

Abstract
BACKGROUND
Mannitol, hypertonic saline, and progesterone may blunt leukocyte recruitment after traumatic brain injury (TBI). We hypothesized that progesterone reduces pericontusional recruitment of leukocytes to a greater extent than either osmotherapy a day after TBI.
METHODS
CD1 mice underwent controlled cortical impact and were treated with osmotherapy (mannitol and hypertonic saline) or progesterone. Thirty-two hours after TBI, live pial microscopy was used to evaluate leukocyte–endothelial interactions and immunohistochemistry was used for the detection of pericontusional tissue polymorphonuclear neutrophils. Neurologic recovery was assessed before sacrifice.
RESULTS
Mannitol resulted in the lowest in vivo leukocyte recruitment compared with progesterone (795 ± 282 vs 1,636 ± 434 LEU/100 μm/minutes, P < .05). Mannitol also displayed lower tissue accumulation of leukocytes as compared with progesterone (5.7 ± 1.7 vs 15.2 ± .1 LEU/mm2, P = .03). However, progesterone resulted in better neurologic recovery than either osmotherapy.
CONCLUSIONS
Leukocyte recruitment to injured brain is lowest with mannitol administration. How different agents alter progression of secondary brain injury will require further evaluation in humans.
Keywords: Intravital microscopy, Neuroinflammation, Traumatic brain injury, Osmotherapy, Progesterone, Polymorphonuclear neutrophil
Nearly 1.7 million cases of traumatic brain injury (TBI) are recorded in the United States yearly, making this disease the leading cause of death and disability among young Americans.1 Brain edema after TBI is thought to result from an impaired and leaking blood brain barrier (BBB) that results from the initial traumatic insult, and also from ongoing cerebrovascular inflammation and tissue damage.2 To date, no therapy exists to curb the persistent cerebral inflammation that follows initial brain injury.
In the last decade, increasing evidence has emerged suggesting that leukocytes, in particular the polymorphonuclear neutrophil (PMN), and endothelial cell interactions in the brain microcirculation may sustain the persistent microvascular disruption that evolves in the hours after injury.1,2 Well described in the systemic circulation, PMNs pass from the vasculature to the tissue by first rolling on the endothelial surface, then adhering, and finally transmigrating through endothelial cell tight junctions to perform their cytotoxic and phagocytic functions in tissue.3 In certain cases of injury, neutrophils may become prematurely activated and their inappropriate and uncontrolled release of cytotoxic substances may occur intravascularly resulting in ongoing vascular and organ injury.4 At the BBB, while not as well described, this may also occur, but in a delayed fashion following TBI.5 This ongoing leukocyte-mediated neurovascular inflammation may potentiate secondary brain injury and thus be an optimal target for therapies to mitigate leukocyte activation.
Clinically, both mannitol (MTL) and hypertonic saline (HTS) are routinely used osmotherapies to reduce elevated intracranial pressure (ICP) in brain injured patients with suspected cerebral edema.6 Both these agents may also possess immunomodulatory properties particularly affecting circulating and tissue-phase leukocytes.7–9 Although not an osmotherapy nor yet used routinely outside of clinical trials, progesterone (PRO) has been found to improve outcome in brain injured patients.10,11 We recently demonstrated reduced leukocyte–endothelial cell interactions in the brain circulation following TBI treated with PRO, which may contribute to its observed neuroprotective effects.12 In this study, we sought to compare the effects of standard osmotherapies (HTS and MTL) with PRO on leukocyte recruitment post-TBI.
Patients and Methods
Animal model—craniotomy and traumatic brain injury
All experiments were performed after approval by the Institutional Animal Care Committee of the University of Pennsylvania. Male CD1 mice (25 to 30 g) were housed in standard facilities for 5 to 7 days before study. On day 1, mice were anesthetized with intraperitoneal ketamine, xylazine, and acepromazine (100, 10, 1 mg/kg, respectively) and were maintained at normal body temperature. A tunneled right external jugular vein line (Laboratory Tubing, SILASTIC) was then placed for repeated long-term administration of intravenous (IV) therapies.13 After scalp incision, a left-sided, 4-mm first craniotomy, centered between the sagittal, bregma, and lambda sutures, was created with a trephine. The left parietotemporal cortex was then injured using a controlled cortical impact (CCI) device (AMS 201; AmScien Instruments, Richmond, VA). This resulted in an injury consistent with severe TBI (3-mm diameter impactor tip, impact velocity of 6 m/seconds, and cortical deformation of 1.0 mm).
Experimental protocol and study groups
Thirty animals were randomized into 5 groups: CCI + HTS (n = 6), CCI + MTL (n = 6), CCI + normal saline (NS: n = 6), CCI + PRO (n = 6), and Sham (craniotomy, no CCI: n = 6) (Fig. 1). Osmotherapy animals were given equiosmolar (5.5 mOsm/kg) IV doses of either HTS (Baxter, Deerfield, IL) or MTL (supplemented with NS: 7.4 mL/kg for prevention of dehydration because of the diuretic effect of MTL) (Baxter). NS animals received 3.2 mL/kg of NS (.9%) (Baxter) IV. All agents were administered immediately and at repeated intervals after CCI. PRO (16 mg/kg in 22.5% cyclodextrin) (Sigma Aldrich, St. Louis, MO) was given intraperitoneally immediately after CCI and at repeated intervals for 32 hours when animals were prepared for intravital microscopy.
Figure 1.
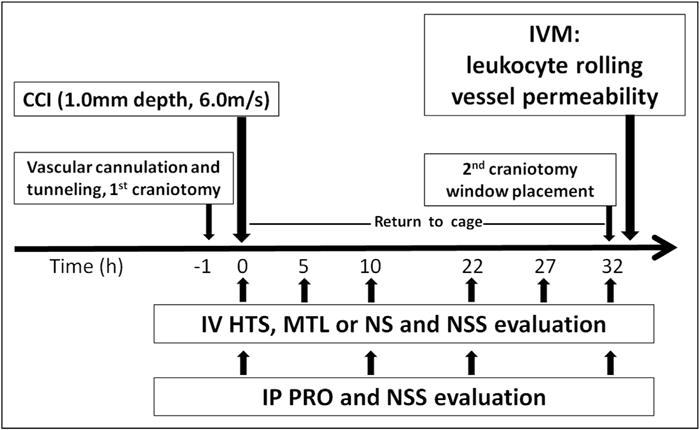
Timeline of experiments. Therapies were administrated at repeated intervals following CCI ranging from 5 to 12 hours, while progesterone was administered every 10 to 12 hours. IP = intraperitoneal.14
Evaluation of neurologic recovery (n = 30)
At the time of each treatment administration, rodent neurologic recovery was subjectively graded by the primary investigator using the Neurological Severity Score.14 The Neurological Severity Score is a composite index of motor, sensory, reflex, and balance tests graded on a scale from 3 to 18.
In vivo assessment of microcirculation (n = 30)
Thirty-two hours after CCI, a second 2.5-mm craniotomy was created in front of the first and covered with a 5-mm coverslip (Fisher Scientific, Waltham, MA). Animals were then transferred to an intravital microscope (ECLIPSE FN1; Nikon Instruments, Melville, NY) and received a 50-μL IV bolus of .3% rhodamine 6G to fluorescently label circulating leukocytes. Footage of the pial microcirculation was recorded under a 590-nm epi-illumination emission filter using a digital camera (QuantEM; Photometrics, Tucson, AR) for subsequent analysis by a blinded observer (Fig. 2A). Nonbranching postcapillary venules (25 to 50-μm diameter) were selected for 1-minute recordings in each animal. Once this recording was completed, 100 mg/kg fluorescein isothiocyanate (FITC)-labeled albumin (Sigma Aldrich) was administered and the same pial regions were visualized under a 488-nm fluorescent filter for determination of albumin leakage immediately outside the selected venules.
Figure 2.
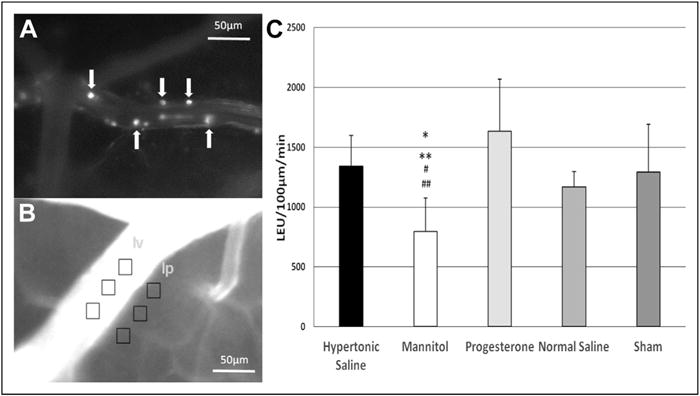
(A) Representative image of the pial microcirculation. White arrows indicate leukocytes rolling on the endothelium. (B) Representative image after FITC–albumin injection. Light intensity (gray) was measured within the vessel (Iv) and outside the vessel (Ip), where vascular permeability was calculated as mean Iv/mean Ip. (C) Leukocyte rolling on endothelium 32 hours after TBI. Scale bar = 50 μm. *P = .01 vs hypertonic saline, **P = .004 vs progesterone, #P = .02 vs normal saline, and ##P = .04 vs Sham.
Quantification of leukocyte–endothelial cell interactions and microvascular permeability (n = 30)
At a separate setting, leukocyte rolling (mean number of labeled cells crossing a 100-μm venular segment: cells/100 μm/minutes) was counted offline by a blinded observer using digital analysis software (NIS-Elements; Nikon Instruments) (Fig. 2A). FITC-labeled albumin recordings were also analyzed and fluorescence (light intensity) was measured in three distinct regions within the vessel (venular intensity, Iv) and outside the vessel wall (perivenular intensity, Ip). The ratio of mean Ip to mean Iv was calculated to determine the mean permeability index for the given vessel (Fig. 2B).
Wet-to-dry assessment of cerebral edema (n = 15)
After sacrifice, whole brains were excised and were weighed immediately (wet weight) and after 72 hours of drying at 70°C (dry weight). The wet-to-dry ratio (dry weight/wet weight) was calculated.
Immunohistochemistry and quantification of brain polymorphonuclear neutrophils (n = 8)
A subgroup of animals underwent transcardial perfusion with 1× PBS (Life technologies, Carlsbad, CA) and 10% neutral buffered formalin (Sigma Aldrich). The brain was excised and immediately postfixed in 10% neutral buffered formalin for up to 7 days. Brains were then dissected into 2-mm thick blocks in coronal plane and processed to paraffin using standard techniques. Analysis was performed using 8 micron-thick whole brain coronal sections at the epicenter of the contusion.
Antigen retrieval was performed on slides immersed in Tris-ethylenediaminetetraacetic acid buffer (pH 8.0) and placed in a microwave pressure cooker for 8 minutes. Sections were subsequently blocked using 2% normal goat serum (Vector Labs, Burlingame, CA) in Optimax buffer (BioGenex, San Ramon, CA) for 1 hour. Incubation with a rat monoclonal antibody specific for the protein Ly-6G (clone 1A8) (BD Biosciences, San Jose, CA) was then performed (1:45 K for 20 hours at 4°C). The Ly-6G protein is found predominantly on neutrophils.15 The primary antibody was then rinsed off and the sections incubated with biotinylated goat anti-rat immunoglobulin G for 30 minutes (Vector Labs), followed by an avidin biotin complex as per the manufacturer’s instructions (Vectastain Universal Elite kit; Vector Labs). Finally, visualization was conducted with DAB peroxidase substrate kit, as per manufacturer’s instructions (Vector Labs).
All stained sections were digitally scanned at 20× magnification and visualized using the associated Image-Scope software (Leica Biosystems, Wetzlar, Germany). The area of the hemisphere, both ipsilateral and contralateral to the contusion, was measured and the number of PMNs (as identified by 1A8 IHC) in this area was reported as PMNs/mm2.
Statistical analysis
All data are presented as mean ± standard deviation. Differences in means between groups were compared using the Student’s t-test or Kruskal–Wallis test. Differences in proportions were compared with the chi-square test using SPSS software (SPSS, Chicago, IL). A P value of less than .05 was considered statistically significant.
Results
In vivo leukocyte rolling and microvascular permeability
Thirty-two hours after injury, labeled leukocytes were visualized clearly as round spheres measuring 10 to 20 μm within the pial venules (Fig. 2A). Animals treated with HTS, PRO, and NS as well as the Sham group demonstrated similar levels of leukocyte rolling (Fig. 2C). However, animals treated with MTL (795 ± 282) demonstrated significantly lower leukocyte rolling than HTS, PRO, NS, and Sham (1,340 ± 259 vs 1,637 ± 434 vs 1,170 ± 127 vs 1,291 ± 401, P < .05 vs MTL, respectively). There were no differences between the groups in microvascular permeability as measured by in vivo leakage of FITC albumin.
Neurologic recovery during post-TBI period
Neurological recovery was assessed every time the animals received IV therapy. Five hours after TBI, Sham animals displayed greater mean NSS (17.1 ± 1.2) than all brain injured groups (P <.05) (Fig. 3). PRO animals tended to recover faster than all brain injured counterparts at 5 hours (14.3 ± 2.0 vs NS: 12.7 ± 1.6, P > .05). At 10 hours, treatment groups showed better recovery than NS. PRO animals displayed significantly greater mean NSS than NS (17.5 ± 1.1 vs 14.5 ± 1.7, P < .05). All animals demonstrated a full neurological recovery at 32 hours after TBI.
Figure 3.
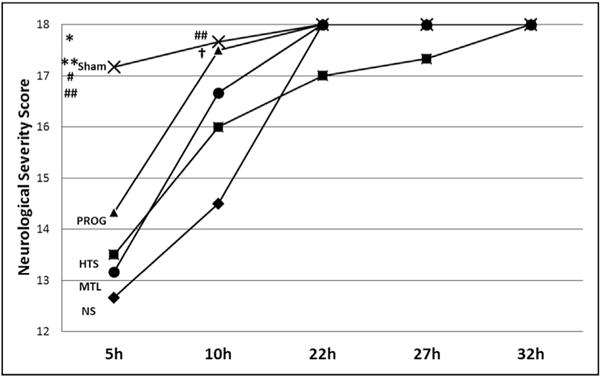
Neurologic recovery at different time points following TBI. All animals showed a full neurologic recovery at 32 hours after TBI. *P = .02 vs progesterone, **P = .001 vs hypertonic saline, #P = .004 vs mannitol, ##P < .003 vs normal saline, and †P = .008 vs normal saline. PRO = progesterone.
Brain tissue edema 32 hours after TBI
The whole brain wet-to-dry ratios were similar across all groups.
Immunohistochemical analysis of brain PMN sequestration
Thirty-two hours after TBI, brains were excised and sections stained with an immunohistochemical marker of PMNs (1A8). Following CCI, multiple IA8 immunoreactive cells were observed in brain parenchyma, primarily in the cortical, pericontusional region with limited migration in to the hippocampus and surrounding white matter. In contrast, 1A8 positive cells were virtually absent in the hemisphere contralateral to injury (Fig. 4C). Labeled cells had the morphologic appearance of PMNs, including multi-segmented nuclei. Fig. 4 demonstrates extensive PMNs in the contusional/pericontusional region 32 hours post-CCI (Fig. 4A) (scale bars 300 μm). Note the contusional lesion can be observed extending through the depth of the cortex. PMN counts in the ipsilateral hemisphere were lowest in the MTL group (5.7 ± 1.7 PMN/mm2) but similar in the other 3 groups. HTS (16.5 ± 1.2 PMN/mm2) and PRO animals (15.2 ± .1 PMN/mm2) demonstrated greater PMN recruitment than MTL counterparts (P < .05). There were no differences in PMN counts when analyzed in the contralateral hemisphere.
Figure 4.
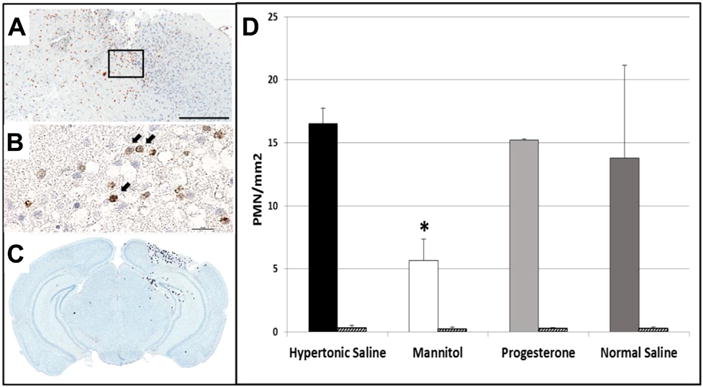
Representative immunohistochemistry images of coronal brain sections demonstrating PMNs in the contusional/pericontusional region as 1A8 positive cells indicated by black arrows (A) (Scale bar = 300 μm) with inset detail at 600× magnification (Scale bar = 20 μm) (B). Mean PMN counts were counted by a blinded observer (C). Solid and patterned bars indicate PMN counts in ipsilateral and contralateral hemispheres respectively (D). *P = .03 vs hypertonic saline, progesterone. (For interpretation of the references to color in this Figure, the reader is referred to the web version of this article.)
Comments
Persistent neuroinflammation following disruption of the BBB is a key mechanism underlying secondary brain injury after TBI. During this process, neutrophils are recruited to the pericontusional area and release proinflammatory cytokines, cytotoxic proteases, and reactive oxygen species, which in turn initiate the immune functions of the native glia.16 Thus, interruption or inhibition of leukocyte activation following TBI presents an attractive target to potentially reduce secondary brain injury.
MTL and HTS are routinely used osmotherapies to decrease interstitial and intracellular fluid volume in patients with severe TBI and lower ICP.6 In addition, to their physicochemical effects, these agents may also possess immunomodulatory properties.17 HTS has been shown to decrease direct PMN activation by reducing surface expression of adhesion receptors as well as their release of proteinases and reactive oxygen species.7–9 Blunting PMN–endothelial cell interactions by HTS has been reported in sepsis, burns, and ischemia–reperfusion models evaluating different organ systems.18–20 In muscle, our group previously reported blunted PMN–endothelial cell interactions in hemorrhagic shock resuscitated with HTS.21 MTL may also alter activated leukocytes by acting as a free radical scavenger,22,23 but little is known on whether it directly reduces PMN activation in the systemic circulation or in tissue. MTL also has other known effects on the microcirculation including alteration in the deformability of erythrocytes and reductions in blood viscosity24,25 resulting in increases in cerebral oxygen delivery.25 In this study, we have shown that MTL may also reduce mobilization of PMNs in the tissue surrounding the brain injury. This was demonstrated both in vivo and with immunohistochemistry in brain sections more than a day after TBI treatment with MTL. We chose this time frame for in vivo observation because previous studies have shown that brain PMN recruitment to areas of trauma is delayed and does not peak before 24 to 48 hours.5,26 Immunohistochemical evaluation found this effect to only occur in MTL-treated animals. No similar effect was seen in either HTS or PRO animals.
PRO in TBI has been investigated extensively in humans and animals.10,11,27 In particular, PRO has been shown to reduce glutamate toxicity in hippocampal neurons and its resultant toxic effect on cellular apoptosis by increasing anti-apoptotic protein, namely blc-2, expression.28 PRO has also been shown to closely regulate cellular expression of aquaporins, which are known to have a direct role in the development of brain edema following injury.29 Our group recently evaluated PRO in a TBI model evaluated at a longer time frame and found that PRO reduced both cerebral swelling as well as pial PMN–endothelial cell interactions as compared with placebo.12 In this study, when comparing PRO with standard osmotherapies, PMN recruitment in MTL animals was significantly lower than that found in PRO counterparts. However, improved neurologic recovery was seen with PRO in the first 10 hours following injury. It remains to be determined how PRO affects neutrophil mobilization following TBI and how this affects the development of secondary brain injury.
Our study has important limitations. First, the number of animals particularly in the immunohistochemical analyses is small and greater numbers may have demonstrated more subtle differences between groups. Second, only the MTL group received fluid resuscitation with NS. This was the result of an earlier modification to the experimental protocol to reflect the clinical practice of our and many other centers that routinely replace MTL-related urine losses with the same volume of isotonic saline when patients receive MTL for ICP correction. Without replacing the considerable natriuresis induced by MTL, patients can develop hypotension and suffer reductions in cerebral perfusion pressure. It is nonetheless possible that HTS and PRO animals were under-resuscitated particularly in the first day following TBI, as animals were observed to have poor oral fluid intake because of poor mobility. Third, the route (IV) and frequency (q5 to 12 hours) of osmotherapy administration favored a greater systemic exposure as compared with the less frequent intraperitoneal administration of PRO (q10 to 12 hours). Although this greater systemic exposure of osmotherpy may have had a greater effect on PMN and endothelium activation, frequent peritoneal injections (every 10 to 12 hours for 2 days) may also have been associated with abdominal septic complications from inadvertent puncture of the gastrointestinal tract that were unmeasured in the PRO group. Fourth, we did not evaluate systemic or cerebral hemodynamics including blood pressure, heart rate, ICP, or cerebral perfusion pressure, which would have allowed a greater ability to determine how different therapies affected parameters that can directly affect microcirculatory interactions.
In conclusion, we have demonstrated in an in vivo study how administration of MTL results in greater blunting of leukocyte–endothelial cell interactions as compared with HTS or PRO following TBI. Although we were able to show this benefit both in vivo and ex vivo, we were unable to demonstrate differences in brain edema formation and accumulation. Additional studies will be needed to determine if this particular effect of MTL is reproducible in human TBI patients and whether the preferential use of one osmotherapy over other therapies is warranted.
Discussion
Discussant: Dr Herb A. Phelan (Dallas, TX). It’s always difficult when one’s results don’t tell a consistent story. The fact that mannitol showed significant attenuation of the PMN-endothelial cell interaction without affecting the clinical picture, while progesterone showed the reverse, is vexing. At its most stripped-down level, these results are explainable for one of three reasons:
The model lacks verisimilitude;
PMN recruitment doesn’t matter for the clinical picture, which is unlikely; or.
PMN recruitment and transmigration do matter but there’s a threshold effect that isn’t reached here.
The authors nicely discussed the limitations of their model in their manuscript, and I will avoid rehashing most of that here. The one caveat about your model I would ask that you speak to, because it leads into my following questions, is whether we can really call this a model of severe TBI. This was a nonlethal model in which there was no need for airway control or ICP management. Brain edema was minimal or absent. All the animals’ exams had normalized by 32 hours after injury. How likely do you think it to be that there wasn’t a strong enough effect to cross a threshold, which manifested as a change in this clinical picture?
If in fact this is a model of milder TBI, do you have any plans to ascertain if these results are seen after a more severe level of injury? Indeed, there’s a strong clinical precedent for this, as the PROTect trials showed different levels of benefit of progesterone for different severities of injury.
Finally, do you have any plans to investigate other avenues of the neuroinflammatory cascade for these same three interventions? As you well know, the redundancy built into the brain’s recovery pathways is considerable, and apoptosis, selectin expression, and myelin promotion are but a few of the multiple parallel pathways that could be affected. It would be interesting to see how this study’s results stand up to other immunomodulatory activities after TBI.
Dr Jose Pascual: I think your points are very well raised, showing that we found something that was opposite to our hypothesis. The model is an important issue. We do know this is a validated CCI model for severe traumatic brain injury. The velocity of the probe that dives within a given depth of brain tissue and at a given speed is specifically validated by Smith and others over 15 years ago as being a replicator of penetrating severe traumatic brain injury.
Perhaps it would have been better to do a lateral fluid percussion model, which may have been more of a blunt or closed traumatic brain injury. But the whole issue of intravital microscopy, where the brain is exposed to the outside world with open craniotomy, would then render that a little bit into question as to whether that’s a better model.
I think we were a little perplexed that the positive controls really did not show significant differences to the negative controls. To that, we postulated that since we are ligating the carotid artery, the jugular vein after the infusion of fluids and the insertion of indwelling catheters that may have made the negative controls also injured, as well as the process of doing two craniotomies on these smaller animals.
I think it’s interesting you mention the PROTect trial, which has now been stopped because of futility, we think. Yet the two previous, phase 1 and subsequent trials, showed that there was a possibility that progesterone as a steroid, as a sex steroid, potentially made a difference. We could not find this here, in contrast to the study we presented last year where we compared progesterone versus placebo, and it did reduce leukocyte mobilization.
Dr Alicia Mangram (Phoenix, AZ). It seems that we keep kind of going around and around. We go to hypertonic saline, we get away from progesterone, now we’re looking at mannitol in the 3%. Where are we going? Do you think it’s kind of a combination? I really have been impressed by the last two papers looking at traumatic brain injury, because I still feel perplexed in what’s the correct management, how aggressive should we be, should we put in monitors, don’t put in monitors?
Your comments.
Dr Jose Pascual: The NIH ceased two trials in 2009 that came out of the ROC consortium, where hypertonic saline was administered in brain-injured patient but also hemorrhagic shock patients in the prehospital setting. That was stopped for futility, but if you look at the data, it looks like patients who received hypertonic saline prehospital may have had a higher mortality. That threw all of that into question.
I think we reviewed five trials that have compared head-to-head mannitol in humans with hypertonic saline. Two of them showed the superiority of hypertonic saline, one of them was equivocal, and the other two showed, out of Europe that mannitol was better.
To summarize, I would say our neuro ICU group, though it’s been, I guess, favoring hypertonic saline in last few years, cannot do that anymore. And what we have chosen to do is if the volume status of the patient is diminished, then we don’t use mannitol. Even though I have worked on hypertonic saline for many years, I think there’s no justification to give hypertonic saline to the euvolemic patient with traumatic brain injury, because there’s no evidence that it’s superior.
Dr Herb Phelan (Dallas, TX). As an editorial comment, one of the things I talk about on rounds a lot when we round on our TBI patients is I made a point about all the redundancy that’s in these pathways. But it’s so hard, as you show in an animal model, okay, this pathway, A equals B, there’s a cause and effect here. But in vivo in humans, it’s just so much more problematic. But I think if we ever do wind up with true immunomodulatory therapies for these, it’s going to be like an HIV cocktail, where it’s going to be a multiple medication regimen, as opposed to the silver bullet that we are all hoping for. I just don’t see it happening.
Acknowledgments
We would like to thank Ms Robin Armstrong for the technical and organizational assistance and Professor Tetsuo Yukioka of Tokyo Medical University for his support and guidance.
Footnotes
The authors declare no conflicts of interest.
Presented at the 66th Annual Meeting of the Southwestern Surgical Congress, April 15, 2014, Scottsdale, Arizona.
References
- 1.Rosenfeld JV, Maas AI, Bragge P, et al. Early management of severe traumatic brain injury. Lancet. 2012;380:1088–98. doi: 10.1016/S0140-6736(12)60864-2. [DOI] [PubMed] [Google Scholar]
- 2.Lukaszewicz AC, Soyer B, Payen D. Water, water, everywhere: sodium and water balance and the injured brain. Curr Opin Anaesthesiol. 2011;24:138–43. doi: 10.1097/ACO.0b013e32834458af. [DOI] [PubMed] [Google Scholar]
- 3.Pascual JL, Khwaja KA, Chaudhury P, et al. Hypertonic saline and the microcirculation. J Trauma. 2003;54(5 Suppl):S133–40. doi: 10.1097/01.TA.0000064526.33647.63. [DOI] [PubMed] [Google Scholar]
- 4.Gando S, Kameue T, Matsuda N, et al. Combined activation of coagulation and inflammation has an important role in multiple organ dysfunction and poor outcome after severe trauma. Thromb Haemost. 2002;88:943–9. [PubMed] [Google Scholar]
- 5.Li S, Marks J, Sanati P, et al. In vivo evolution of microvascular inflammation after traumatic brain injury: an intravital microscopy study. Crit Care Med. 2011;39:1. [Google Scholar]
- 6.Hinson HE, Stein D, Sheth KN. Hypertonic saline and mannitol therapy in critical care neurology. J Intensive Care Med. 2013;28:3–11. doi: 10.1177/0885066611400688. [DOI] [PubMed] [Google Scholar]
- 7.Rizoli SB, Kapus A, Fan J, et al. Immunomodulatory effects of hypertonic resuscitation on the development of lung inflammation following hemorrhagic shock. J Immunol. 1998;161:6288–96. [PubMed] [Google Scholar]
- 8.Yada-Langui MM, Anjos-Valotta EA, Sannomiya P, et al. Resuscitation affects microcirculatory polymorphonuclear leukocyte behavior after hemorrhagic shock: role of hypertonic saline and pentoxifylline. Exp Biol Med (Maywood) 2004;229:684–93. doi: 10.1177/153537020422900713. [DOI] [PubMed] [Google Scholar]
- 9.Angle N, Cabello-Passini R, Hoyt DB, et al. Hypertonic saline infusion: can it regulate human neutrophil function? Shock. 2000;14:503–8. [PubMed] [Google Scholar]
- 10.Wright DW, Kellermann AL, Hertzberg VS, et al. ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann Emerg Med. 2007;49:391–402. doi: 10.1016/j.annemergmed.2006.07.932. 402.e1–2. [DOI] [PubMed] [Google Scholar]
- 11.Xiao G, Wei J, Yan W, et al. Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: a randomized controlled trial. Crit Care. 2008;12:R61. doi: 10.1186/cc6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pascual JL, Murcy MA, Li S, et al. Neuroprotective effects of progesterone in traumatic brain injury: blunted in vivo neutrophil activation at the blood-brain barrier. Am J Surg. 2013;206:840–5. doi: 10.1016/j.amjsurg.2013.07.016. discussion, 845–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.David Wasserman OM, Ayala Julio. Glucose Clamping the Conscious Mouse: A Laboratory Course. Available at: http://www.mc.vanderbilt.edu/documents/mmpc/files/2012%20Lab%20Manual.pdf. Accessed February 12, 2014.
- 14.Schaar KL, Brenneman MM, Savitz SI. Functional assessments in the rodent stroke model. Exp Transl Stroke Med. 2010;2:13. doi: 10.1186/2040-7378-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daley JM, Thomay AA, Connolly MD, et al. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 16.Morganti-Kossmann MC, Satgunaseelan L, Bye N, et al. Modulation of immune response by head injury. Injury. 2007;38:1392–400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Soustiel JF, Vlodavsky E, Zaaroor M. Relative effects of mannitol and hypertonic saline on calpain activity, apoptosis and polymorphonuclear infiltration in traumatic focal brain injury. Brain Res. 2006;1101:136–44. doi: 10.1016/j.brainres.2006.05.045. [DOI] [PubMed] [Google Scholar]
- 18.Vachharajani V, Vital S, Russell J, et al. Hypertonic saline and the cerebral microcirculation in obese septic mice. Microcirculation. 2007;14:223–31. doi: 10.1080/10739680601139153. [DOI] [PubMed] [Google Scholar]
- 19.Nolte D, Bayer M, Lehr HA, et al. Attenuation of postischemic microvascular disturbances in striated muscle by hyperosmolar saline dextran. Am J Physiol. 1992;263:H1411–6. doi: 10.1152/ajpheart.1992.263.5.H1411. [DOI] [PubMed] [Google Scholar]
- 20.Barone M, Jimenez F, Huxley VH, et al. Morphologic analysis of the cerebral microcirculation after thermal injury and the response to fluid resuscitation. Acta Neurochir Suppl. 1997;70:267–8. doi: 10.1007/978-3-7091-6837-0_83. [DOI] [PubMed] [Google Scholar]
- 21.Pascual JL, Ferri LE, Seely AJ, et al. Hypertonic saline resuscitation of hemorrhagic shock diminishes neutrophil rolling and adherence to endothelium and reduces in vivo vascular leakage. Ann Surg. 2002;236:634–42. doi: 10.1097/00000658-200211000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong CH, Liu TZ, Chye SM, et al. Sevoflurane-induced oxidative stress and cellular injury in human peripheral polymorphonuclear neutrophils. Food Chem Toxicol. 2006;44:1399–407. doi: 10.1016/j.fct.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 23.Alvarez B, Ferrer-Sueta G, Radi R. Slowing of peroxynitrite decomposition in the presence of mannitol and ethanol. Free Radic Biol Med. 1998;24:1331–7. doi: 10.1016/s0891-5849(98)00005-7. [DOI] [PubMed] [Google Scholar]
- 24.Rosner MJ, Coley I. Cerebral perfusion pressure: a hemodynamic mechanism of mannitol and the postmannitol hemogram. Neurosurgery. 1987;21:147–56. doi: 10.1227/00006123-198708000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Burke AM, Quest DO, Chien S, et al. The effects of mannitol on blood viscosity. J Neurosurg. 1981;55:550–3. doi: 10.3171/jns.1981.55.4.0550. [DOI] [PubMed] [Google Scholar]
- 26.Clark RS, Schiding JK, Kaczorowski SL, et al. Neutrophil accumulation after traumatic brain injury in rats: comparison of weight drop and controlled cortical impact models. J Neurotrauma. 1994;11:499–506. doi: 10.1089/neu.1994.11.499. [DOI] [PubMed] [Google Scholar]
- 27.Deutsch ER, Espinoza TR, Atif F, et al. Progesterone’s role in neuroprotection, a review of the evidence. Brain Res. 2013;1530:82–105. doi: 10.1016/j.brainres.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 28.Nilsen J, Brinton RD. Impact of progestins on estrogen-induced neuroprotection: synergy by progesterone and 19-norprogesterone and antagonism by medroxyprogesterone acetate. Endocrinology. 2002;143:205–12. doi: 10.1210/endo.143.1.8582. [DOI] [PubMed] [Google Scholar]
- 29.Schumacher M, Guennoun R, Ghoumari A, et al. Novel perspectives for progesterone in hormone replacement therapy, with special reference to the nervous system. Endocr Rev. 2007;28:387–439. doi: 10.1210/er.2006-0050. [DOI] [PubMed] [Google Scholar]