

Abstract
Glioblastoma is the most common malignant brain tumor and has a poor prognosis. Tachykinin receptor neurokinin-1 (NK1R) is a promising target in glioblastoma therapy because of its overexpression in human glioblastoma. NK1R agonists promote glioblastoma cell growth, whereas NK1R antagonists efficiently inhibit cell growth both in vitro and in vivo. However, the molecular mechanisms involved in these effects are incompletely understood. β-Arrestins (ARRBs) serve as scaffold proteins and adapters to mediate intracellular signal transduction. Here we show that the ARRB1-mediated signaling pathway is essential for NK1-mediated glioblastoma cell proliferation. ARRB1 knockdown significantly inhibited NK1-mediated glioblastoma cell proliferation and induced G2/M phase cell cycle arrest. ARRB1 knockdown cells showed remarkable down-regulation of CDC25C/CDK1/cyclin B1 activity. We also demonstrated that ARRB1 mediated prolonged phosphorylation of ERK1/2 and Akt in glioblastoma cells induced by NK1R activation. ERK1/2 and Akt phosphorylation are involved in regulating CDC25C/CDK1/cyclin B1 activity. The lack of long-term ERK1/2 and Akt activation in ARRB1 knockdown cells was at least partly responsible for the delayed cell cycle progression and proliferation. Moreover, we found that ARRB1-mediated ERK1/2 and Akt phosphorylation regulated the transcriptional activity of both NF-κB and AP-1, which were involved in cyclin B1 expression. ARRB1 deficiency increased the sensitivity of glioblastoma cells to the treatment of NK1R antagonists. Taken together, our results suggest that ARRB1 plays an essential role in NK1R-mediated cell proliferation and G2/M transition in glioblastoma cells. Interference with ARRB1-mediated signaling via NK1R may have potential significance for therapeutic strategies targeting glioblastoma.
Keywords: arrestin, cell cycle, cell proliferation, cyclin-dependent kinase (CDK), glioblastoma, neurokinin-1 receptor
Introduction
Glioblastoma, classified as a grade IV astrocytoma, is the most common malignant brain tumor worldwide (1). The average survival of glioblastoma patients is less than 1 year when treated with current standard therapeutic strategies, including surgery, radiation, and chemotherapy (2). The high rate of mortality is due to the glioblastoma properties of rapid proliferation, deregulated apoptosis, and ready infiltration into neighboring tissues (3, 4). Therefore, it is critical to understand the molecular mechanisms of glioblastoma progression and develop novel strategies of treatment.
The MAS gene was the first link between G protein-coupled receptors (GPCR)3 and cell transformation (5). Over the years, cumulative evidence has demonstrated a link between GPCR and tumorigenesis (6, 7). Several of them are involved in glioblastoma cell proliferation, metastasis, and glioma stem cell differentiation through initiating an array of signaling pathways, representing some promising targets in glioblastoma therapies. One such GPCR is the tachykinin receptor NK1 (8–10). Studies have demonstrated the overexpression of NK1R in human glioblastoma tissues and derived cell lines (11–13). NK1R has two endogenous peptide agonists, substance P (SP) and hemokinin-1 (HK-1). These NK1R agonists promote glioblastoma cell proliferation and migration as well as cytokine secretion (10, 14–16), whereas the antagonists of NK1R efficiently inhibit glioma cell growth in vitro and in vivo (17, 18). Moreover, SP exerts anti-apoptosis effects induced by drugs and antibodies through the NK1-mediated Akt pathway in various cell types (10, 19, 20). These preclinical studies suggest NK1R as an important regulator of glioblastoma, being characterized as a potential target in glioblastoma therapy (16, 18). However, high doses seem to be required to demonstrate the tumor-suppressive effect (21, 22).
Classic GPCRs, including NK1R, exert their functions through the activation of the G protein-mediated cell signaling pathway to release intracellular second messengers (23–25). β-Arrestins (ARRB, including ARRB1 and ARRB2) were originally known to negatively regulate GPCR signaling. Activated GPCR recruited ARRB from the cytoplasm to the cell membrane, and the receptor became desensitized and internalized, which caused the attenuation of receptor signaling. However, recent studies showed that ARRBs also function to mediate diverse cell signaling pathways independent of G protein (26, 27). They act as scaffold proteins that bind various kinases and proteins, such as MAPK, E3 ubiquitin ligases, MDM2, and PI3K-Akt NF-κB (28–32). NK1R can couple the Gs or Gq protein to transduce signaling events as a result of rapid cellular responses to agonist treatment (23). Besides its role in NK1R desensitization and sequestration, studies showed that ARRB also functions as a signal transducer, changing the spatiotemporal distribution of NK1R-mediated signaling pathways. This was first implied by a chimeric NK1R-ARRB1 fusion protein that caused robust constitutive activation of ERK1/2 independent of PKC activity (33). DeFea et al. (34) reported that SP stimulates the formation of a scaffolding complex comprising internalized NK1R, ARRB, Src, and ERK1/2 in rat kidney epithelial cells and that the formation of the ARRB-containing complex facilitates the proliferative and antiapoptotic effects of SP.
Malignant tumor cells benefit from GPCR signaling to proliferate, undergo anti-apoptosis, and invade into surrounding tissues (35–37). ARRBs facilitate these processes by mediating the activation of ERK1/2 and PI3K/Akt signaling pathways, which are well characterized regarding their roles in tumor development (27, 38). For example, in non-small-cell lung cancer, ARRB1 was shown to regulate nicotine-induced epithelial cell proliferation by activating the ERK1/2 and Rb-Raf-1pathways, subsequently causing the recruitment of E2F1, Rb, and Raf-1 to E2F-responsive proliferative promoters (39, 40). ARRB2 was shown to mediate the anti-apoptotic effect of the angiotensin II type 1A (AT1A) receptor through the ERK/p90RSK and PI3K/AKT pathways, which converge to inactivate the pro-apoptotic protein Bcl-2-associated death promoter (BAD) (41). However, in granule neuron precursors, the sonic hedgehog (Shh)-mediated signaling pathway negatively regulates its mitogenic potential through ARRB1 by enhancing the cyclin-dependent kinase (CDK) inhibitor p27 (42). In glioblastoma cells, NK1R stimulation can promote DNA synthesis and cell proliferation by inducing the phosphorylation of Akt and ERK1/2 (43). Mandell et al. (44) described a correlation between grade II and III glioblastoma and ARRB1 phosphorylation at Ser412. However, whether ARRB regulated NK1-induced signaling and what role it played in NK1-mediated glioblastoma cell proliferation remained unknown. In this study, we show that the ARRB1-mediated signaling pathway is essential for NK1-induced glioblastoma cell proliferation and G2/M transition in the cell cycle.
Results
ARRB1 mediates NK1R signaling in human glioblastoma cells
The distribution of NK1R has been demonstrated in human glioblastoma tissues by several studies (11–13). ARRB1/2 are important signal transducers of NK1R (33, 34, 45). However, the role of ARRB1/2 in NK1R signaling in human glioblastoma is unclear. We detected ARRB1/2 protein by Western blot in human glioblastoma samples (grade III/IV) and human glioblastoma cells (U251 and U87). All eight glioblastoma samples expressed full-length NK1R (∼48 kDa), six of them at a high level. It was interesting that all detected samples showed a high level of ARRB1 and a low level of ARRB2 (Fig. 1A). Both U251 and U87 cells expressed high levels of full-length NK1R and ARRB1, but only U251 expressed significant ARRB2 (Fig. 1B). These results encouraged us to investigate the possible role of NK1R and ARRBs in human glioblastoma progression.
Figure 1.
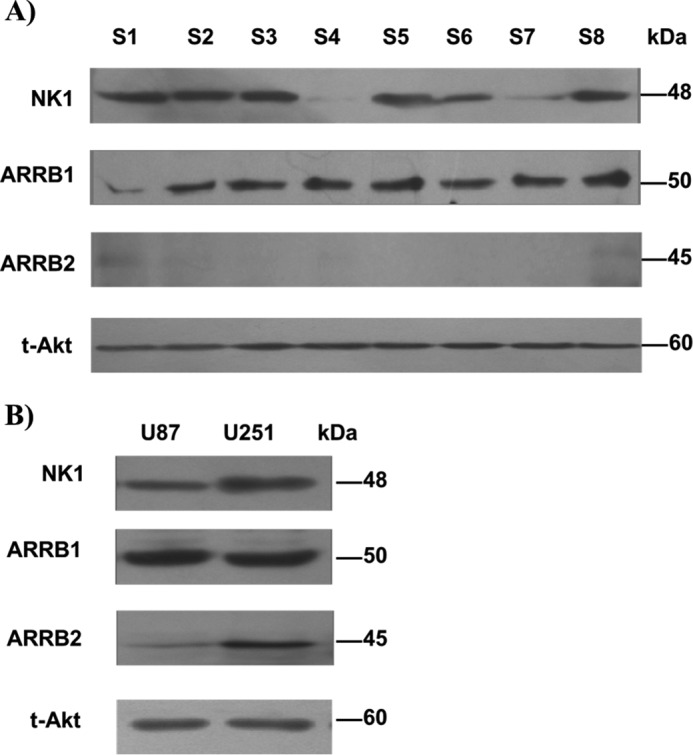
Expression of NK1R, ARRB1, and ARRB2 in human glioblastoma. A and B, Western blot of total lysates extracted from human glioblastoma tissue samples (A) and the human glioblastoma cell lines U251 and U87 (B), probed with anti-NK1R (1:5000), anti-ARRB1 (1:1000), or anti-ARRB2 (1:1000) antibodies. Total Akt served as a control.
We generated stable cell lines expressing specific shRNA against ARRB1 or ARRB2 (U251-ARRB1-sh1/sh2, U251-ARRB2-sh1/sh2, and U87-ARRB1-sh1/sh2). Cells stably transfected with a scramble shRNA construct (U251-sh-c) were used as a control. The shRNA constructs efficiently reduced the expression of the target protein in U251 (Fig. 2, A–D) and U87 cells (Fig. 2, E and F). First we examined the role of ARRB1and ARRB2 in NK1-mediated signaling in glioblastoma cells. We compared ERK1/2 and Akt phosphorylation in U251-ARRB1-sh1, U251-ARRB2-sh1, and U251-sh-c cells. With the treatment of hHK-1, ERK1/2 phosphorylation in U251-sh-c cells rapidly reached a peak at 5–10 min and then decreased. After 90 min, ERK1/2 phosphorylation reached a second peak and lasted until 150 min. U73122 is a phospholipase C inhibitor and inhibits hHK-1-induced rapid ERK1/2 and Akt phosphorylation in U251 cells (15). When U251-sh-c cells were pretreated with U73122, ERK phosphorylation at 5–10 min was inhibited but remained strong at 90–150 min. On the other hand, when ARRB1 expression was knocked down (U251-ARRB1-sh1/sh2), hHK-1 still induced rapid ERK1/2 phosphorylation at 5–10 min but not the second peak (Fig. 3A).
Figure 2.

Lentivirus-mediated ARRB knockdown in U251 and U87 cells. Cells were transfected by lentivirus shRNA against ARRB1, ARRB2, or sh-c. The protein and mRNA levels of the target protein were detected by Western blot and QPCR. A and B, the protein and mRNA levels of ARRB1in U251, U251-Sh-c, U251-ARRB1-sh1, and U251-ARRB1-sh2. C and D, the protein and mRNA levels of ARRB2 in U251, U251-Sh-c, U251-ARRB2-sh1, and U251-ARRB2-sh2. D and E, the protein and mRNA levels of ARRB1 in U87, U87-Sh-c, U87-ARRB1-sh1, and U87-ARRB1-sh2. Total Akt served as a control. The QPCR data were normalized to β-actin levels. The results are means ± S.E. for three independent experiments in triplicate. ***, p < 0.001 versus scramble shRNA.
Figure 3.

ARRB1 mediated the phosphorylation of ERK1/2 and Akt induced by NK1R activation in U251 cells. A, hHK-1 stimulated time-dependent phosphorylation of ERK1/2 (anti-pERK1/2, 1:1000). The phospholipase C inhibitor U73122 (10 μm) inhibited hHK-1-stimulated ERK1/2 phosphorylation at 5–10 min, whereas ARRB1 shRNA inhibited ERK1/2 phosphorylation after 60 min. Treatment of U73122 and ARRB1 shRNA in combination totally inhibited hHK-1-stimulated ERK1/2 phosphorylation. ARRB2 shRNA had little effect on hHK-1-stimulated ERK1/2 phosphorylation. Total ERK1/2 served as a control (anti-ERK1/2, 1:1000). B, hHK-1 stimulated time-dependent phosphorylation of Akt (anti-pAkt, 1:1000). U73122 inhibited hHK-1-stimulated Akt phosphorylation at 5–10 min, whereas ARRB1 shRNA inhibited Akt phosphorylation after 60 min. Treatment of U73122 and ARRB1 shRNA in combination totally inhibited hHK-1-stimulated Akt phosphorylation. ARRB2 shRNA had little effect on hHK-1-stimulated Akt phosphorylation. Total Akt served as a control (anti-Akt, 1:1000). Densitometry of phosphorylated ERK1/2 and Akt level was calculated by Quantity One software (corrected with total ERK1/2 or Akt). Using untreated cells as 1.0, the data are presented as means ± S.E. for three independent experiments.
Similarly, the kinetic of Akt phosphorylation was detected. hHK-1 rapidly provoked significant Akt phosphorylation and sustained it as long as 150 min in U251-sh-c cells. Treatment with U73122 in U251-sh-c cells inhibited the rapid Akt phosphorylation induced by hHK-1. In ARRB1 knockdown cells, Akt phosphorylation occurred at 5 min but reduced rapidly after 15 min (Fig. 3B).
When ARRB1 knockdown cells were treated with U73122, no ERK1/2 or Akt phosphorylation was detected. Deficiency of ARRB2 (U251-ARRB2-sh1) had little effect on hHK-1-induced ERK and Akt phosphorylation (Fig. 3, A and B). Taken together, these results suggest that, in glioblastoma cells, Gq and ARRB1 mediate the activation of ERK1/2 and Akt induced by NK1R in a very different way. Gq-dependent activity occurred rapidly and was maintained for a short time, whereas ARRB1-dependent activity was responsible for the slow and prolonged activation of ERK1/2 and Akt.
ARRB1 modulates glioma cell proliferation in response to NK1R activation
NK1R has been shown to promote glioma cell proliferation. We investigated the role of ARRB1 in this process. The ARRB1 gene was targeted by two independent shRNA constructs to avoid possible off-target effects in U251 and U87 cells (Fig. 2, A, B, E, and F). In a colony formation assay, hHK-1 stimulated the proliferation of U251 and U87 cells transfected with scramble shRNA constructs (U251-sh-c and U87-sh-c) dose-dependently. ARRB1 knockdown in U251 and U87 cells significantly inhibited hHK-1-induced cell proliferation (Fig. 4A). To support the data of the colony formation assay, a CellTiter-Glo assay was performed, and the cell growth curve was measured. The results showed that hHK-1-induced glioma cell proliferation was dose- and time- dependent and suppressed by ARRB1 knockdown (Fig. 4, B and C). These results demonstrate a critical role of ARRB1 in NK1-mediated glioma cell proliferation, and this role is not cell line-dependent.
Figure 4.

ARRB1 is required for NK1R-mediated U251 and U87 cell proliferation. hHK-1 was added to cells transfected with shRNA against ARRB1 (Sh-1 or Sh-2) or sh-c. A, dose-dependent cell proliferation induced by hHK-1 was measured by colony formation assay (the top panel is representative of colony images from the indicated cell type, and the bottom panel shows quantitative analysis of the colony formation assay). B and C, CellTiter-Glo assay (B) and growth curve (C) of the indicated types of cells treated with 1 μm hHK-1 (determined by CellTiter-Glo assay). The results are means ± S.E. for three independent experiments performed in triplicate. ctl, control.
ARRB1 knockdown induced G2/M phase arrest and down-regulation of cyclin B1, CDC25C, and CDK1 in glioblastoma cells
ARRB1 knockdown significantly reduced glioblastoma cell proliferation mediated by NK1R stimulation. We compared cell cycle progression in scramble or ARRB1 shRNA-transfected glioblastoma cells. Cells were treated with hHK-1 for 48 h and stained with PI for FACS analysis to determine the cell cycle (Fig. 5, A and B). The percentage of cells at G2/M phase was 8.4% in U251-sh-c, 21.4% in U251-ARRB1-sh1 (p < 0.05 versus U251-sh-c), and 19.4% in U251-ARRB1-sh2 (p < 0.05 versus U251-sh-c). Similarly, the percentage of cells at G2/M phase was 14.1% in U87-sh-c, 30.7% in U87-ARRB1-sh1 (p < 0.001 versus U87-sh-c), and 36.2% in U87-ARRB1-sh2 (p < 0.001 versus U87-sh-c). The CDK1-cyclin B1 complex is the primary regulator of G2/M transition in the cell cycle (46). We further examined cyclin B1 and CDK1 levels in wild-type and ARRB1 knockdown glioblastoma cells synchronized by double thymidine block. In ARRB1 knockdown U251 or U87 cells, the cyclin B1 protein level was significantly decreased, whereas the phosphorylation of CDK1 at the Tyr15 residue (an inactive form of CDK1) increased remarkably. CDC25C was responsible for CDK1 activation via dephosphorylation at Tyr15 (47). A Western blot assay showed that CDC25C remained phosphorylated at Ser216 (an inactive form of CDC25C) in ARRB1 knockdown glioblastoma cells (Fig. 5, C and D). These results together suggest that ARRB1 knockdown induces G2/M phase arrest in glioma cells treated with hHK-1, down-regulating the cyclin B1 protein level as well as the activity of CDC25C and CDK1. These data are consistent with the decreased cell growth rate shown in Fig. 4.
Figure 5.

ARRB1 knockdown induced G2/M phase arrest and down-regulation of cyclin B1, CDC25C, and CDK1 in glioblastoma cells. U251 and U87 cells expressing shRNA against ARRB1 (sh-1 and sh-2) or sh-c were treated with hHK-1. A, FACS analysis of the cell cycle. ctl, control. B, the proportions of cells in G2/M phase are presented as mean ± S.E. from three independent experiments. ***, p < 0.001 versus sh-c. Synchronized cells were harvested to analyze protein expression. C, Western blot analysis of cyclin B1 (anti-cyclin B1, 1:1000), pCDK1(Tyr15) (anti-pCDK1(Tyr15), 1:1000), and pCDC25C(Ser261) (anti-pCDC25C(Ser216), 1:1000). Total Akt served as a control. D, densitometry of cyclinB1, pCDC25C(Ser216), and pCDK1(Tyr15) levels was calculated by Quantity One software (corrected with GAPDH). The results are means ± S.E. for three independent experiments.
ARRB1 regulates cyclin B1 and CDK1 via NK1-mediated ERK1/2 and Akt signaling
ERK1/2 and Akt play important roles in regulating the cell cycle (48, 49). Because ARRB1 was demonstrated to mediate the prolonged phosphorylation of ERK1/2 and Akt induced by NK1 activation, we examined whether ERK1/2 and Akt phosphorylation is involved in G2/M arrest as well as down-regulation of CDC25C/CDK1/cyclin B1 activity in ARRB1 knockdown glioblastoma cells (Fig. 5). We used the MEK inhibitor PD98059 and the PI3K inhibitor LY294002, which efficiently suppressed the phosphorylation of ERK1/2 or Akt induced by NK1 activation in U251 and U87 cells, respectively (15). hHK-1 promoted U251 and U87 cell proliferation by ∼30% after an incubation of 72 h. When PD98059 (50 μm) or LY294002 (50 μm) was added, the proliferative effect was subdued (Fig. 6A). FACS analysis showed that the cell cycle was arrested at G2/M phase (Fig. 6, B and C). Additionally, the cyclin B1 protein level was significantly reduced, and pCDC25C(Ser216) and pCDK1(Tyr15) were notably increased (Fig. 6, D and E). These results indicate that ERK1/2 and Akt phosphorylation are involved in the regulation of CDC25C/CDK1/cyclin B1 activity. Because ARRB1 prolonged ERK1/2 and Akt phosphorylation induced by NK1R activation in glioblastoma cells, we suppose that the lack of long-term ERK1/2 and Akt activation is at least partly responsible for the down-regulation of cyclin B1 expression and CDC25C and CDK1 activity as well as G2/M arrest in ARRB1 knockdown glioblastoma cells.
Figure 6.

ARRB1 regulated cyclin B1/CDC25C/CDK1 through the ERK1/2 and Akt signaling pathways. A, treatment with PD98059 (PD, 50 μm) and LY294002 (LY, 50 μm) inhibited U251 and U87 cell proliferation induced by hHK-1 (measured by CellTiter-Glo assay). ctl, control. ***, p < 0.001 versus untreated control; ###, p < 0.001 versus hHK-1-treated control. B, FACS analysis of PD98059- and LY294002-induced G2/M phase arrest. C, the proportions of cells in G2/M phase are presented as means ± S.E. from three independent experiments. ***, p < 0.001 versus hHK-1 treated control. D, synchronized U251-sh-c and U87-sh-c cells were treated with PD98059 and LY294002 in the presence of hHK-1 and collected at the indicated time points. Cyclin B1, pCDK1(Tyr15), and pCDC25C(Ser261) levels were analyzed by Western blotting. Total Akt served as the protein loading control. E, densitometry of cyclin B1, pCDC25C(Ser216), and pCDK1(Tyr15) levels normalized to total Akt levels. The results are means ± S.E. for three independent experiments.
NF-κB and AP-1 are involved in ARRB1-regulated cyclin B1 expression
The cyclin B1 level is periodic during the cell cycle, and the degradation is under the control of ubiquitin-mediated proteolysis. Bortezomib is a specific inhibitor of the proteasome that was shown to stabilize mitotic cyclins and prevent cell cycle progression (50). Treatment with bortezomib did not rescue the cyclin B1 protein level in U251-ARRB1-sh1, implying that the reduction of cyclin B1 may not be through the proteasome-mediated protein degradation pathway (Fig. 7A). Several transcription factors, including NF-κB and AP-1, have been shown to enhance the transcription of cyclin B1. hHK-1 induced p65 and c-Jun phosphorylation in U251 cells, and ARRB1 knockdown significantly suppressed this effect (Fig. 7, B and C). Similarly, in a reporter gene assay, hHK-1 increased the luciferase activity driven by NF-κB and AP-1, and this effect was remarkably reduced in ARRB1 knockdown cells (Fig. 7, D and E). Then we asked whether NF-κB and AP-1 were involved in the reduction of cyclin B1 expression in ARRB1 knockdown cells. CAPE and pyrrolidine dithiocarbamate (PDTC) are inhibitors of NF-κB. Curcumin and tanshinone IIA are inhibitors of AP-1. The cyclin B1 protein level significantly decreased in U251 cells treated with NF-κB and AP-1 inhibitor (Fig. 7F). Furthermore, NK1-mediated U251 cell proliferation was inhibited, and the cell cycle arrested at G2/M phase (Fig. 7, G–I). These results suggest that NF-κB and AP-1 play a part in regulating cyclin B1 expression. It was shown that NF-κB and AP-1 activation induced by hHK-1 is inhibited by PD98059 and LY294002 (Fig. 7D). Taking these results together, we suppose that ARRB1-mediated ERK1/2 and Akt phosphorylation regulate the transcriptional activity of NF-κB and AP-1, which are involved in cyclin B1 expression along with the G2/M transition in glioma cells.
Figure 7.

NF-κB and AP-1 are involved in ARRB1-regulated cyclin B1 expression. A, treatment with the proteasome inhibitor bortezomib did not rescue the cyclin B1 protein level in U251-ARRB1-sh1. B, top panel, hHK-1 stimulated time-dependent phosphorylation of p65 and c-JUN. Bottom panel, ARRB1 knockdown suppressed hHK-1-stimulated p65 and c-JUN phosphorylation. Total Akt served as a control. C, densitometry of p-p65 and p-c-JUN normalized to total Akt levels. The results are means ± S.E. for three independent experiments. D and E, the activity of luciferase driven by NF-κB and AP-1 was inhibited by ARRB1 knockdown as well as PD98059 (PD) and LY294002 (LY) treatment in U-251-sh-c. ctl, control. F, treatment with NF-κB and AP-1 inhibitors suppressed the expression of cyclin B1 in U251-sh-c. Cells were treated as follows: CAPE (25 μm), pyrrolidine dithiocarbamate (PDTC) (100 μm), curcumin (Cur, 25 μm), and tanshinone IIA (TIIA, 25 μm). Densitometric analysis of cyclin B1 protein expression was normalized to total Akt. The results are means ± S.E. for three independent experiments. G, treatment with NF-κB and AP-1 inhibitors reduced the proliferation of U251 cells by the treatment of hHK-1 (CellTiter-Glo assay). H, FACS analysis of NF-κB and AP-1 inhibitor-induced G2/M phase arrest. I, the proportions of cells in G2/M phase are presented as means ± S.E. from three independent experiments. ***, p < 0.001 versus untreated control; **, p < 0.05 versus untreated control; ###, p < 0.001 versus hHK-1-treated control.
Knockdown of ARRB1 increases the sensitivity of glioblastoma cells to NK1 antagonist treatment and cell apoptosis
NK1 receptor agonists promote glioblastoma cell proliferation, whereas NK1 antagonists inhibit tumor cell proliferation in vitro and in vivo (10, 17, 18). Because ARRB1 knockdown reduced NK1-mediated glioblastoma cell proliferation, we examined the effect of ARRB1 knockdown on the antiproliferative activity of NK1 antagonists. Two NK1 receptor antagonists, L732138 and SPA, were used (51). Both antagonists dose-dependently reduced the surviving fraction of WT glioblastoma cells transfected with scramble shRNA. Accordingly, the surviving fraction was markedly reduced, and the IC50 values were significantly lowered in ARRB1 knockdown cells (Fig. 8A). Antagonists of NK1 treatment blocked the glioblastoma cell cycle at G2/M phase, and this effect was greatly enhanced in ARRB1 knockdown cells (Fig. 8, B and C). Consistent with this, TUNEL staining showed that ARRB1 knockdown aggravated cell apoptosis induced by L732138 and SPA (Fig. 8, D and E). These data suggest that the absence of ARRB1 increases the sensitivity of glioblastoma cells and cell apoptosis to treatment with NK1 antagonists.
Figure 8.

ARRB1 knockdown increased the sensitivity of glioblastoma cells to NK1R antagonist treatment and cell apoptosis. A, IC50 of human glioblastoma cells with L732138 and SPA treatment (measured by CellTiter-Glo assay). B, FACS analysis of L732138- and SPA-induced G2/M phase arrest. ctl, control. C, the proportions of cells in G2/M phase are presented as means ± S.E. from three independent experiments. D, TUNEL staining for apoptosis induced by L732138 and SPA in scramble shRNA- and ARRB1-sh1-transfected cells. E, the percentages of positively stained cells are presented as means ± S.E. from three independent experiments. ***, p < 0.001.
Discussion
NK1R has been closely associated with human glioblastoma because of the elevated expression level in tumor tissues (11–13). Together with endogenous peptide agonists, NK1R exerts oncogenic activity by promoting glioblastoma DNA synthesis and cell proliferation (43). Moreover, we reported previously that NK1 reactivation stimulates glioblastoma cell migration by increasing MMP-2 and MT1-MMP levels (15). In support of a crucial role of NK1R in glioblastoma progression, studies have demonstrated that the NK1-mediated ERK1/2 and Akt signaling pathways are involved in providing these survival benefits (14). However, our knowledge about the mechanism for these effects is still inadequate. In this study, we show for the first time that ARRB1 contributes to NK1R-mediated human glioblastoma progression by regulating cell cycle G2/M phase as well as cell proliferation. Our data demonstrate that Gq and ARRB1 mediate ERK1/2 and Akt signaling by NK1R activation in glioblastoma cells. Gq-dependent signaling occurred rapidly and was maintained for a short time, whereas ARRB1-dependent signaling was slow and prolonged. ARRB1 knockdown suppressed NK1R-mediated glioblastoma cell proliferation and increased cell apoptosis caused by NK1R antagonist treatment. The ARRB1-mediated NK1-signaling pathway regulated cyclin B1 expression and CDC25C and CDK1 activity. Lack of ARRB1 induced glioblastoma cell cycle arrest at G2/M phase.
ARRB1 was originally reported to modulate GPCR desensitization and internalization for uncoupling GPCR-mediated G protein signaling. More recent studies show that ARRB1 also serves as scaffolding protein to transduce the G protein-independent signaling pathway (26, 27). Previously, ARRB was shown to form a signalosome with NK1R, Src, and ERK1/2 to provide ARRB-dependent ERK1/2 signaling in the transfected cell lines HEK293-NK1 and KNRK-NK1 (34). In glioblastoma cells, NK1R-mediated signaling pathways have been described, including MAPK and Akt phosphorylation (15). However, little was known about the role of ARRBs in NK1-mediated signaling pathways in glioblastoma cells. In this study, we show for the first time that, in glioblastoma cells, Gq and ARRB1 mediate ERK1/2 and Akt signaling by ligand-activated NK1R in temporally distinct fashions: the Gq-dependent pathway is immediate and transient, and the ARRB1-dependent pathway is slow but much more persistent (Fig. 3). The samples and cell lines in this study showed low levels of ARRB2 expression, except U251 (Fig. 1). Although the protein level of ARRB1 and ARRB2 was comparable in U251 cells, ARRB2 knockdown seemed to have little effect on NK1-mediated signaling pathways (Fig. 3).
The ERK1/2 and Akt pathways have been related to the prosurvival effect of NK1R in previous studies. Considering the role of ARRB1 in NK1-mediated ERK1/2 and Akt signaling, we further investigated the role of ARRB1 in NK1-mediated glioblastoma cell proliferation. ARRB1 promotes cell growth by facilitating receptor-mediated tumor cell signaling, such as prostaglandin E2, GLP-1, and nicotine receptor (39, 52, 53). In this study, the expression of ARRB1 was knocked down by a stably transfected ARRB1-specific shRNA construct in U251 and U87 glioblastoma cells. As shown in Fig. 4, the data of the CellTiter-Glo assay, colony formation, and cell growth curve measurement supported that ARRB1 knockdown significantly delayed the glioblastoma cell proliferation induced by hHK-1 treatment. Interestingly, cell cycle analysis showed that ARRB1 knockdown caused glioblastoma cell arrest at G2 phase. In line with the observations, the cyclin B1 protein level was significantly decreased, whereas phosphorylated CDK1 at Tyr15 and phosphorylated CDC25C at Ser216 were notably increased in ARRB1 knockdown U251 cells (Fig. 5, C and D). Actually, only a few reports are about the correlation between ARRB1 and cell cycle progression. Girnita et al. (29) reported that ARRB1 plays a critical role in IGF-1R induced cell growth by mediating ERK1/2 signaling and that ARRB1 knockdown caused G1 phase accumulation in the cell cycle similar to ERK inhibitor treatment. However, in granule neuron precursors, the sonic hedgehog-mediated signaling pathway negatively regulated its mitogenic potential through ARRB1 by enhancing the CDK inhibitor p27, which eventually drives cell cycle exit (42). Here we show for the first time that ARRB1 regulates cyclinB1/CDC25C/CDK1 activity as well asG2/M transition in glioblastoma cells through ERK1/2 and Akt signaling. Therefore, the role of ARRB1 in cell cycle regulation could be cell type-dependent. The complex of cyclin B1 and CDK1 phosphorylates hundreds of substrate proteins to promote G2/M transition (46). Cyclin B1/CDK1 deregulation is forcefully associated with malignant transformation (54–56). Cyclin B1 or CDK1 depletion inhibits cell proliferation by inducing G2/M arrest and apoptosis in human tumor cells (54). CDK1 activity is regulated by CDC25C phosphatase, which activates CDK1 by dephosphorylation at Thr14 and Tyr15 (47). Our results show for the first time that ARRB1 knockdown reduces cyclin B1 expression as well as the activity of CDC25C and CDK1 in glioblastoma cells.
Deregulated signaling of ERK1/2 and Akt has been shown to promote tumor cell proliferation and survival (14, 41, 57). We examined the correlation of ARRB1 deficiency-induced cell cycle arrest with the ERK1/2 and Akt signaling pathway. It was demonstrated that PD98059 and LY294002 treatment abolished hHK-1-induced glioblastoma cell proliferation, arresting cells at G2/M phase (Fig. 6, A–C). In parallel, the cyclin B1 protein level was reduced, CDC25C remained phosphorylated at Ser216, and dephosphorylation of CDK1 at Tyr15 was markedly postponed (Fig. 6, D and E). Actually, the role of ERK1/2 and Akt signaling in cell cycle regulation has been studied (48, 49). ERK1/2 loss in epithelial cells reduces cyclin B1 expression and induces G2/M arrest while disrupting a gene regulatory network centered on the cyclin B1-CDK1 complex (58). ERK1/2 phosphorylates two of the four phosphorylation sites of the cytoplasmic retention sequence of cyclin B1 that are necessary for nuclear localization of cyclin B1 and mitosis progression (59). Either PI3K inhibitors or the absence of Akt protein in cells was shown to induce a delay in G2/M transition, which can be rescued by overexpressing Akt (60). Here ARRB1 knockdown was shown to reduce the sustained activation of ERK1/2 and Akt by NK1 stimulation in glioblastoma cells. ERK1/2 and Akt inhibition caused down-regulation of CDC25C/CDK1/cyclin B1 along with a G2/M arrest similar to ARRB1 reduction. These results suggest that the impaired longevity and duration of ERK1/2 and Akt activation in ARRB1 knockdown cells is at least partly responsible for the delayed cell cycle and cell proliferation.
The NF-κB and AP-1 signaling pathway regulates cyclin B1 expression, which is key to the control of G2/M transition (61, 62). Both the NF-κB and AP-1 binding sites have been identified in the cyclin B1 promoter region (62, 63). NF-κB-dependent transcription is necessary for G2/M progression (64). The AP-1 inhibitor curcumin preferentially arrested cells in G2/M phase of the cell cycle (65). Previously, we reported that the NK1 receptor mediated the activation of NF-κB and AP-1 through MAPK and Akt signaling in glioblastoma cells (15). We show here that ARRB1 knockdown significantly suppresses hHK-1-induced p65 and c-Jun phosphorylation in U251 cells (Fig. 7). In a reporter gene assay, hHK-1 increased the luciferase activity driven by NF-κB and AP-1, and this effect was remarkably reduced in ARRB1 knockdown cells. Then we asked whether NF-κB and AP-1 were involved in the reduction of cyclin B1 expression. Treatment with NF-κB and AP-1 inhibitors significantly inhibited NK1-mediated U251 cell proliferation and caused a cell cycle arrest in G2/M phase. Moreover, the protein level of cyclin B1 decreased (Fig. 7). These results suggest that NF-κB and AP-1 activation are involved in regulating cyclin B1 expression. hHK-1-induced NF-κB and AP-1 activation was inhibited by PD98059 and LY294002 in U251-sh-c cells (Fig. 7D). Taking these results together, we consider that ARRB1-mediated ERK1/2 and Akt phosphorylation regulates the transcriptional activity of NF-κB and AP-1 as well as cyclin B1 expression in glioblastoma cells.
The agonists of NK1R play a protective role in cell apoptosis and the tissue damage process through ERK1/2 and Akt activation (10, 19, 20). NK1 antagonists showed anti-tumor activity in both in vitro and in vivo models (17, 18). Here we show that NK1 antagonist treatment (L732138 and SPA) induced glioblastoma cell apoptosis and inhibited cell proliferation. The effects were potentiated in ARRB1 knockdown cells (Fig. 8). These data show that ARRB1 knockdown enhanced the sensitivity of glioblastoma cells to treatment with NK1 antagonists by aggravating cell apoptosis and G2/M phase arrest. Although NK1R has been suggested as a potential target for glioblastoma therapy, some preclinical studies showed that higher doses of NK1R antagonist seemed to be required for the tumor-suppressive effect. Aprepitant is a Food and Drug Administration-approved NK1R antagonist used in the clinic to prevent chemotherapy-induced nausea and vomiting. How the ARRB1 expression level influences the antitumor effect of such drugs in glioblastoma patients deserves further clinical study.
In summary, we demonstrated in this study that the ARRB1-mediated ERK1/2 and Akt signaling pathway, in response to NK1R activation, plays an important role in regulating G2/M phase transition and cell proliferation in glioblastoma cells. The results afford a new perspective to understand the molecular mechanism underlying NK1-stimulated glioblastoma cell proliferation. This may be of particular significance in NK1-mediated glioblastoma development. Our data also show that a lower ARRB1 protein level improves the ability of NK1 antagonists to inhibit glioblastoma cell proliferation. Thus, inhibition of NK1-ARRB1-mediated signaling is important for the glioblastoma therapeutic strategy of targeting the NK1 receptor.
Experimental procedures
Reagents and antibodies
DMEM and FBS were from Invitrogen. PD98059, LY294002, the CellTiter-Glo reagent, and the luciferase assay system were from Promega (Madison, WI). The protease inhibitor mixture and phosphatase inhibitor mixtures were from Roche Applied Science. U73122, propidium iodide (PI), and L-732138 were from Sigma-Aldrich (St. Louis, MO). Curcumin, tanshinone IIA, and caffeic acid phenethyl ester (CAPE) were from Sangon (Shanghai, China).
The antibodies against phospho-ERK1/2 (4370, 1:2000), ERK1/2 (4695, 1:2000), pAkt (4060, 1:1000), phospho-p65 (3033, 1:1000), phospho-c-JUN (3270, 1:1000), pCDK1 (9111, 1:1000), pCDC25C (4901, 1:1000), ARRB2 (3857, 1:1000), and cyclin B1 (4135, 1:2000) were purchased from Cell Signaling Technology Inc. (Danvers, MA). The antibodies against ARRB1 (ab32099, 1:1000) and NK1R (ab183713, 1:2000) were from Abcam (Cambridge, UK). The antibodies against AKT (AA326, 1:1000), HRP-conjugated goat anti-rabbit antibodies (A0208, 1:1000), and HRP-conjugated goat anti-mouse antibodies (A0216, 1:1000) were from Beyotime (Jiangsu, China). The antibody against Cyclin B1 was mouse monoclonal, and all other antibodies were rabbit monoclonal. All antibodies were characterized before immunoblotting studies by serial dilutions to determine optimal conditions and negative controls to ensure specificity.
QPCR primers, RNA isoPlus, and real-time quantitative PCR reagents were from Takara Biotechnology (Dalian, China). The ECL detection system and BCA protein assay kit were from Pierce. Bortezomib was from Selleck (Shanghai, China).
Human HK1 (TGKASQFFGLM-NH2) and [d-Arg1, d-Trp5,7,9, Leu11]substance P (SPA) were synthesized using the Fmoc (N-(9-fluorenyl)methoxycarbonyl) method on a solid-phase peptide synthesis system and purified by HPLC as described previously (15). The identity of the peptide was confirmed using electrospray ionization (ESI)-TOF mass spectrometry. Peptides were determined to be>95% pure by reverse-phase high-performance liquid chromatography using a C18 column as the solid phase and an H2O/acetonitrile gradient as the solution phase.
Cell culture and human tissue samples
U251 MG, U87 MG, and HEK293T cells were from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and maintained in DMEM supplemented with 10% FBS and antibiotics (100 units/ml penicillin, 100 μg/ml streptomycin). All cells were cultured in a humidified atmosphere of CO2/air (5:95%) at 37 °C.
All human tissues were collected from the Department of Neurosurgery, Second Affiliated Hospital of Lanzhou University. Approval for this study was obtained from the ethics committee of the Second Affiliated of Hospital of Lanzhou University. The samples included eight World Health Organization grade III-IV tumors. Written informed consent was obtained from all subjects prior to their participation in this study. Freshly resected tissue samples were immediately frozen in liquid nitrogen. For each sample, at least one slice was stained with hematoxylin and eosin to control the percentage of tumor cells. Only samples with a histologically estimated tumor cell content of 70% or more were used for subsequent experiments.
shRNA-mediated gene suppression
Knockdown of ARRB1 or ARRBB2 expression in U251 and U87 cells was performed by stable transfection of an shRNA construct in the lentiviral pLVX-shRNA1 vector. For lentiviral production, 293T cells were seeded in a 10-cm dish with 30% confluence and co-transfected with 7 μg of plasmid, 36 μl of Lenti-X HTX packaging mix, and 7.5 μl of Xfect polymer by using the Lenti-X HTX packaging system (Clontech) for 12 h. The transfection medium was then removed, and 10 ml of fresh basis medium was added. The virus was collected 48 h after adding the fresh medium. The virus-containing medium was centrifuged at 500 × g for 10 min and passed through a sterile 0.45-μm filter.
For stable cell line selection, cells were infected with lentivirus for 24 h and further selected with puromycin (0.5 μg/ml) for 2 weeks. Then the mRNA and protein expression of ARRB1 and ARRB2 in the infected cells was validated by QPCR and Western blot assays. A scramble shRNA (sh-c) was used as a control (Origene). Oligonucleotides encoding hairpin precursors were as follows: ARRB1-sh1 (5′-AAGGAAGAGGCTGATGACACTGTGGCACC-3′), ARRB1-sh2 (5′-AAGGCTTGCGGTGTGGACTATGAAGTCAA-3′), ARRB2-sh1 (5′-AAGGAACTCTGTGCGGCTGGTGATCCGAA-3′), ARRB2-sh2 (5′-CACACGCCACTTCCTCATGTCTGACCGGT-3′), and sh-c, (5′-GCACTACCAGAGCTAACTCAGATAGTACT-3′). All experiments were performed in stably transfected cells if not specified otherwise.
Quantitative PCR analysis
Total RNA was isolated using RNA isoPlus according to the instructions of the manufacturer. First-strand cDNA was synthesized from 1 μg of RNA using PrimeScript RT reagent. QPCR was performed using SYBR Premix Ex TaqII reagent according to the instructions of the manufacturer and analyzed on an Applied real-time system (Agilent, MX3005P). The PCR reaction was performed as follows: 1 min at 95 °C, 20 s at 60 °C, 40 cycles of 5 s at 95 °C, followed by 20 s at 60 °C. β-Actin was used as internal control to normalize mRNA expression. Sh-c-transfected cells were defined as 1.0 to determine the -fold-change in the expression levels. Each QPCR analysis was performed three times in triplicate. The QPCR primer sequences for ARRB1, ARRB2, and β-actin were as follows: β-actin, 5′-AGCGAGCATCCCCCAAAGTT-3′ and 5′-GGGCACGAAGGCTCATCATT-3′; ARRB1, 5′-TGGAGAACCCATCAGCGTCAAC-3′ and 5′-AGGCAGATGTCTGCATACTGGC-3′; and ARRB2, 5′-CTGACTACCTGAAGGACCGCAA-3′ and 5′-GTGGCGATGAACAGGTCTTTGC-3′.
Cell lysate preparation and Western blots
Cells were seeded into 12-well plates at a density of 300,000/well and grown for 24 h. To detect ERK1/2 and Akt phosphorylation, cells were treated with 1 μm hHK-1 at 37 °C for the indicated times after starvation in serum-free DMEM for 4 h. Subsequently, the cells were washed three times with ice-cold PBS. Cell lysates were prepared by adding 75 μl of radioimmune precipitation assay buffer supplemented with proteinase and phosphatase inhibitors. The lysates were then centrifuged at 20,000 × g for 10 min at 4 °C. The supernatant was collected, and the protein concentration was determined using a BCA protein assay kit. Equal amounts of protein were separated by 10% polyacrylamide gel, transferred to a PVDF membrane, and blocked in 5% nonfat milk in TBST for 2 h. The membranes were probed with the specific primary antibodies overnight at 4 °C, followed by incubation with HRP-conjugated secondary antibodies for 2 h at room temperature. Finally, the target proteins were detected with an enhanced chemiluminescence detection system and visualized by Kodak film.
Cell proliferation assay
Cell proliferation was measured by CellTiter-Glo luminescence assay according to the instructions of the manufacturer. To determine the effect hHK-1 on glioma cell proliferation, cells were seeded in a 96-well plate at a density of 1000 cells/well and starved in serum-free DMEM for 4 h. Cells were treated with hHK-1 at the indicated dosage and time. To determine the effect of NK1 antagonists on glioma cell proliferation, cells were seeded in 96-well plates at a density of 10,000 cells/well. Cells were treated with L732138 or SPA for 72 h. 100 μl of CellTiter-Glo substrate was added and incubated for 10 min. Luminescence was recorded on a FlexStation III plate reader. The experiments were repeated at least three times with five replicates. Untreated cells (U251-sh-c or U87-sh-c) were defined as 1.0. Cell proliferation was expressed as -fold induction relative to the untreated control.
Colony formation assay
Glioma cells were seeded in a 6-well plate at a density of 500 cells/well and treated with hHK-1. After an incubation of 2 weeks, cells were washed with PBS, fixed with 90% ethanol for 30 min, and stained with crystal violet solution for 30 min. The dishes were photographed, and total colony numbers were counted under a microscope. Colonies with >50 cells were scored. Control shRNA-transfected cells were used to evaluate the colony formation efficiency.
Luciferase reporter assays for NF-κB and AP-1 activity
The luciferase reporter gene assay was measured as described previously (15). Briefly, cells were transfected with the reporter plasmid pNF-κB-luc or pAP-1-luc with Lipofectamine 2000 following the instructions of the manufacturer. 6 h later, the transfected cells were trypsinized and seeded in a 96-well plate at a density of 30,000 and cultured for another 24 h. Then the cells were exposed to 1 μm hHK-1 for 8 h at 37 °C. Untreated cells were used as a control. For the inhibitory assays, the cells were pretreated with different inhibitors for 30 min, followed by treatment with 1 μm hHK-1. Luciferase activities were measured using a luciferase assay system. The untreated control was defined as 1.0. The luciferase activity was expressed as -fold induction relative to the untreated control.
Cell synchronization and cell cycle assay
Cells were synchronized by double thymidine block. Briefly, cells were plated and treated with 2 mm thymidine for 16 h. Then cells were washed with PBS twice to remove thymidine and cultured for 8 h. Thymidine was resupplemented and incubated for another 16 h. Cells were synchronized at the G1/S boundary, released, and subsequently collected for the indicated time points.
Cells were treated with hHK-1 or chemical inhibitors as indicated. 106 cells were harvested, washed three times with PBS, and fixed in 70% ethanol at 4 °C overnight. The fixed cells were incubated with 100 μg/ml RNase for 30 min at 37 °C and stained with PI (50 μg/ml) in the dark for 30 min at 4 °C. Labeled cells were analyzed with FACSCalibur (BD Biosciences). 105 cells were analyzed for each sample, and every experiment was repeated at least three times. The DNA content and cell cycle were analyzed using Modfit LT version 3.0 software.
TUNEL assay
The TUNEL assay was carried out using a One Step TUNEL apoptosis assay kit according to the instructions of the manufacturer. Briefly, after 72-h treatment with SPA or L732138, the cells were fixed by 4% paraformaldehyde. The fixed cells were washed in PBS, permeabilized with 0.1% Triton X-100 for 2 min on ice, and then incubated with 50 μl of TUNEL solution for 60 min at 37 °C in a humidified chamber in the dark. The positively stained cells were counted under a fluorescence microscope in five random fields, and the average numbers were considered the apoptotic index.
Data analysis
GraphPad Prism 5.01 software was used to perform curve-fitting and statistical analysis. Statistical significance between two or more groups was calculated by one-way ANOVA followed by Tukey's post test.
Author contributions
L. Y. M. and R. W. conceived and designed the research. Y. X. Z., X. F. L., G. Q. Y., H. H., X. Y. S., J. Y. L., X. K. M., T. X. Z., W. L. Y., and X. W. Z. performed the experiments. L. Y. M. and Y. X. Z. wrote the manuscript. R. W., H. H., and X. K. M. revised the manuscript.
This work was supported by the National Nature Science Foundation of China (Grants 81302798, 81473095, and 21432003), the Project of Healthy and Family Planning Commission of Gansu (Grant GSWSKY-2014-31/2015-58), the Doctoral Research Fund of Lanzhou University Second Hospital (Grant ynbskyjj2015-1-02), and Fundamental Research Funds for the Central Universities (Grants lzujbky-2015-154 and lzujbky-2016-ct01). The authors declare that they have no conflicts of interest with the contents of this article.
- GPCR
- G protein-coupled receptor(s)
- SP
- substance P
- ARBB
- β-arrestin
- CDK
- cyclin-dependent kinase
- PI
- propidium iodide
- CAPE
- caffeic acid phenethyl ester
- SPA
- [d-Arg1, d-Trp5,7,9, Leu11]substance P
- QPCR
- quantitative PCR
- sh-c
- scramble shRNA.
References
- 1. American Cancer Society (2015) Cancer Facts and Figures 2015, American Cancer Society, Atlanta, GA [Google Scholar]
- 2. Adamson D. C., Rasheed B. A., McLendon R. E., and Bigner D. D. (2010) Central nervous system. Cancer Biomark. 9, 193–210 [DOI] [PubMed] [Google Scholar]
- 3. Chen J., McKay R. M., and Parada L. F. (2012) Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell 149, 36–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wen P. Y., and Kesari S. (2008) Malignant gliomas in adults. N. Engl. J. Med. 359, 492–507 [DOI] [PubMed] [Google Scholar]
- 5. Young D., Waitches G., Birchmeier C., Fasano O., and Wigler M. (1986) Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell 45, 711–719 [DOI] [PubMed] [Google Scholar]
- 6. Dorsam R. T., and Gutkind J. S. (2007) G-protein-coupled receptors and cancer. Nat. Rev. Cancer 7, 79–94 [DOI] [PubMed] [Google Scholar]
- 7. O'Hayre M., Degese M. S., and Gutkind J. S. (2014) Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr. Opin. Cell Biol. 27, 126–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo W., Sharif T. R., and Sharif M. (1996) Substance P-induced mitogenesis in human astrocytoma cells correlates with activation of the mitogen-activated protein kinase signaling pathway. Cancer Res. 56, 4983–4991 [PubMed] [Google Scholar]
- 9. Łazarczyk M., Matyja E., and Lipkowski A. (2007) Substance P and its receptors: a potential target for novel medicines in malignant brain tumour therapies (mini-review). Folia Neuropathol. 45, 99–107 [PubMed] [Google Scholar]
- 10. Akazawa T., Kwatra S. G., Goldsmith L. E., Richardson M. D., Cox E. A., Sampson J. H., and Kwatra M. M. (2009) A constitutively active form of neurokinin 1 receptor and neurokinin 1 receptor-mediated apoptosis in glioblastomas. J. Neurochem. 109, 1079–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hennig I. M., Laissue J. A., Horisberger U., and Reubi J. C. (1995) Substance-P receptors in human primary neoplasms: tumoral and vascular localization. Int. J. Cancer 61, 786–792 [DOI] [PubMed] [Google Scholar]
- 12. Berger A., and Paige C. J. (2005) Hemokinin-1 has substance P-like function in U-251 MG astrocytoma cells: a pharmacological and functional study. J. Neuroimmunol. 164, 48–56 [DOI] [PubMed] [Google Scholar]
- 13. Ogo H., Kuroyanagi N., Inoue A., Nishio H., Hirai Y., Akiyama M., DiMaggio D. A., Krause J. E., and Nakata Y. (1996) Human astrocytoma cells (U-87 MG) exhibit a specific substance P binding site with the characteristics of an NK-1 receptor. J. Neurochem. 67, 1813–1820 [DOI] [PubMed] [Google Scholar]
- 14. Yamaguchi K., Kugimiya T., and Miyazaki T. (2005) Substance P receptor in U373 MG human astrocytoma cells activates mitogen-activated protein kinases ERK1/2 through Src. Brain Tumor Pathol. 22, 1–8 [DOI] [PubMed] [Google Scholar]
- 15. Mou L., Kang Y., Zhou Y., Zeng Q., Song H., and Wang R. (2013) Neurokinin-1 receptor directly mediates glioma cell migration by up-regulation of matrix metalloproteinase-2 (MMP-2) and membrane type 1-matrix metalloproteinase (MT1-MMP). J. Biol. Chem. 288, 306–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palma C. (2006) Tachykinins and their receptors in human malignancies. Curr. Drug Targets 7, 1043–1052 [DOI] [PubMed] [Google Scholar]
- 17. Palma C., Bigioni M., Irrissuto C., Nardelli F., Maggi C. A., and Manzini S. (2000) Anti-tumour activity of tachykinin NK1 receptor antagonists on human glioma U373 MG xenograft. Br. J. Cancer 82, 480–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Palma C., and Maggi C. A. (2000) The role of tachykinins via NK1 receptors in progression of human gliomas. Life Sci. 67, 985–1001 [DOI] [PubMed] [Google Scholar]
- 19. Koon H. W., Zhao D., Zhan Y., Moyer M. P., and Pothoulakis C. (2007) Substance P mediates antiapoptotic responses in human colonocytes by Akt activation. Proc. Natl. Acad. Sci. U.S.A. 104, 2013–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Backman L. J., and Danielson P. (2013) Akt-mediated anti-apoptotic effects of substance P in anti-Fas-induced apoptosis of human tenocytes. J. Cell. Mol. Med. 17, 723–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cordier D., Forrer F., Bruchertseifer F., Morgenstern A., Apostolidis C., Good S., Müller-Brand J., Mäcke H., Reubi J. C., and Merlo A. (2010) Targeted α-radionuclide therapy of functionally critically located gliomas with 213Bi-DOTA-[Thi8, Met(O2)11]-substance P: a pilot trial. Eur. J. Nucl. Med. Mol. Imaging 37, 1335–1344 [DOI] [PubMed] [Google Scholar]
- 22. Cordier D., Forrer F., Kneifel S., Sailer M., Mariani L., Mäcke H., Müller-Brand J., and Merlo A. (2010) Neoadjuvant targeting of glioblastoma multiforme with radiolabeled DOTAGA-substance P: results from a phase I study. J. Neurooncol. 100, 129–136 [DOI] [PubMed] [Google Scholar]
- 23. Mou L., Xing Y., Kong Z., Zhou Y., Chen Z., and Wang R. (2011) The N-terminal domain of human hemokinin-1 influences functional selectivity property for tachykinin receptor neurokinin-1. Biochem. Pharmacol. 81, 661–668 [DOI] [PubMed] [Google Scholar]
- 24. Nakajima Y., Tsuchida K., Negishi M., Ito S., and Nakanishi S. (1992) Direct linkage of three tachykinin receptors to stimulation of both phosphatidylinositol hydrolysis and cyclic AMP cascades in transfected Chinese hamster ovary cells. J. Biol. Chem. 267, 2437–2442 [PubMed] [Google Scholar]
- 25. Sagan S., Chassaing G., Pradier L., and Lavielle S. (1996) Tachykinin peptides affect differently the second messenger pathways after binding to CHO-expressed human NK-1 receptors. J. Pharmacol. Exp. Ther. 276, 1039–1048 [PubMed] [Google Scholar]
- 26. Ahn S., Nelson C. D., Garrison T. R., Miller W. E., and Lefkowitz R. J. (2003) Desensitization, internalization, and signaling functions of β-arrestins demonstrated by RNA interference. Proc. Natl. Acad. Sci. U.S.A. 100, 1740–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. DeWire S. M., Ahn S., Lefkowitz R. J., and Shenoy S. K. (2007) β-Arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 [DOI] [PubMed] [Google Scholar]
- 28. Jean-Charles P. Y., Rajiv V., and Shenoy S. K. (2016) Ubiquitin-related roles of β-arrestins in endocytic trafficking and signal transduction. J. Cell. Physiol. 231, 2071–2080 [DOI] [PubMed] [Google Scholar]
- 29. Girnita L., Shenoy S. K., Sehat B., Vasilcanu R., Vasilcanu D., Girnita A., Lefkowitz R. J., and Larsson O. (2007) β-Arrestin and Mdm2 mediate IGF-1 receptor-stimulated ERK activation and cell cycle progression. J. Biol. Chem. 282, 11329–11338 [DOI] [PubMed] [Google Scholar]
- 30. Shenoy S. K., and Lefkowitz R. J. (2011) β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 32, 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cianfrocca R., Tocci P., Semprucci E., Spinella F., Di Castro V., Bagnato A., and Rosanò L. (2014) β-Arrestin 1 is required for endothelin-1-induced NF-κB activation in ovarian cancer cells. Life Sci. 118, 179–184 [DOI] [PubMed] [Google Scholar]
- 32. Shenoy S. K., Drake M. T., Nelson C. D., Houtz D. A., Xiao K., Madabushi S., Reiter E., Premont R. T., Lichtarge O., and Lefkowitz R. J. (2006) β-Arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. J. Biol. Chem. 281, 1261–1273 [DOI] [PubMed] [Google Scholar]
- 33. Jafri F., El-Shewy H. M., Lee M. H., Kelly M., Luttrell D. K., and Luttrell L. M. (2006) Constitutive ERK1/2 activation by a chimeric neurokinin 1 receptor-β-arrestin1 fusion protein: probing the composition and function of the G protein-coupled receptor “signalsome.” J. Biol. Chem. 281, 19346–19357 [DOI] [PubMed] [Google Scholar]
- 34. DeFea K. A., Vaughn Z. D., O'Bryan E. M., Nishijima D., Déry O., and Bunnett N. W. (2000) The proliferative and antiapoptotic effects of substance P are facilitated by formation of a β-arrestin-dependent scaffolding complex. Proc. Natl. Acad. Sci. U.S.A. 97, 11086–11091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rozengurt E. (1999) Autocrine loops, signal transduction, and cell cycle abnormalities in the molecular biology of lung cancer. Curr. Opin. Oncol. 11, 116–122 [DOI] [PubMed] [Google Scholar]
- 36. Voisin T., El Firar A., Fasseu M., Rouyer-Fessard C., Descatoire V., Walker F., Paradis V., Bedossa P., Henin D., Lehy T., and Laburthe M. (2011) Aberrant expression of OX1 receptors for orexins in colon cancers and liver metastases: an openable gate to apoptosis. Cancer Res. 71, 3341–3351 [DOI] [PubMed] [Google Scholar]
- 37. Rozengurt E. (2002) Neuropeptides as growth factors for normal and cancerous cells. Trends Endocrinol. Metab. 13, 128–134 [DOI] [PubMed] [Google Scholar]
- 38. Schulte G., and Shenoy S. K. (2011) β-Arrestin and dishevelled coordinate biased signaling. Proc. Natl. Acad. Sci. U.S.A. 108, 19839–19840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dasgupta P., Rizwani W., Pillai S., Davis R., Banerjee S., Hug K., Lloyd M., Coppola D., Haura E., and Chellappan S. P. (2011) ARRB1-mediated regulation of E2F target genes in nicotine-induced growth of lung tumors. J. Natl. Cancer Inst. 103, 317–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dasgupta P., Rastogi S., Pillai S., Ordonez-Ercan D., Morris M., Haura E., and Chellappan S. (2006) Nicotine induces cell proliferation by β-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J. Clin. Invest. 116, 2208–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kendall R. T., Lee M. H., Pleasant D. L., Robinson K., Kuppuswamy D., McDermott P. J., and Luttrell L. M. (2014) Arrestin-dependent angiotensin AT1 receptor signaling regulates Akt and mTor-mediated protein synthesis. J. Biol. Chem. 289, 26155–26166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Parathath S. R., Mainwaring L. A., Fernandez-L. A., Guldal C. G., Nahlé Z., and Kenney A. M. (2010) β-Arrestin-1 links mitogenic sonic hedgehog signaling to the cell cycle exit machinery in neural precursors. Cell Cycle 9, 4013–4024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Palma C., Nardelli F., Manzini S., and Maggi C. A. (1999) Substance P activates responses correlated with tumour growth in human glioma cell lines bearing tachykinin NK1 receptors. Br. J. Cancer 79, 236–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mandell J. W., Glass G., Gianchandani E. P., Locke C. N., Amos S., Bourne T. D., Schiff D., and Papin J. A. (2009) Dephosphorylation of β-arrestin 1 in glioblastomas. J. Neuropathol. Exp. Neurol. 68, 535–541 [DOI] [PubMed] [Google Scholar]
- 45. Yamaguchi R., Yamamoto T., Sakamoto A., Ishimaru Y., Narahara S., Sugiuchi H., and Yamaguchi Y. (2016) Substance P enhances tissue factor release from granulocyte-macrophage colony-stimulating factor-dependent macrophages via the p22phox/β-arrestin 2/Rho A signaling pathway. Blood Cells Mol. Dis. 57, 85–90 [DOI] [PubMed] [Google Scholar]
- 46. Krek W., and Nigg E. A. (1991) Mutations of p34cdc2 phosphorylation sites induce premature mitotic events in HeLa cells: evidence for a double block to p34cdc2 kinase activation in vertebrates. EMBO J. 10, 3331–3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. O'Farrell P. H. (2001) Triggering the all-or-nothing switch into mitosis. Trends Cell Biol. 11, 512–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chambard J. C., Lefloch R., Pouysségur J., and Lenormand P. (2007) ERK implication in cell cycle regulation. Biochim. Biophys. Acta 1773, 1299–1310 [DOI] [PubMed] [Google Scholar]
- 49. Warfel N. A., and Kraft A. S. (2015) PIM kinase (and Akt) biology and signaling in tumors. Pharmacol. Ther. 151, 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bavi P., Uddin S., Ahmed M., Jehan Z., Bu R., Abubaker J., Sultana M., Al-Sanea N., Abduljabbar A., Ashari L. H., Alhomoud S., Al-Dayel F., Prabhakaran S., Hussain A. R., and Al-Kuraya K. S. (2011) Bortezomib stabilizes mitotic cyclins and prevents cell cycle progression via inhibition of UBE2C in colorectal carcinoma. Am. J. Pathol. 178, 2109–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Muñoz M., and Coveñas R. (2011) NK-1 receptor antagonists: a new paradigm in pharmacological therapy. Curr. Med. Chem. 18, 1820–1831 [DOI] [PubMed] [Google Scholar]
- 52. Chun K. S., Lao H. C., and Langenbach R. (2010) The prostaglandin E2 receptor, EP2, stimulates keratinocyte proliferation in mouse skin by G protein-dependent and β-arrestin1-dependent signaling pathways. J. Biol. Chem. 285, 39672–39681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Talbot J., Joly E., Prentki M., and Buteau J. (2012) β-Arrestin1-mediated recruitment of c-Src underlies the proliferative action of glucagon-like peptide-1 in pancreatic β INS832/13 cells. Mol. Cell. Endocrinol. 364, 65–70 [DOI] [PubMed] [Google Scholar]
- 54. Yuan J., Yan R., Krämer A., Eckerdt F., Roller M., Kaufmann M., and Strebhardt K. (2004) Cyclin B1 depletion inhibits proliferation and induces apoptosis in human tumor cells. Oncogene 23, 5843–5852 [DOI] [PubMed] [Google Scholar]
- 55. Santamaría D., Barrière C., Cerqueira A., Hunt S., Tardy C., Newton K., Cáceres J. F., Dubus P., Malumbres M., and Barbacid M. (2007) Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448, 811–815 [DOI] [PubMed] [Google Scholar]
- 56. Diril M. K., Ratnacaram C. K., Padmakumar V. C., Du T., Wasser M., Coppola V., Tessarollo L., and Kaldis P. (2012) Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc. Natl. Acad. Sci. U.S.A. 109, 3826–3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gesty-Palmer D., Chen M., Reiter E., Ahn S., Nelson C. D., Wang S., Eckhardt A. E., Cowan C. L., Spurney R. F., Luttrell L. M., and Lefkowitz R. J. (2006) Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J. Biol. Chem. 281, 10856–10864 [DOI] [PubMed] [Google Scholar]
- 58. Dumesic P. A., Scholl F. A., Barragan D. I., and Khavari P. A. (2009) Erk1/2 MAP kinases are required for epidermal G2/M progression. J. Cell Biol. 185, 409–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Walsh S., Margolis S. S., and Kornbluth S. (2003) Phosphorylation of the cyclin b1 cytoplasmic retention sequence by mitogen-activated protein kinase and Plx. Mol. Cancer Res. 1, 280–289 [PubMed] [Google Scholar]
- 60. Shtivelman E., Sussman J., and Stokoe D. (2002) A role for PI 3-kinase and PKB activity in the G2/M phase of the cell cycle. Curr. Biol. 12, 919–924 [DOI] [PubMed] [Google Scholar]
- 61. Yin D. L., Liang Y. J., Zheng T. S., Song R. P., Wang J. B., Sun B. S., Pan S. H., Qu L. D., Liu J. R., Jiang H. C., and Liu L. X. (2016) EF24 inhibits tumor growth and metastasis via suppressing NF-κB dependent pathways in human cholangiocarcinoma. Sci. Rep. 6, 32167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Trembley J. H., Chen Z., Rodrigues C. M., Kren B. T., and Steer C. J. (2000) Genomic organization and promoter characterization of the rat cyclin B1 gene. Gene 255, 93–104 [DOI] [PubMed] [Google Scholar]
- 63. Liu F., Li X., Wang C., Cai X., Du Z., Xu H., and Li F. (2009) Downregulation of p21-activated kinase-1 inhibits the growth of gastric cancer cells involving cyclin B1. Int. J. Cancer 125, 2511–2519 [DOI] [PubMed] [Google Scholar]
- 64. Berges C., Fuchs D., Opelz G., Daniel V., and Naujokat C. (2009) Helenalin suppresses essential immune functions of activated CD4+ T cells by multiple mechanisms. Mol. Immunol. 46, 2892–2901 [DOI] [PubMed] [Google Scholar]
- 65. Cheng C., Jiao J. T., Qian Y., Guo X. Y., Huang J., Dai M. C., Zhang L., Ding X. P., Zong D., and Shao J. F. (2016) Curcumin induces G2/M arrest and triggers apoptosis via FoxO1 signaling in U87 human glioma cells. Mol. Med. Rep. 13, 3763–3770 [DOI] [PMC free article] [PubMed] [Google Scholar]