression system, which has the significant advantage that it retains as closely as possible the native conformation of the identified proteins (membrane topology, glycosylation status, etc.). Hence, we produced a set of plasmids, each of which encoded a protein identified in our bioinformatics screen. These plasmids also encoded a cytoplasmic version of GFP as a marker for successful cell transfection. These plasmids were transiently transfected into HEK293T cells, which were subsequently labeled with an ACSA-2-allophycocyanin (APC) conjugate for use in flow cytometry. Only cells transfected with plasmids encoding the protein ATP1B2 showed significant co-labeling between GFP and ACSA-2-APC (Fig. 5).
Figure 5.

Flow cytometry-based experiments identify ATP1B2 as a target for ACSA-2. HEK293T cells (which do not bind ACSA-2 under normal conditions) were transfected with plasmids encoding for proteins identified by our bioinformatic screen (see Table 3). These plasmids also expressed soluble GFP as a marker for successful plasmid transfection. Cells were then stained with an ACSA-2-APC conjugate and analyzed by flow cytometry. From the list of proteins identified in Table 3, only cells expressing ATP1B2 showed strong co-labeling for ACSA-2 and GFP (28.2% of cells). A representative experiment is presented in the figure. This experiment was repeated twice using independent samples (effectively 1 technical replicate per sample) on separate days with the same results. At least 60,000 cells were analyzed per sample. Lines in each plot delineate gates; numbers represent the proportion of cells in each particular gate.
Second, to validate the HEK293T cell-based experiments, we decided to carry out the reverse experiment and attempted to knock down ATP1B2 expression using an shRNA-based approach, which we hypothesized would reduce ACSA-2 staining. We performed this experiment using primary cultured astrocytes. We attempted protein knockdown using four independent shRNAs (see Table 4 for details). These shRNAs were transfected into cells using electroporation. The plasmids also encoded a cytoplasmic version of GFP as a marker for successful cell transfection. After 7 days of shRNA expression, transfected cells were FACS-sorted. We first assessed shRNA efficiency by assessing Atp1b2 mRNA expression using qPCR. The most efficient shRNA construct (Table 4, TL500159A) knocked down mRNA expression by 57%, when compared with a scrambled control (p = 0.022) (Fig. 6A). We then used this shRNA construct to assess the effects of ATP1B2 knockdown on ACSA-2 binding, using flow cytometry. As expected from the qPCR data, the average drop in the number of ACSA-2-positive cells was 67% (p = 0.0002), when compared with a scrambled shRNA control (Fig. 6B). As an additional test of shRNA specificity for Atp1b2, we also monitored the expression of the astrocyte-specific plasma membrane protein GLAST at both the mRNA and protein levels (Fig. 6, A and C). No nonspecific effects of shRNA expression were seen. Taken together, our overexpression and knockdown experiments unambiguously identify the ACSA-2 target to be ATP1B2.
Table 4.
shRNAs used for ATP1B2 knockdown experiments
shRNA constructs specific for mouse ATP1B2 were purchased from Origene. In addition to encoding shRNAs, these constructs also contained cytosolic GFP to act as a marker for cell transfection. Constructs were amplified using a Midiprep kit (Macherey-Nagel) and verified by sequencing. Constructs were then transfected into cells using the Neon electroporation system. A scrambled shRNA sequence was used as a negative control.
| shRNA target | shRNA identifier | shRNA sequence |
|---|---|---|
| ATP1B2 | TL500159A | CATGTTCAGAAGCTCAACAAGTTCTTGGA |
| ATP1B2 | TL500159B | CCAAGATGGTCATCCAGAAAGAGAAGAAG |
| ATP1B2 | TL500159C | CCAAGACTGAGAACCTTGATGTCATTGTC |
| ATP1B2 | TL500159D | CGTGTGGCCTTCAAACTCCGGATCAACAA |
| Scrambled shRNA | TR30021 | GCACTACCAGAGCTAACTCAGATAGTACT |
Figure 6.

ACSA-2 targets ATP1B2 on the plasma membrane of primary astrocytes. A, primary astrocytes expressing ATP1B2 shRNA for 7 days showed a 57% drop in the level of Atp1b2 mRNA, as judged by qPCR. This result was statistically significant (p = 0.022) when compared with a scrambled control shRNA. The level of an independent marker gene Slc1a3 (which encodes for the plasma membrane transporter GLAST) did not change upon expression of either shRNA. Results are from four sets of samples, prepared independently on four different days. Each sample was run using four technical replicates. a.u., arbitrary units. Graphs are plots of average ± S.D. (error bars). B, primary astrocytes expressing a scrambled control shRNA readily bind ACSA-2, as measured using flow cytometry (left). However, when primary astrocytes were transfected with shRNAs targeting Atp1b2, staining with ACSA-2 was markedly reduced (center). 7 days post-transfection, the average drop in the number of ACSA-2-positive cells for the most effective shRNA construct was 67% when compared with the scrambled control (quantified in the summary plot; right). This difference was statistically significant (p = 0.0002). C, astrocytes transfected with the same shRNAs as used in A (left, scrambled control; center, anti-Atp1b2) readily bind an antibody targeting the astrocyte plasma membrane protein GLAST, indicating the specificity of the shRNA for Atp1b2. Experiments were repeated three times with independent samples (effectively 1 technical replicate per sample) with similar results. Flow-cytometry plots show representative experiments. At least 100,000 cells were analyzed per sample. Graphs are plots of average ± S.D.
Unbiased cell isolation using ACSA-2 is possible because ATP1B2 is a ubiquitously expressed astrocyte protein
To assess the utility of ACSA-2 as a tool for general astrocyte biology, we next investigated the expression pattern of ATP1B2, using a combination of immunoblotting and immunohistochemistry. Using protein homogenates prepared from mice of different developmental ages, we first investigated the developmental profile of ATP1B2 using immunoblotting. Because the ACSA-2 antibody does not work well in blotting experiments (see “Discussion”), we used an anti-ATP1B2 antibody (from Abcam), which was previously validated for use in this type of experiment. Fig. 7A shows that ATP1B2 expression mirrors astrocyte development, starting at embryonic day 18.5 (E18.5) and increasing until postnatal days 10–20, after which high levels of expression are maintained into adulthood.
Figure 7.

ATP1B2 is highly expressed in most regions of the CNS from late embryonic stages to adulthood. A, ATP1B2 was detectable from late embryonic stages (E18.5) with levels increasing until P10–P20, after which high levels were maintained into adulthood. B, ATP1B2 was expressed at high levels in all brain regions tested, except for the olfactory lobe and the pituitary gland. Trace amounts of ATP1B2 were present in the olfactory lobe, but levels were below the detection limit in the pituitary. Blots were performed using two independent sets of tissue samples. For each tissue sample, a minimum of three separate blots were run with identical results.
In the adult brain, ATP1B2 is expressed at high levels throughout the majority of the brain, including diverse regions, such as the midbrain, cerebellum, pons, hippocampus, striatum, thalamus, medulla, frontal cortex, cortex, and occipital lobe (Fig. 7B). Levels of ATP1B2 were lower in the olfactory bulb and were below the limit of detection in the pituitary gland.
Labeling of multiple brain regions with ACSA-2 was confirmed using immunohistochemistry. As can be seen from a standard widefield fluorescence image, ACSA-2 staining is visible throughout the brain, although immunostaining levels are highly variable, with low levels of staining in areas such as the cortex and hippocampus but high staining in the cerebellum (Fig. 8, top). Higher-magnification confocal images of the visual cortex, cerebellum, hippocampal CA3 region, and thalamus show that ACSA-2 staining appears in a diffuse pattern throughout the parenchyma. Crucially, dual staining with ACSA-2 and a specific anti-ATP1B2 antibody showed perfect colocalization in all brain regions sampled. Under no conditions did we see separation of the two signals, arguing strongly that the in vivo target of ACSA-2 is ATP1B2 (Fig. 8, bottom panels). Closer inspection of the (diffuse) staining pattern produced by ACSA-2 in the visual cortex is consistent with localization to fine astrocyte processes penetrating through the parenchyma. These processes should contain astrocyte-specific glutamate transporters (20), and co-staining with an antibody against GLT-1 resulted in a very similar staining pattern (although there was rarely evidence for colocalization). In contrast, staining against a known neuronal marker (MAP2) was visually very different (Fig. 9A). Together, these data strongly indicate that the principal cells expressing ACSA-2/ATP1B2 in the mouse CNS are astrocytes (as indicated by our immunoisolation data). To confirm this, we also performed confocal imaging in the corpus callosum (where cell density is known to be lower, giving less of the diffuse parenchymal staining seen in Fig. 8). Our findings clearly show that ACSA-2 staining outlines the cell body and processes of astrocytes, consistent with localization to the plasma membrane. However, ATP1B2 must be present in distinct subcellular domains to the glutamate transporter GLT-1, because no major overlap between the proteins was detected (Fig. 9B).
Figure 8.
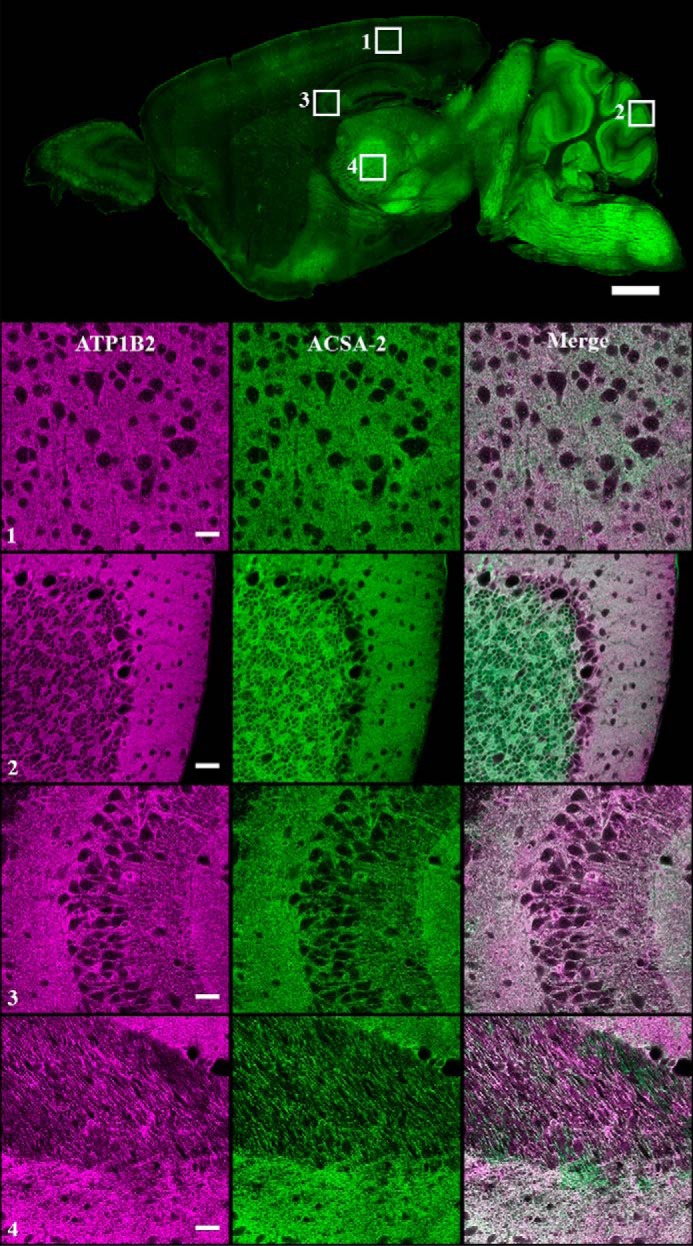
Colocalization of ACSA-2 and ATP1B2 signals in mouse brain. ACSA-2 was used to stain sagittal sections of mouse brain. A low-magnification widefield image taken on a slide scanner (top) shows ACSA-2 to be expressed throughout the brain at various levels. To accurately assess the degree of ATP1B2 and ACSA-2 colocalization, a laser-scanning confocal microscope was used to take images at higher magnification in four different brain regions indicated by the numbered white boxes (1, visual cortex; 2, cerebellum; 3, CA3 region of the hippocampus; 4, thalamus). ATP1B2 (magenta) and ACSA-2 (green) show nearly perfect overlap (bottom panels). For all images, acquisition parameters were individually optimized for maximum dynamic range to allow easier visualization of protein localization. Tissue sections from three animals were analyzed with the same results. Scale bars, 1000 μm (low magnification) and 30 μm (high magnification).
Figure 9.
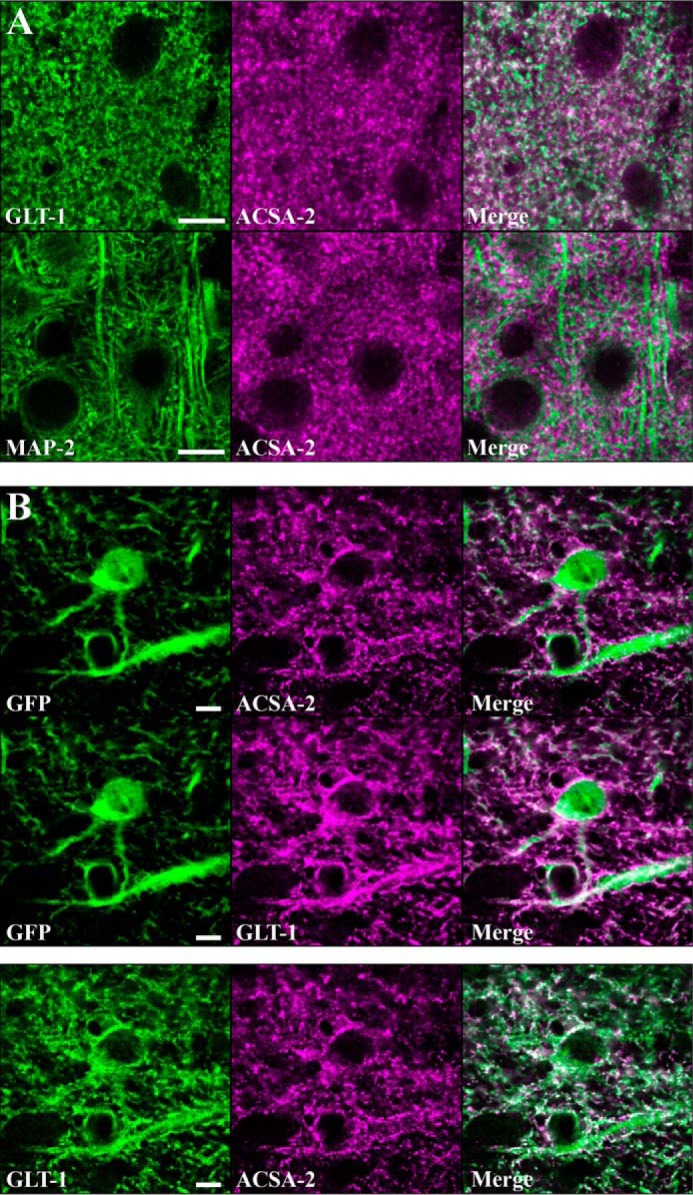
ATP1B2 is expressed in discrete subcellular domains on the plasma membrane of astrocytes. A, top panels, astrocytes were stained with ACSA-2 and an antibody against an astrocyte-specific membrane protein, GLT-1. Imaging in the visual cortex revealed the staining patterns to be highly similar although non-overlapping. Bottom panels, co-staining of ACSA-2 together with a marker of neuronal microtubules (MAP2) showed highly dissimilar staining. Scale bar, 10 μm. B, top panels, tissue sections from an Aldh1l1-EGFP mouse. Images were taken in the corpus callosum, where the lower cell density allows the clear identification of individual astrocytes based on GFP expression. Both ACSA-2 and GLT-1 signal clearly localize to astrocytes. Note that ACSA-2 signal is detected around astrocyte cell bodies and extending into astrocyte processes. Scale bar, 5 μm. Bottom panels, both GLT-1 and ACSA-2 signal colocalize to the same cell, albeit in non-overlapping domains. Scale bar, 5 μm. Tissue sections from two animals were analyzed with the same results for both sets of stainings.
To summarize this set of experiments, we identified the ACSA-2 binding partner to be ATP1B2 and showed that it is a specific marker of astrocytes throughout development and across brain regions. Hence, ACSA-2 should prove an ideal general tool for astrocyte labeling and isolation from wild-type brain.
ATP1B2 is stably expressed on a wide range of reactive astrocytes
All neurological disorders show reactive astrogliosis (1). However, it is now obvious that astrogliosis is an extremely heterogeneous reaction, with different insults producing very different transcriptional responses (4). Obviously, an ideal tool for astrocyte labeling and isolation would also reliably label reactive astrocytes in a variety of neuropathologies. To test the utility of ACSA-2 in injury and disease models, we chose to analyze astrocyte response to a wide range of acute and chronic pathologies, including stab wound injury and spinal cord lesion, stroke, bacterial infection, and Alzheimer's disease.
ATP1B2 expression on astrocytes following stab wound injury
As a model of acute trauma, we made a stab wound injury in the mouse visual cortex with a 30-gauge needle; previous reports have shown strong reactive astrogliosis that peaks 1 week after such a procedure (18). Under similar conditions, the site of needle insertion in our experiments was obvious, with damage to the surface of the cortex immediately apparent. Fluorescence microscopy revealed a heavy infiltration of microglia directly into the injury site (as judged by IBA1 staining). Astrocytes, on the other hand, seemed largely lost at the site of injury, although pronounced astrogliosis (as judged by up-regulation of GFAP expression) was evident throughout the cortex (including sites well away from the injury; Fig. 10A). Interestingly, ACSA-2 staining was not diminished, even in the region directly proximal to the injury (and was still expressed on the small number of astrocytes remaining in the area where the needle was inserted, with an expression pattern similar to that seen in healthy tissue; compare with Fig. 8).
Figure 10.
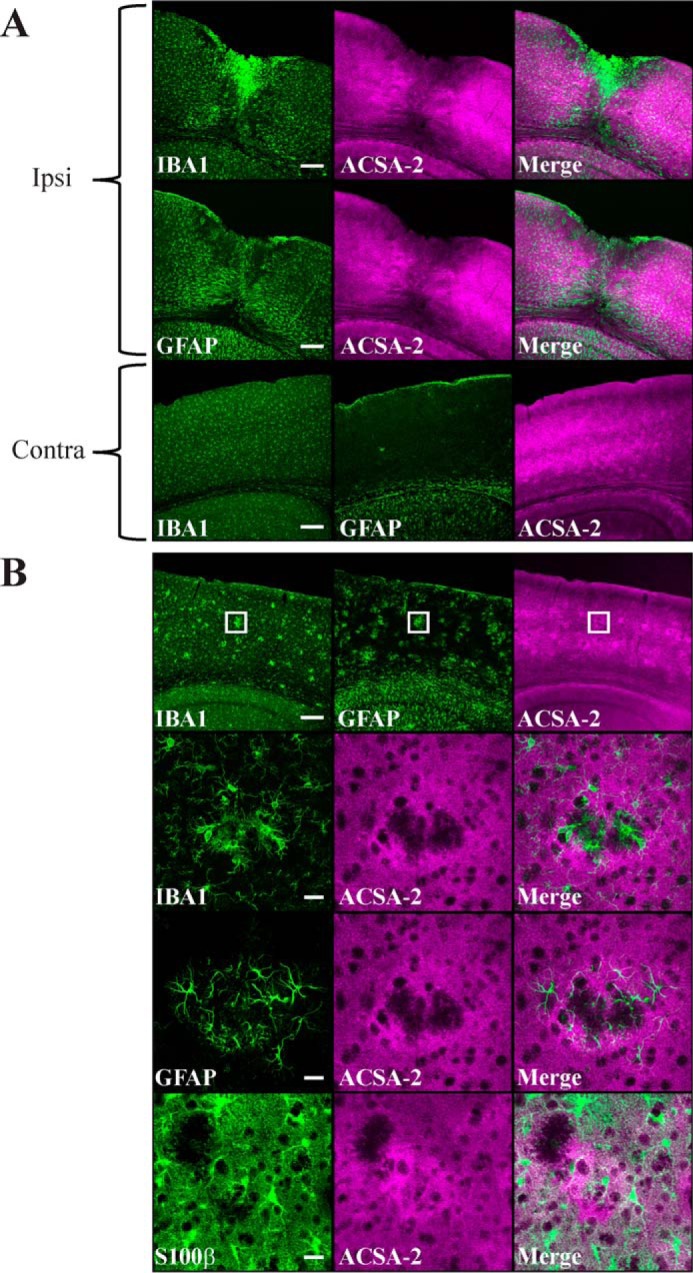
ACSA-2 staining is retained in reactive astrocytes. A, IBA1 (microglia), GFAP (reactive astrocytes), and ACSA-2 staining 5 days after a stab wound injury to the cortex. A low-magnification confocal image shows the pronounced migration of microglia ipsilateral (ipsi) to the injury. Astrocytes in the immediate vicinity of the injury appear to be lost, although those that remain appear to be highly reactive (as judged by up-regulation of GFAP expression throughout the ipsilateral cortex in comparison with the contralateral (contra) side). Note that ACSA-2 staining is still widespread even on reactive astrocytes, including those immediately adjacent to the site of injury. Tissue sections from three animals were analyzed with the same results. Scale bars, 200 μm. B, staining for IBA1, GFAP, S100β (astrocytes), and ACSA-2 in the cortex of an AppNL-G-F knock-in model of Alzheimer's disease at 6 months of age. Animals at this age show clear amyloid plaques in the cortex, which cause typical accumulation of microglia and local reactive astrogliosis, as seen in the low-magnification confocal images (top). Note that the ACSA-2 signal is consistent across the cortex. The boxed region is shown at higher magnification in the bottom panels. Plaques cause local aggregates of microglia to form in the tissue, which exclude astrocytes (as judged using an independent marker for astrocyte cell bodies, S100β). Astrocytes immediately adjacent to plaques show high levels of reactivity (as judged by up-regulation of GFAP expression) but do not lose immunoreactivity for ACSA-2. Tissue sections from two animals were analyzed with the same results. Scale bars, 200 μm (low magnification) and 20 μm (high magnification).
ATP1B2 expression on astrocytes in an Alzheimer's amyloid model
As a model of chronic neurodegenerative disease, we chose to use the AppNL-G-F model of Alzheimer's type amyloidosis. This particular mouse model starts to show cortical amyloidosis at 2 months of age, with heavy deposition of amyloid plaques by 7 months (21). Consistent with the literature, we saw plaques across the cortex at 6 months of age. These plaques could be stained with thioflavin and were always surrounded by activated microglia (as judged by simultaneous immunohistochemistry with IBA1; data not shown). As such, we used the appearance of microglial clusters as a proxy for the presence of local amyloid plaques. As reported previously, the presence of amyloid plaques also resulted in local reactive astrogliosis (as measured by up-regulation of GFAP expression). The widespread astrogliosis shown in Fig. 10B effectively mirrored the deposition of plaques across the cortex. Although astrogliosis is effectively accompanied by the appearance of large regions of ACSA-free signal in the cortex, it is important to note that these areas are filled with clusters of activated microglia. Astrocytes themselves are excluded from these areas (as judged by an independent marker for astrocyte cell bodies, S100β). Importantly, however, ACSA-2 signal was not lost in the reactive astrocytes immediately surrounding microglial clusters.
ATP1B2 expression on astrocytes in response to spinal cord injury, stroke, and bacterial infection
In an attempt to further validate the use of ACSA-2 as a tool to study reactive astrocytes, we analyzed publicly available sets of microarray and RNA-seq data for differential transcript expression in astrocytes following spinal cord injury (22), stroke (middle cerebral artery occlusion), and bacterial infection (peripheral administration of lipopolysaccharide) (4). In all cases, astrocytes isolated from the injury model showed elevated levels of Gfap transcript expression, when compared with appropriate controls. However, levels of Atp1b2 transcript remained constant across all samples. Hence, these experiments suggest that ATP1B2 levels remain relatively constant in reactive astrocytes, making ACSA-2 a viable tool for cell labeling (and isolation).
Discussion
Astrocytes are one of the major cell types in the mammalian brain with a wide range of reported functions.
Long thought to play primarily passive supporting roles in the nervous system (for instance regulating extracellular levels of potassium ions and neurotransmitters), recent evidence has highlighted their importance in the formation, function, and elimination of synapses (23), as well as formation of the blood-brain barrier and control of cerebral blood flow (23). However, despite these advances, our mechanistic understanding of astrocyte development and function, as well as how these cells interact with both neurons and the cerebral vasculature, is still rudimentary. For instance, it is unclear to what degree CNS astrocytes perform the same functions or whether defined astrocyte subtypes are functionally specialized (24, 25). It is also unclear whether astrocytes are differentially sensitive to injury and disease (potentially explaining the strong regional predisposition of these cells to conditions such as Alzheimer's) or how astrocytes respond to different neuropathologies (4, 26). Experimental approaches designed to address these questions include molecular characterization of astrocytes (transcriptome and proteome) following cell isolation (9, 16, 27–29) or reduced complexity cell culture systems to study the role of astrocytes in either blood-brain barrier function or synaptic transmission (3).
The studies listed above have all made use of the direct (prospective) isolation of astrocytes from postnatal mouse brain, given concerns that recovered astrocytes were actually more stem cell-like than bona fide astrocytes. Although FACS-based techniques to sort astrocytes from transgenic animals (expressing fluorescent proteins under the control of cell type-specific promoters) have proved popular (9, 16, 28), they suffer from a number of caveats, including the need to establish and maintain specific mouse colonies and required access to specialized cell-sorting equipment. This is especially true when considering the use of sorting methods to isolate astrocytes from transgenic disease models, which means extra breeding steps and considerations of genetic load on the molecular profile of astrocytes.
An alternative option would be to use labeling of specific cell surface markers for astrocyte isolation (with either FACS or MACS-based methods) (3, 11), which would have the added benefit of also allowing simple, direct isolation of astrocytes from models of CNS injury and disease. To this end, we decided to carry out the first comprehensive characterization of the ACSA-2 antibody. Using a range of evidence, we demonstrate that the principal cell type targeted by ACSA-2 is indeed astrocytes. First, ACSA-2 can be used for isolation of a general population of ultrapure astrocytes from both young and adult mouse brain, which is only possible if it targets a specific plasma membrane protein on these cells (Figs. 1–4). Second, using overexpression and knockdown methods in reduced-complexity tissue culture systems, we clearly identified the unknown ACSA-2 epitope as ATP1B2, which has previously been reported as an astrocyte protein (Figs. 5 and 6). Third, we mapped ATP1B2 localization through the mouse CNS, using a combination of immunoblotting and immunohistochemistry, showing clear localization to the plasma membrane of astrocytes, alongside the known astrocyte marker protein GLT-1 (Figs. 7–9). Finally, we showed ACSA-2/ATP1B2 to be stably expressed in reactive astrocytes across a range of diverse neuropathologies (Fig. 10). This information has been summarized schematically (Fig. 11).
Figure 11.
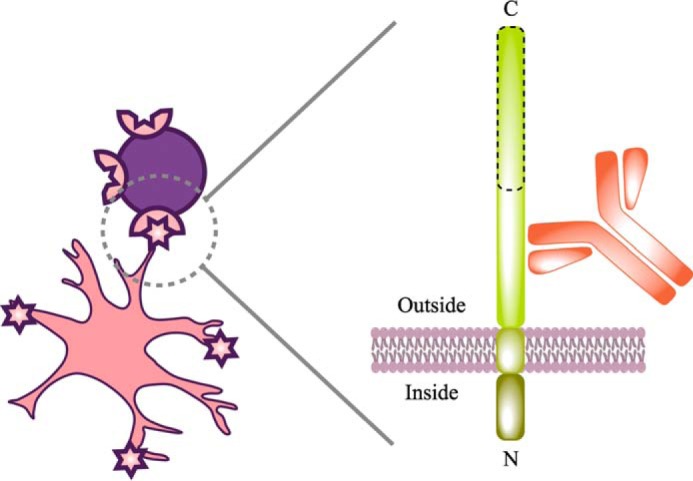
Schematic of cell labeling with ACSA-2. This schematic attempts to summarize our understanding of ACSA-2 binding to astrocytes. Multiple lines of experimental evidence suggest that the target of ACSA-2 is ATP1B2. ATP1B2 is a single-pass transmembrane protein, consisting of a short N-terminal intracellular region (which is protected from binding by ACSA-2 by the shielding effect of the plasma membrane) and a longer extracellular C-terminal domain. Although detailed epitope mapping was not undertaken, immunoblotting experiments, performed immediately after cell isolation, indicate that ATP1B2 is sensitive to papain, because the apparent molecular mass of the protein decreases from 55 to 25 kDa (data not shown). The proteolytically sensitive region is outlined with a dashed line in this schematic. As such, ACSA-2 must be binding ATP1B2 on an intact, membrane-proximal region, as illustrated, to be useful for immunoisolation.
Labeling and immunoisolation of astrocytes using ACSA-2 is possible due to the expression of the protein on the cell body of astrocytes as well as on the processes (which are generally lost during tissue dissociation) (Fig. 9). However, immunoisolation is highly efficient (from both young and old brains), generally yielding 300,000 ultrapure astrocytes per mouse cortex dissociated, with an average viability (post-isolation) of 86%. Interestingly, in our hands, the yield of cells was actually higher than with ACSA-1 purification and immunopanning against integrin β5 (both using young postnatal animals; data not shown).
Although levels of absolute ACSA-2 expression differ widely across the brain, as judged by both Western blotting and immunohistochemistry (Figs. 7 and 8), we have used ACSA-2 to isolate astrocytes from brain regions showing comparatively low levels of expression (cortex and hippocampus), suggesting that even low levels of protein are sufficient for isolation (hippocampal data not shown).
Purity checks (using quantitative RT-PCR and flow cytometry) on isolated astrocytes were highly consistent with each other. Using common cell type-specific markers to measure purity, both techniques showed the preparation to be highly enriched in astrocytes (typically > 95% as judged by flow cytometry). Concomitant with astrocyte enrichment, other cell types were lost. In fact, in our hands, purity was at least equal to ACSA-1 purification and immunopanning against integrin β5. Our ACSA-2-purified samples were largely devoid of contaminating microglia and endothelia. Neurons and oligodendrocytes were both < 2% of the total cells isolated (although additional myelin removal steps were necessary when using adult material, which will obscure the true degree to which ACSA-2 also purifies oligodendrocytes) (see Fig. 1 and “Experimental procedures”). The major source of residual contamination using our standard protocols (although still typically < 4%) was from markers associated with NG2+ cells (particularly in preparations from adult brain, as judged by qPCR) (Figs. 2 and 3). Given that this was confirmed at both the mRNA and protein level for young astrocytes and at the mRNA level for adult astrocytes, we think that it probably represents a genuine result. By traditional criteria, this would indicate a contamination in the sample preparation. However, it is becoming increasingly obvious that cells are defined by multiple (complex) gene signatures (which vary according to developmental time and brain region) (30, 31). In a recent independent single-cell RNA-seq study (19), it was shown that Cspg4 is expressed in a small fraction of astrocytes, confirming our PCR data. Hence, it is entirely possible that a small subpopulation of (adult) astrocytes express both ATP1B2 and CSPG4 (NG2) (31). This would also be the most parsimonious explanation for the small overlap in the proportion of cells immunopositive for the astrocyte glutamate transporter GLAST and NG2 in flow-cytometry experiments (98.9% versus 3.7%, respectively; Fig. 3). The origin and/or function of this population remains enigmatic, because recent work suggests that in the postnatal mouse forebrain and spinal cord, NG2+ cells show restricted lineage potential and are committed to generating only oligodendrocytes (32). However, during development (33) and after certain injuries (e.g. cortical stab wound and cryoinjury) (34, 35), NG2+ cells also contribute to generating astrocytes. Hence, the NG2+ population in our cell preparation may represent a set of cells transitioning along the “NG2+ cell to astrocyte” pathway, potentially acting as a population of “primed cells” ready to respond to CNS damage by terminal differentiation into astrocytes.
One potential limitation of ACSA-2 compared with ACSA-1 is that it can only be used for the labeling and isolation of mouse astrocytes (whereas ACSA-1 can be used for the purification of astrocytes from mouse, rat, and human tissue). However, ACSA-1 (which targets GLAST on the astrocyte) has the major limitation that GLAST levels are reported to be lowered in several rodent models of neuropathology (including Alzheimer's and stroke) (reviewed in Ref. 36), which is a major disadvantage, considering the large role these animal models play in modern biomedical research.
To assess the potential applicability of ACSA-2 in injury models, we conducted a range of experiments in different models of neurological injury and disease because it has been proposed that astrocytes respond with distinct transcriptional changes in response to specific insults (4). Our data from stab wound and spinal cord injury, Alzheimer's type amyloidosis, stroke, and bacterial infection show ATP1B2 levels to be stable across a range of injuries, which all promote reactive astrogliosis (as measured by GFAP up-regulation). Although the data from spinal cord injury, stroke, and bacterial infection were bulk population studies of astrocytes, in our opinion, the manipulations used (cord lesion, middle cerebral artery occlusion, and lipopolysaccharide application) are sufficient to induce widespread astrogliosis in the CNS regions studied, minimizing the risk that loss of ATP1B2 was underreported (4, 22).
Our results stand in contrast to those obtained by Kantzer (37). In this work, Kantzer looked at levels of ATP1B2 expression in a mouse model deficient in ATPase family gene 3-like 2 (Afg3l2), which is essential for integrity of the mitochondrial respiratory chain (38). Kantzer observed loss of ACSA-2 signal in this model, which was interpreted as being due to the actual loss of astrocytes from the tissue. In our minds, however, it is unsurprising that inhibiting ATP production results in such a drastic and unspecific phenotype as cell death (and loss of ACSA signal). In contrast, our more nuanced injury models produced significant astrogliosis without significant loss of astrocytes. Hence, we propose that ACSA-2 will also prove to be a useful tool for labeling and isolating astrocytes from models of CNS injury and disease, although we cannot exclude the possibility that ATP1B2 is lost under certain specific experimental paradigms (meaning that each injury or disease model should be checked using experiments similar to those detailed above).
Although we believe ACSA-2 to be an extremely valuable addition to the toolbox for astrocyte research, there are a number of considerations that need to be taken care of when planning any experiments.
First, one needs to consider the population of astrocytes to be labeled (and isolated). Although our experiments show ATP1B2 to be broadly expressed throughout CNS development (Figs. 7), we cannot entirely exclude the possibility that specific brain regions show variation in ATP1B2 levels over development, which may make them refractory to cell isolation. This is exemplified by reports of ACSA-2 staining in the developing cerebellum (37), in which ATP1B2 expression is positively linked to foliation during aging.
Second, one needs to consider the type of experiments that ACSA-2 will be used for. Whereas ACSA-2 obviously works well for in situ astrocyte labeling and immunoisolation, several lines of evidence indicate that the ACSA-2 antibody is extremely sensitive to the conformation of ATP1B2. First, it does not work well in Western blotting applications (37).8 Attempts to identify the protein target of ACSA-2 based on immunoprecipitation experiments were largely unsuccessful, with only a small fraction (< 10%) of the protein pulled down from tissue extracts (despite screening a range of detergent types and concentrations; data not shown). Second, we observed profound differences when using mild heating for epitope retrieval during immunohistochemistry; ACSA-2 staining decreased considerably, whereas anti-ATP1B2 antibody staining was enhanced (not shown). At this time, we cannot exclude possible changes in glycosylation status during isolation (39), because ATP1B2 is known to have a carbohydrate content of at least 30% (40), which explains why the protein is detected at a much higher weight on immunoblots than predicted from its amino acid sequence (∼55 kDa versus 33.5 kDa), with multiple species visible as smeared bands (as often seen with proteins that are post-translationally modified (41)). Furthermore, ACSA-2 staining is known to be (partially) sensitive to enzymatic removal of carbohydrate groups (37).
We believe this justifies our extensive use of cell-based (HEK293T) overexpression studies to keep proteins in their native conformation (including potentially important glycosylation modifications). These flow-cytometry experiments identified ATP1B2 as the sole target of ACSA-2. Given that ACSA-2 does not stain human samples (untransfected HEK293T cells in Fig. 5 and Miltenyi Biotec data), we discount the possibility that ATP1B2 is actually part of a larger molecular complex in cells, which acts as the final target of the antibody. The discrepancy between the relative GFP and ACSA-2 levels in our overexpression experiments is at present unclear, particularly because GFP expression was driven using an internal ribosome entry site, which is generally less efficient than promoter-driven protein expression (ATP1B2) (42). However, the most parsimonious explanation is that GFP is protected from papain digestion when cells are being recovered from tissue culture plates before staining, whereas a fraction of the total (overexpressed) ATP1B2 present on the cell surface can be cleaved, rendering it undetectable. In any case, the identification of ATP1B2 was validated independently using shRNA-mediated gene silencing in primary astrocytes.
The conformational sensitivity of the antibody is not too surprising, given that the antibody was raised by injection into rats of whole astrocytes isolated by FACS from Gfap-EGFP mice. Interestingly, many of the mRNAs identified in 90% plus of astrocytes encode known plasma membrane proteins (e.g. GLAST and GLT-1); this implies that ATP1B2 is the dominant antigen on the astrocyte.
Interestingly, the findings we report here are entirely consistent with previous work on ATP1B2 (initially referred to as AMOG (adhesion molecule on glia)), suggesting that the protein is an extracellularly exposed membrane glycoprotein (43, 44), showing widespread expression on astrocytes in mouse brain, with particularly high levels in cerebellum (45), and which functions as a cell adhesion molecule with a critical role in neurite outgrowth (40, 46).
In summary, we have identified the ACSA-2 target to be the glycoprotein ATP1B2. We further show that ATP1B2 shows widespread distribution in the brain throughout development, acting as an astrocyte marker. Taken together, these facts make ACSA-2/ATP1B2 an excellent tool for rapid astrocyte labeling and isolation, allowing subsequent in vitro experiments. Furthermore, when combined with additional myelin-removal steps, ACSA-2 allows astrocyte isolation from the adult brain to unparalleled levels of purity. Finally, ATP1B2 expression appears to remain stable during reactive astrogliosis in a variety of acute and chronic conditions. Hence, ACSA-2 appears to be an ideal tool for the rapid and cost-effective one-step purification of mouse astrocytes under a wide range of circumstances, without the need to resort to expensive fluorescently labeled mouse lines and FACS sorting. As such, we think ACSA-2 is likely to become a major tool for astrocyte research.
Experimental procedures
All animal procedures were performed in accordance with the regulations of the institutional animal care and use committee of KU Leuven. Animals were kept in a specific pathogen-free facility with controlled humidity, temperature, and light conditions; water and food were provided ad libitum.
Purification of ultrapure astrocytes from young and adult animals
Astrocyte isolation by MACS
Tissue dissociation
9 mice (either P6–P8 pups or approximately P56 adults) were used per experiment. Mice used in these experiments were on a wild-type CD1 background or were Aldh1l1-EGFP BAC transgenics (maintained as heterozygotes) on a CD1 background. Both males and females were used. Tissue was dissociated using the Neural Tissue Dissociation Kit P (papain) (Miltenyi Biotec) with slight modifications of the manufacturer's instructions (except when samples were used for GLAST staining, in which case the Neural Tissue Dissociation Kit T (trypsin) (Miltenyi Biotec) was used). Following enzymatic digestion, cells were released using mechanical trituration with 10-ml serological pipettes (3 rounds of 10 strokes each). Freshly dissociated cells were then passed through a 20-μm Nitex filter (SEFAR) to remove remaining tissue clumps. Myelin removal using equilibrium density centrifugation was then performed. Basically, 90% Percoll PLUS (Life Sciences) in 1× Hanks' balanced salt solution (HBSS) with calcium and magnesium (Sigma) was added to the cell suspension to give a final Percoll concentration of 24%. DNase I (Worthington) was then added (1250 units per 10 ml of suspension), and the suspension was mixed and then spun down at 300 × gav for 11 min at room temperature in a Hettich 320R universal centrifuge (with minimal brake). The cell-containing pellet was resuspended in 0.5% BSA (Sigma) in PBS without calcium and magnesium (Thermo Fisher Scientific). The P7 cell suspension was processed directly for astrocyte isolation, whereas the P56 suspension was subjected to additional myelin removal.
Additional myelin removal
An additional myelin removal step was crucial when using material from P56 animals. Myelin Removal Beads II (Miltenyi Biotec) were used according to the manufacturer's instructions (usually with LS magnetic columns (Miltenyi Biotec)). However, when more stringent conditions were required (e.g. during some control experiments for flow cytometry), LD magnetic columns were used (Miltenyi Biotec). The flow-through was collected and used for subsequent astrocyte isolation.
Astrocyte isolation
The ACSA-2 kit (Miltenyi Biotec) was used for positive selection of astrocytes according to the standard protocol, using two runs of enrichment on consecutive MS columns (Miltenyi Biotec). The purification procedure is summarized in schematic form in Fig. 1.
Purity testing of astrocytes
qPCR
RNA isolation and reverse transcription
RNA from cells was isolated using a RNeasy Plus Micro kit (Qiagen), and cDNA was made using a Moloney murine leukemia virus reverse transcriptase kit (Life Technologies). Both kits were used according to the manufacturer's instructions.
Primers
Primers were custom-designed to span exon-exon junctions with a final product size of 70–130 base pairs. All primers were synthesized by Integrated DNA Technologies.
PCR procedure
Quantitative PCR was performed using the LightCycler FastStart DNA MasterPLUS SYBR Green I kit (Roche Applied Science) on a LightCycler 480 Real-Time PCR System (Roche Applied Science). The efficiency of primer amplification was tested before experiments, using standard methods of serial dilution and linear regression analysis. Only primer pairs with a single peak on the melting curve and amplification efficiency of 90–110% were used for further analysis (Table 1). Two housekeeping genes, Tbp (TATA box-binding protein) and Cyc1 (cytochrome c1), were used for normalization. Relative quantification was done as described previously (47).
Flow cytometry
Antibody staining
Antibodies against a range of cell type-specific markers were used in our experiments (Table 2). All antibodies were used at company-recommended dilutions, except for anti-O1-eFluor660, which was used at a dilution of 1:80. In general, all cells were stained directly after isolation, except when anti-NG2 was used. In the case of NG2 staining, cells were allowed to recover in MACS Neuro Medium (Miltenyi Biotec) supplemented with B27 (Thermo Fisher Scientific) for 3 h at 37 °C with gentle nutation. Because THY1.2 was reported to be present on astrocytes as well as neurons (28), staining was performed using a preparation of astrocytes isolated from the Aldh1l1-EGFP mouse line, to distinguish between the two cell types.
Flow cytometry procedure
Flow cytometry was performed on a BD FACSCanto I (BD Biosciences), controlled using FACSDiva software (version 6.1.3). Compensations were done using single-stained controls and setting the median fluorescence of positive and negative populations at the same level. Gates were set on unstained controls or FMO (fluorescence minus one) controls. In the case of O4- and O1-stained samples, gates were set on astrocytes isolated using very stringent myelin depletion (see above). Debris and cell clumps were discriminated using forward scattering (FSC) and side scattering (SSC) gating. Flow experiments were performed on live, non-fixed cells (with the dye 7-AAD dye being used to discriminate dead cells).
Identification of ATP1B2 as the ACSA-2 epitope
Bioinformatics analysis
To identify genes that are exclusively expressed in at least 90% of astrocytes, we downloaded the raw mRNA count data published in the single-cell survey of mouse hippocampus and somatosensory cortex from the Linnarsson laboratory (19). This data set is publicly available.
A normalization step to reads per million was applied, correcting for the library size. A template vector was then created containing a 1 for each cell classified as an astrocyte by Zeisel et al. (19) and a 0 for all other cells (48). Using this template vector, the Pearson correlation was calculated for the normalized reads per million values of each gene across all cells. This results in a list where genes that are consistently expressed in astrocytes and not (or to a lesser extent) in non-astrocytic cells rank high. This ranked list was augmented with the mean expression value of each individual gene in astrocytes and non-astrocytes as well as the log -fold change between these. Additionally, UniprotKB was mined to identify genes in which the encoded protein product contains a transmembrane region (KW-1133) and associates with the cell membrane (KW-1003). According to these criteria, we identified putative astrocyte-specific plasma membrane proteins, which were used for further testing (and which are summarized in Table 3).
Cloning of identified genes
RNA isolation
RNA from purified cells was isolated using the RNeasy Plus Micro kit (Qiagen), according to the manufacturer's instructions. Total recovered RNA was measured on a NanoDrop 1000 (Thermo Fisher Scientific).
Reverse transcription
RNA was reverse-transcribed using Moloney murine leukemia virus reverse transcriptase (Life Technologies), according to the manufacturer's instructions.
Amplification of specific ORFs
PCR primers were designed to flank ORFs of genes of interest as well as to incorporate restriction sites on both ends (see Table 3). PCR was performed using Q5 Hot Start high-fidelity DNA polymerase (New England Biolabs), according to the manufacturer's protocol. PCR products were then cleaned up using the QIAquick PCR purification kit (Qiagen).
Restriction digestion of PCR-amplified ORFs and gel purification
Amplicons were digested using appropriate restriction enzymes (Table 3). Digested products were then resolved using electrophoresis on a 1% agarose gel. Bands were visualized using Midori Green (NIPPON Genetics) and cut out under blue light trans-illumination. DNA was purified from gel pieces using the Zymoclean Gel DNA recovery kit (Zymo Research), using the recommended protocol.
Ligation into the pCAGIG expression vector
The pCAGIG vector was obtained from Addgene and was cut using appropriate restriction enzymes (Table 3). Ligation with amplicons was performed using T4 ligase (Promega), according to standard protocols. Amplicons were mixed with linearized vector in a 3:1 molar ratio (not exceeding 10 ng/μl DNA concentration). The mix was incubated at 4 °C overnight followed by 70 °C for 10 min.
Plasmid amplification, purification, and sequencing
Plasmids were amplified in Top10 cells using standard methods. Plasmids were then subsequently purified using a QIAprep Spin Miniprep kit (Qiagen). Cloned ORFs were sequenced and verified against their PubMed entry (Table 3). Single-nucleotide polymorphisms that did not change the amino acid sequence were tolerated.
Overexpression of ORF-encoding plasmids, ACSA-2 staining, and flow cytometry
Heterologous expression of identified genes in a HEK293T system
HEK293T (human embryonic kidney) cells (sourced from ATCC) were maintained according to standard protocols (49) and were mycoplasma-free. One day before transfection with plasmids, cells were reseeded and plated at a density of 6.25 × 105 cells/well in a 6-well plate. Cells were then transfected using Lipofectamine LTX reagent (Thermo Fisher Scientific), according to the manufacturer's instructions. After 4–6 h, Lipofectamine was removed and replaced with DMEM-based growth medium. Cells were left for 48 h (with a further medium change after 24 h) before experiments.
Staining with ACSA-2-APC conjugate
Transiently transfected HEK293T cells were recovered from the 6-well plates using gentle enzymatic digestion (Neural Tissue Dissociation Kit P, Miltenyi Biotec) adapted to use a tenth of the recommended amount of papain. Enzyme was inactivated using 10 volumes of HBSS with calcium and magnesium (Sigma). Cells were recovered by low-speed centrifugation and then labeled with ACSA-2-APC (Miltenyi Biotec), according to the manufacturer's instructions. The vital dye 7-AAD (eBioscience) was added to the final cell suspension to allow selection (based on viability) in flow cytometry.
Flow cytometry
Flow cytometry was performed on a BD FACSCanto I (BD Biosciences) controlled using FACSDiva software (version 6.1.3). All gatings were set based on unstained and FMO controls. Compensations were set based on single-color controls. Debris and cell clumps were initially gated out on the basis of FSC and SSC plots, allowing selection of only the population of interest. Further doublets were gated out using FSC-width/FSC-area plots. Flow cytometry was performed on live, non-fixed cells (with the dye 7-AAD being used to discriminate dead cells). Data from a minimum of 50,000 live cells were collected for each construct tested. Data were plotted to show the distribution of GFP- and ACSA-2-positive cells.
Primary astrocyte cultures, ATP1B2 knockdown, qPCR, and flow cytometry
Primary astrocyte cultures
Primary cultures were prepared as described previously (50). Essentially, CD1 mouse pups (postnatal days 1 and 2) were sacrificed using cervical dislocation, and brains were removed, allowing isolation of cortical material. Cortices were dissociated using the papain-based neural tissue dissociation kit (P) (Miltenyi Biotec) with slight modifications of the manufacturer's instructions. Following enzymatic digestion of the extracellular matrix, cells were released using mechanical trituration. Dissociated cells were then passed through a 20-μm Nitex filter (SEFAR) to remove tissue clumps. Cells were seeded into a tissue culture plate and maintained for 7–14 days in a DMEM-based medium at 37 °C in a standard incubator. Following sufficient astrocyte stratification, cultures were washed three times with PBS to remove contaminating oligodendrocytes and microglia. Cultures were then maintained in standard conditions for a further 24 h before use.
ATP1B2 knockdown
Primary astrocytes were recovered using 0.05% trypsin-EDTA (Gibco). Cells in suspension were recovered using low-speed centrifugation and resuspended in PBS (without calcium or magnesium) (Thermo Fisher Scientific). Cells were then transfected with shRNA constructs (Table 4) using the Neon electroporation system (100-μl tips; Thermo Fisher Scientific), according to the manufacturer's instructions. The best transfection efficiency was obtained using the following parameters: 1300-V pulses of 10-ms duration repeated three times. Following transfection, cells were added to DMEM-based medium and plated onto tissue culture dishes. Seven days were allowed for robust shRNA expression, mRNA degradation, and protein turnover.
qPCR
Cells were recovered from plates using trypsin-EDTA (as above). shRNA-transfected cells were then sorted (based on GFP expression) using a BD FACSARIA III sorter, equipped with a 100-μm nozzle. RNA isolation, reverse transcription, and SYBR Green-based qPCR were then performed as described (see above). Primers used in qPCR are listed in Table 1. Relative quantification was done using Tbp and Cyc1 for normalization. A Shapiro-Wilk test was used to test for normal distribution of data. This was followed by an F-test to assess the equality of variances between experimental groups. Finally, a two-tailed t test (two-sample assuming equal variances) was used to judge whether any differences were statistically significant.
Flow cytometry
Cells were recovered from plates using gentle enzymatic digestion with trypsin (as above), to avoid proteolytic cleavage of the astrocyte glutamate transporter GLAST. Cells were then stained with ACSA-2-APC or anti-GLAST-APC and used in flow cytometry experiments (using protocols described above). Flow cytometry was run on live (7-AAD-negative), non-fixed cells using a FACSCanto I (as described). The number of cells positive or negative for ACSA-2 or GLAST was expressed as the percentage of total transfected (GFP+) cells. Following data acquisition, statistical analysis was performed as described for gene expression levels following shRNA-mediated knockdown.
Western blotting
Isolation of defined brain regions
Brains were harvested from anesthetized C57BL/6 mice aged to postnatal day 56 and were then sliced into 2-mm-thick coronal sections. Individual brain regions were identified in sections and dissected out. Regions from different slices were pooled when appropriate. Samples were then stored at −80 °C until use.
Developmental profiling
Embryos were removed from the uterus of anesthetized C57BL/6 mice. In the case of E9.5 mice, the whole head was taken. For embryos at later developmental stages, the brain was dissected out under a stereomicroscope. Postnatal animals were killed by cervical dislocation (with adults first having been anesthetized with nembutal), and tissue was recovered. Samples were stored at −80 °C until use.
Sample preparation
All samples were homogenized using a motor-driven glass Teflon homogenizer at a speed of 900 rpm. The homogenization buffer was ice-cold sucrose (320 mm sucrose, 4 mm HEPES (pH 7.4; NaOH)) supplemented with the protease inhibitors pepstatin and PMSF (51). After homogenization, samples were passed through 20- and 27-gauge needles to remove residual clumps of tissue. Protein determination was done using a modified Lowry assay (52) with BSA as a standard.
SDS-PAGE and Western blotting
SDS-PAGE was carried out with a Tricine-based buffer system to ensure maximum resolution in the molecular weight range of interest (53). Immunoblotting was carried out using standard semi-dry electrophoretic techniques (54). Ponceau S was used post-transfer to check for equal protein loading across lanes. Membranes were then blocked and probed with antibodies (using standard protocols). A specific anti-ATP1B2 antibody (rabbit monoclonal; Abcam, ab185207; lot GR157624-1) was used at a dilution of 1:10,000. This antibody was validated by Abcam using a peptide competition assay, in which a peptide corresponding to a C-terminal fragment of ATP1B2 was preincubated with the antibody before use in Western blotting. Peroxidase-labeled secondary antibodies were from Jackson ImmunoResearch Laboratories and were used at a final dilution of 1:10,000. Detection was performed by enhanced chemiluminescence with a LAS reader (Fuji). Images were further processed using ImageJ (National Institutes of Health). Figures were prepared using Adobe Photoshop CS4 and Adobe Illustrator CS4.
Immunohistochemistry
Tissue fixation and slice preparation
P21 animals were used for immunohistochemistry. Animals were transcardially perfused with 4% paraformaldehyde. Brains were then removed and post-fixed overnight in 4% paraformaldehyde. Vibratome sections of 50-μm thickness were cut using a Leica VT1000S.
Antibody staining
Before staining, sections were generally blocked for 1 h at room temperature in 0.2% Triton X-100/PBS supplemented with 10% normal goat serum, although in a limited number of cases (Fig. 9), sections were blocked in 0.2% Triton X-100/PBS supplemented with 10% donkey serum. Sections were incubated with primary antibodies (diluted in 0.2% Triton X-100/PBS supplemented with 1% normal goat serum or 1% donkey serum) overnight at 4 °C. Sections were then given three 10-min washes in PBS before incubation with secondary antibodies (diluted in 0.2% Triton X-100/PBS) for 2–3 h at room temperature. Sections were then given three 10-min washes in PBS, before mounting using Vectashield (Vector Laboratories). Widefield images were taken using a Zeiss Slide scanner (Axio Scan Z1) with a PL APO ×20/0.8 objective, controlled using Zen 2 data acquisition software. Confocal images were taken using a Leica SP8 microscope with an HC PL APO CS2 ×40/1.30 objective (Fig. 8) or with an HC PL APO CS2 ×63/1.40 objective with zoom setting at 2 (Fig. 9A) or at 4 (Fig. 9B). The Leica SP8 microscope was controlled using Las X version 2.0 data acquisition software. Images were then imported into ImageJ (National Institutes of Health) and adjusted for brightness and contrast before saving in a (non-compressed) TIFF file format. Final figures were prepared using Adobe Photoshop CS6. Images are representative of observations made in at least two animals.
The specific primary antibodies used for staining include rat monoclonal ACSA-2 (1:200; Miltenyi Biotec), guinea pig anti-GFP (1:300; Synaptic Systems, 132004), rabbit anti-GFP (1:300; Synaptic Systems, 132002), rabbit anti-ATP1B2 (1:200; Abcam, Ab185207), guinea pig anti-GLT-1 (1:200; EMD Millipore, AB1783) and rabbit anti-MAP-2 (1:300; Synaptic Systems, 188003).
Secondary antibodies were initially raised in goat or donkey. Goat antibodies include anti-rat Alexa Fluor 488 (1:600; Abcam, ab150157), anti-rat Alexa Fluor 647 (1:600; Jackson ImmunoResearch Laboratories, 112-605-143), anti-guinea pig Alexa Fluor 488 (1:600; Jackson ImmunoResearch Laboratories, 106-545-003), and anti-rabbit Cy3 (1:600; Jackson ImmunoResearch Laboratories, 111-165-003).
Donkey antibodies include anti-guinea pig Alexa Fluor 488 (1:200; Jackson ImmunoResearch Laboratories, 706-545-148), anti-rabbit CY3 (1:200; Jackson ImmunoResearch Laboratories, 711-165-152), anti-rat Alexa Fluor 647 (1:200; Jackson ImmunoResearch Laboratories, 712-605-153), and anti-rabbit 488 (1:600; Invitrogen, A21206).
Expression of ACSA-2/ATP1B2 in mouse models of acute and chronic injury
Stab wound injury
To create a stab wound injury in the mouse visual cortex, 8-week-old C57BL/6J animals were used. Animals were anesthetized with 5% inhaled isoflurane in oxygen for induction and maintained with 1.5–2% isoflurane in oxygen during surgery. Animals were placed in a stereotaxic frame (Harvard Apparatus), the head was shaved, and the skin was disinfected with alcohol. Body temperature was maintained at 37 °C throughout the surgical procedure using a heating pad. A midline incision was made through the scalp, and the skin was retracted. A hole was made over the left cerebral hemisphere using a drill until the dura was exposed. A 30-gauge needle (Hamilton Neuro Syringes, model 7002 KH) was inserted at the following coordinates: M/L, +2.0; A/P, −2.3; D/V, −1.8. The needle was left in place for 1 min and then slowly retracted over a 1-min period. The skin incision was closed with sutures, antibiotic cream (2% fucidin) was applied to the wound, and animals were allowed to recover in their cage. Animals were perfused 5 days after injury (see above for details of the perfusion procedure), because it has been previously reported that astrocyte reactivity after stab wound injury peaks at this time (18).
Alzheimer's disease model
Experiments were done in 6-month-old AppNL-G-F animals (C57BL/6J background). This mouse line has been reported to have heavy deposits of amyloid β-peptide in cortical areas at this age (21).
Immunohistochemistry
Animals were perfused, and vibratome sections were produced (as described above). The specific primary antibodies used for staining include rat ACSA-2 (1:200; Miltenyi Biotec), rabbit anti-IBA1 (1:600; Synaptic Systems, 234003), and guinea pig anti-GFAP (1:300; Synaptic Systems, 173004). The secondary antibodies include donkey anti-rabbit 488 (Invitrogen, A21206), donkey anti-guinea pig Cy3 (Jackson ImmunoResearch Laboratories, 706-165-148), and donkey anti-rat 647 (Jackson ImmunoResearch Laboratories, 712-605-153). All secondary antibodies were used at a dilution of 1:600. Images were acquired using a Leica SP8 confocal microscope. Low-magnification images (Fig. 10A) were taken using a HCX PL APO CS ×10/0.40 objective using a zoom setting of 0.75. High-magnification images (Fig. 10B) were taken using an HC PL APO CS2 ×63/1.40 oil objective. Images were then imported into ImageJ (National Institutes of Health) and adjusted for brightness and contrast before saving in a (non-compressed) TIFF file format. Final figures were prepared using Adobe Photoshop CS6. Images are representative of observations made in at least two animals.
Bioinformatics analysis of reactive astrocytes after stroke, bacterial infection, and spinal cord injury
The expression of Atp1b2 and Gfap mRNA in reactive astrocytes after MCAO and LPS treatments was analyzed using previously published microarray data (4). The normalized expression of the Affymetrix Mouse Genome 430 version 2.0 Array probes corresponding to Atp1b2 (1435148_at and 1422009_at) and Gfap (1440142_s_at, 1426508_at, and 1426509_s_at) were analyzed using the GEO2R web application. The expression of Atp1b2 and Gfap mRNA in reactive astrocytes following spinal cord injury was based on previously published RNA-seq data (22) following GEO expression analysis (see above and GEO accession number GSE76097).
Author contributions
M. G. H. conceived and supervised the study. M. F. and T. G. B. performed single-cell RNA-seq data analysis. M. Y. B. performed experiments described in Figs. 1–6. F. d. V. contributed work to Figs. 8–10. S. I. D. performed part of the work in Fig. 10. C. L. performed the experiments for Fig. 7. T. Saito and T. Saido produced the AppNL-G-F model of Alzheimer's type amyloidosis used in Fig. 10. All authors contributed to data analysis and manuscript writing.
Acknowledgments
The Leica SP8 confocal microscope used in this study was provided by Inframouse (VIB-KU Leuven), through a Hercules type 3 grant (ZW09-03). We thank Dr. Enrico Radaelli for help with dissections of brains and Dr. Carlos Dotti for advice and critical reading of the manuscript. M. G. H. thanks Dr. Ben Barres for generous support, advice, and the gift of several reagents during the early phase of this work. M. G. H. and F. d. V. also acknowledge Ian Hutchinson, Bruce Anstey, and John McGuinness. Artwork for Fig. 1 was made by David Pennington (Pennington Art).
The authors declare that they have no conflicts of interest with the contents of this article.
M. Y. Batiuk and M. G. Holt, unpublished data.
- MACS
- magnetic-activated cell sorting
- PE
- phycoerythrin
- qPCR
- quantitative PCR
- E
- embryonic day
- P
- postnatal day
- RNA-seq
- RNA-sequencing
- HBSS
- Hanks' balanced salt solution
- FSC
- forward scattering
- SSC
- side scattering
- 7-AAD
- 7-aminoactinomycin D
- APC
- allophycocyanin
- EGFP
- enhanced GFP
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- FMO
- fluorescence minus one.
References
- 1. Barres B. A. (2008) The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440 [DOI] [PubMed] [Google Scholar]
- 2. McCarthy K. D., and de Vellis J. (1980) Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J. Cell Biol. 85, 890–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Foo L. C., Allen N. J., Bushong E. A., Ventura P. B., Chung W. S., Zhou L., Cahoy J. D., Daneman R., Zong H., Ellisman M. H., and Barres B. A. (2011) Development of a method for the purification and culture of rodent astrocytes. Neuron 71, 799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zamanian J. L., Xu L., Foo L. C., Nouri N., Zhou L., Giffard R. G., and Barres B. A. (2012) Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doetsch F., Caillé I., Lim D. A., García-Verdugo J. M., and Alvarez-Buylla A. (1999) Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97, 703–716 [DOI] [PubMed] [Google Scholar]
- 6. Haas R. J., Werner J., and Fliedner T. M. (1970) Cytokinetics of neonatal brain cell development in rats as studied by the “complete 3H-thymidine labelling” method. J. Anat. 107, 421–437 [PMC free article] [PubMed] [Google Scholar]
- 7. Skoff R. P., and Knapp P. E. (1991) Division of astroblasts and oligodendroblasts in postnatal rodent brain: evidence for separate astrocyte and oligodendrocyte lineages. Glia 4, 165–174 [DOI] [PubMed] [Google Scholar]
- 8. Bardehle S., Krüger M., Buggenthin F., Schwausch J., Ninkovic J., Clevers H., Snippert H. J., Theis F. J., Meyer-Luehmann M., Bechmann I., Dimou L., and Götz M. (2013) Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat. Neurosci. 16, 580–586 [DOI] [PubMed] [Google Scholar]
- 9. Cahoy J. D., Emery B., Kaushal A., Foo L. C., Zamanian J. L., Christopherson K. S., Xing Y., Lubischer J. L., Krieg P. A., Krupenko S. A., Thompson W. J., and Barres B. A. (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wilhelm A., Volknandt W., Langer D., Nolte C., Kettenmann H., and Zimmermann H. (2004) Localization of SNARE proteins and secretory organelle proteins in astrocytes in vitro and in situ. Neurosci. Res. 48, 249–257 [DOI] [PubMed] [Google Scholar]
- 11. Jungblut M., Tiveron M. C., Barral S., Abrahamsen B., Knöbel S., Pennartz S., Schmitz J., Perraut M., Pfrieger F. W., Stoffel W., Cremer H., and Bosio A. (2012) Isolation and characterization of living primary astroglial cells using the new GLAST-specific monoclonal antibody ACSA-1. Glia 60, 894–907 [DOI] [PubMed] [Google Scholar]
- 12. Schubert V., Bouvier D., and Volterra A. (2011) SNARE protein expression in synaptic terminals and astrocytes in the adult hippocampus: a comparative analysis. Glia 59, 1472–1488 [DOI] [PubMed] [Google Scholar]
- 13. Nolte C., Matyash M., Pivneva T., Schipke C. G., Ohlemeyer C., Hanisch U. K., Kirchhoff F., and Kettenmann H. (2001) GFAP promoter-controlled EGFP-expressing transgenic mice: a tool to visualize astrocytes and astrogliosis in living brain tissue. Glia 33, 72–86 [PubMed] [Google Scholar]
- 14. Zuo Y., Lubischer J. L., Kang H., Tian L., Mikesh M., Marks A., Scofield V. L., Maika S., Newman C., Krieg P., and Thompson W. J. (2004) Fluorescent proteins expressed in mouse transgenic lines mark subsets of glia, neurons, macrophages, and dendritic cells for vital examination. J. Neurosci. 24, 10999–11009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Regan M. R., Huang Y. H., Kim Y. S., Dykes-Hoberg M. I., Jin L., Watkins A. M., Bergles D. E., and Rothstein J. D. (2007) Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J. Neurosci. 27, 6607–6619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang Y., Vidensky S., Jin L., Jie C., Lorenzini I., Frankl M., and Rothstein J. D. (2011) Molecular comparison of GLT1+ and ALDH1L1+ astrocytes in vivo in astroglial reporter mice. Glia 59, 200–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huettner J. E., and Baughman R. W. (1986) Primary culture of identified neurons from the visual cortex of postnatal rats. J. Neurosci. 6, 3044–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sirko S., Irmler M., Gascón S., Bek S., Schneider S., Dimou L., Obermann J., De Souza Paiva D., Poirier F., Beckers J., Hauck S. M., Barde Y. A., and Götz M. (2015) Astrocyte reactivity after brain injury: the role of galectins 1 and 3. Glia 63, 2340–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zeisel A., Muñoz-Manchado A. B., Codeluppi S., Lönnerberg P., La Manno G., Juréus A., Marques S., Munguba H., He L., Betsholtz C., Rolny C., Castelo-Branco G., Hjerling-Leffler J., and Linnarsson S. (2015) Brain structure: cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 [DOI] [PubMed] [Google Scholar]
- 20. Voutsinos-Porche B., Knott G., Tanaka K., Quairiaux C., Welker E., and Bonvento G. (2003) Glial glutamate transporters and maturation of the mouse somatosensory cortex. Cereb. Cortex 13, 1110–1121 [DOI] [PubMed] [Google Scholar]
- 21. Saito T., Matsuba Y., Mihira N., Takano J., Nilsson P., Itohara S., Iwata N., and Saido T. C. (2014) Single App knock-in mouse models of Alzheimer's disease. Nat. Neurosci. 17, 661–663 [DOI] [PubMed] [Google Scholar]
- 22. Anderson M. A., Burda J. E., Ren Y., Ao Y., O'Shea T. M., Kawaguchi R., Coppola G., Khakh B. S., Deming T. J., and Sofroniew M. V. (2016) Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Y., and Barres B. A. (2010) Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr. Opin. Neurobiol. 20, 588–594 [DOI] [PubMed] [Google Scholar]
- 24. Molofsky A. V., Kelley K. W., Tsai H. H., Redmond S. A., Chang S. M., Madireddy L., Chan J. R., Baranzini S. E., Ullian E. M., and Rowitch D. H. (2014) Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature 509, 189–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Doyle J. P., Dougherty J. D., Heiman M., Schmidt E. F., Stevens T. R., Ma G., Bupp S., Shrestha P., Shah R. D., Doughty M. L., Gong S., Greengard P., and Heintz N. (2008) Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 135, 749–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rodríguez-Arellano J. J., Parpura V., Zorec R., and Verkhratsky A. (2016) Astrocytes in physiological aging and Alzheimer's disease. Neuroscience 323, 170–182 [DOI] [PubMed] [Google Scholar]
- 27. Grosche A., Hauser A., Lepper M. F., Mayo R., von Toerne C., Merl-Pham J., and Hauck S. M. (2016) The proteome of native adult Muller glial cells from murine retina. Mol. Cell Proteomics 15, 462–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lovatt D., Sonnewald U., Waagepetersen H. S., Schousboe A., He W., Lin J. H., Han X., Takano T., Wang S., Sim F. J., Goldman S. A., and Nedergaard M. (2007) The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J. Neurosci. 27, 12255–12266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sharma K., Schmitt S., Bergner C. G., Tyanova S., Kannaiyan N., Manrique-Hoyos N., Kongi K., Cantuti L., Hanisch U. K., Philips M. A., Rossner M. J., Mann M., and Simons M. (2015) Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 18, 1819–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Beckervordersandforth R., Tripathi P., Ninkovic J., Bayam E., Lepier A., Stempfhuber B., Kirchhoff F., Hirrlinger J., Haslinger A., Lie D. C., Beckers J., Yoder B., Irmler M., and Götz M. (2010) In vivo fate mapping and expression analysis reveals molecular hallmarks of prospectively isolated adult neural stem cells. Cell Stem Cell 7, 744–758 [DOI] [PubMed] [Google Scholar]
- 31. Zhang Y., Chen K., Sloan S. A., Bennett M. L., Scholze A. R., O'Keeffe S., Phatnani H. P., Guarnieri P., Caneda C., Ruderisch N., Deng S., Liddelow S. A., Zhang C., Daneman R., Maniatis T., et al. (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kang S. H., Fukaya M., Yang J. K., Rothstein J. D., and Bergles D. E. (2010) NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68, 668–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang W., Zhao N., Bai X., Karram K., Trotter J., Goebbels S., Scheller A., and Kirchhoff F. (2014) Novel NG2-CreERT2 knock-in mice demonstrate heterogeneous differentiation potential of NG2 glia during development. Glia 62, 896–913 [DOI] [PubMed] [Google Scholar]
- 34. Hampton D. W., Asher R. A., Kondo T., Steeves J. D., Ramer M. S., and Fawcett J. W. (2007) A potential role for bone morphogenetic protein signalling in glial cell fate determination following adult central nervous system injury in vivo. Eur. J. Neurosci. 26, 3024–3035 [DOI] [PubMed] [Google Scholar]
- 35. Tatsumi K., Takebayashi H., Manabe T., Tanaka K. F., Makinodan M., Yamauchi T., Makinodan E., Matsuyoshi H., Okuda H., Ikenaka K., and Wanaka A. (2008) Genetic fate mapping of Olig2 progenitors in the injured adult cerebral cortex reveals preferential differentiation into astrocytes. J. Neurosci. Res. 86, 3494–3502 [DOI] [PubMed] [Google Scholar]
- 36. Maragakis N. J., and Rothstein J. D. (2004) Glutamate transporters: animal models to neurologic disease. Neurobiol. Dis. 15, 461–473 [DOI] [PubMed] [Google Scholar]
- 37. Kantzer C. G. (2015) Astrocyte Cell Surface Marker Phenotyping: Identification of Multipotent ACSA-2−/GLAST+ Cerebellar Progenitor Cells, Ph.D. thesis, University of Cologne [Google Scholar]
- 38. Maltecca F., Magnoni R., Cerri F., Cox G. A., Quattrini A., and Casari G. (2009) Haploinsufficiency of AFG3L2, the gene responsible for spinocerebellar ataxia type 28, causes mitochondria-mediated Purkinje cell dark degeneration. J. Neurosci. 29, 9244–9254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. He P., Grotzke J. E., Ng B. G., Gunel M., Jafar-Nejad H., Cresswell P., Enns G. M., and Freeze H. H. (2015) A congenital disorder of deglycosylation: biochemical characterization of N-glycanase 1 deficiency in patient fibroblasts. Glycobiology 25, 836–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Antonicek H., Persohn E., and Schachner M. (1987) Biochemical and functional characterization of a novel neuron-glia adhesion molecule that is involved in neuronal migration. J. Cell Biol. 104, 1587–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. White J. K., Stewart A., Popoff J. F., Wilson S., and Blackwell J. M. (2004) Incomplete glycosylation and defective intracellular targeting of mutant solute carrier family 11 member 1 (Slc11a1). Biochem. J. 382, 811–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bouabe H., Fässler R., and Heesemann J. (2008) Improvement of reporter activity by IRES-mediated polycistronic reporter system. Nucleic Acids Res. 36, e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gloor S., Antonicek H., Sweadner K. J., Pagliusi S., Frank R., Moos M., and Schachner M. (1990) The adhesion molecule on glia (AMOG) is a homologue of the beta subunit of the Na,K-ATPase. J. Cell Biol. 110, 165–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gloor S., Nasse K., Essen L. O., and Appel F. (1992) Production and secretion in CHO cells of the extracellular domain of AMOG/β2, a type-II membrane protein. Gene 120, 307–312 [DOI] [PubMed] [Google Scholar]
- 45. Pagliusi S. R., Schachner M., Seeburg P. H., and Shivers B. D. (1990) The adhesion molecule on glia (AMOG) is widely expressed by astrocytes in developing and adult mouse brain. Eur. J. Neurosci. 2, 471–480 [DOI] [PubMed] [Google Scholar]
- 46. Müller-Husmann G., Gloor S., and Schachner M. (1993) Functional characterization of β isoforms of murine Na,K-ATPase: the adhesion molecule on glia (AMOG/β2), but not β1, promotes neurite outgrowth. J. Biol. Chem. 268, 26260–26267 [PubMed] [Google Scholar]
- 47. Hellemans J., Mortier G., De Paepe A., Speleman F., and Vandesompele J. (2007) qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 8, R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pavlidis P., and Noble W. S. (2001) Analysis of strain and regional variation in gene expression in mouse brain. Genome Biol. 2, RESEARCH0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Holt M., Varoqueaux F., Wiederhold K., Takamori S., Urlaub H., Fasshauer D., and Jahn R. (2006) Identification of SNAP-47, a novel Qbc-SNARE with ubiquitous expression. J. Biol. Chem. 281, 17076–17083 [DOI] [PubMed] [Google Scholar]
- 50. Slezak M., Korostynski M., Gieryk A., Golda S., Dzbek J., Piechota M., Wlazlo E., Bilecki W., and Przewlocki R. (2013) Astrocytes are a neural target of morphine action via glucocorticoid receptor-dependent signaling. Glia 61, 623–635 [DOI] [PubMed] [Google Scholar]
- 51. Holt M., Riedel D., Stein A., Schuette C., and Jahn R. (2008) Synaptic vesicles are constitutively active fusion machines that function independently of Ca2+. Curr. Biol. 18, 715–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Peterson G. L. (1977) A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem. 83, 346–356 [DOI] [PubMed] [Google Scholar]
- 53. Schägger H., and von Jagow G. (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 54. Towbin H., Staehelin T., and Gordon J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 76, 4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]