

Abstract
The gene encoding the AT-rich interaction domain-containing protein 1B (ARID1B) has recently been shown to be one of the most frequently mutated genes in patients with intellectual disability (ID). The phenotypic spectrums associated with variants in this gene vary widely ranging for mild to severe nonspecific ID to Coffin–Siris syndrome. In this study, we evaluated three children from a consanguineous Emirati family affected with ID and dysmorphic features. Genomic DNA from all affected siblings was analyzed using CGH array and whole-exome sequencing (WES). Based on a recessive mode of inheritance, homozygous or compound heterozygous variants shared among all three affected children could not be identified. However, further analysis revealed a heterozygous variant (c.4318C>T; p.Q1440*) in the three affected children in an autosomal dominant ID causing gene, ARID1B. This variant was absent in peripheral blood samples obtained from both parents and unaffected siblings. Therefore, we propose that the most likely explanation for this situation is that one of the parents is a gonadal mosaic for the variant. To the best of our knowledge, this is the first report of a gonadal mosaicism inheritance of an ARID1B variant leading to familial ID recurrence.
Keywords: ARID1B, non-Mendelian inheritance, whole-exome sequencing, gonadal mosaicism
INTRODUCTION
Intellectual disability (ID), also called intellectual development disorder (IDD), is a neurodevelopmental disorder defined by significant impairment of cognitive and adaptive functions in early childhood. To date, many genes and loci across the genome have been implicated in ID. Haploinsufficiency for the AT-rich interaction domain-containing protein 1B (ARID1B) has been reported to be responsible for non-syndromic ID (ARID1B-related ID, (MRD12) OMIM 614562), and Coffin–Siris syndrome [Hoyer et al., 2012; Santen et al., 2012]. ARID1B encodes a component of the Brahma-associated factor (BAF) complex, also known as the mammalian SWItch/sucrose non-fermentable (SWI/SNF) complex. The BAF-complex is an ATP-dependent chromatin remodelling complex, which modifies chromatin structure and facilitates access of transcription factors to DNA [Hurlstone et al., 2002; Nie et al., 2003; Hargreaves and Crabtree, 2011]. Recent discoveries demonstrated a crucial role for the BAF complex in neurodevelopment and cancer [Lessard et al., 2007; Singhal et al., 2010; Flores-Alcantar et al., 2011; Santen et al., 2012; Kadoch and Crabtree, 2013; Ronan et al., 2013; Wu and Roberts, 2013; Biegel et al., 2014; Santen et al., 2014]. Mutations and chromosomal abnormalities affecting genes encoding subunits of the BAF complex were found to be responsible for several syndromic and non-syndromic forms of ID and cancers [Hoyer et al., 2012; Santen et al., 2012; Ronan et al., 2013; Wu and Roberts, 2013; Biegel et al., 2014; Santen et al., 2014]. In this study, we report a novel variant in the ARID1B gene explaining a sporadic occurrence of ID and dysmorphic features in three siblings with consanguineous Emirati parents likely due to gonadal mosaicism in one of the parents.
PATIENTS AND METHODS
Patients and Ethics Statement
We ascertained a consanguineous Emirati family with three affected children showing intellectual disability and dysmorphic features. This study has been approved by Al-Ain Medical Human Research Ethics Committee according to the national regulations (protocol number 10/09). The parents in this family provided a signed informed written consent and agreed in writing for the use of photographs in medical publication.
Molecular Analysis
Peripheral blood samples were collected from members of the affected family in EDTA tubes. DNA samples were analyzed first by CGH array. The whole exome capture and next-generation sequencing, including library construction, DNA capturing, sequencing, and data analysis, were carried out by Baylor-Hopkins Center for Mendelian Genomics (www.mendeliangenomics.org) and the variant filtering analysis was performed using the PhenoDB Variant Analysis tool [Sobreira et al., 2015]. We captured the CCDS exonic regions and flanking intronic regions totaling ~51 Mb by using the Agilent SureSelect XT kit and performed paired end 100 bp reads with the Illumina HiSeq2500 platform. We aligned each read to the 1000 genomes phase 2 (GRCh37) human genome reference with the Burrows–Wheeler Alignment (BWA) version 0.5.10-tpx [Li and Durbin, 2009]. Local realignment around indels and base call quality score recalibration were performed using the Genome Analysis Toolkit (GATK) [McKenna et al., 2010] version 2.3-9-ge5ebf34. Variant filtering was done using the Variant Quality Score Recalibration (VQSR) method [DePristo et al., 2011]. For SNVs the annotations of MQRankSum, HaplotypeScore, QD, FS, MQ, ReadPosRankSum were used in the adaptive error model (six max Gaussians allowed, worst 3% used for training the negative model). HapMap3.3 and Omni2.5 were used as training sites with HapMap3.3 used as the truth set. SNVs were filtered to obtain all variants up to the 99th percentile of truth sites (1% false negative rate). For indels the annotations of QD, FS, ReadPosRankSum were used in the adaptive error model (four max Gaussians allowed, worst 12% used for training the negative model, indels that had annotations more than 10 standard deviations from the mean were excluded from the Gaussian mixture model). A set of curated indels obtained from the GATK resource bundle (Mills_and_1000G_gold_standard.indels.b37.vcf) were used as training and truth sites. Indels were filtered to obtain all variants up to the 99th percentile of truth sites (1% false negative rate). The mean percentage of the target bases with at least 30× coverage was 90.5%. Sanger DNA sequencing was carried out to evaluate the segregation of the variants identified by WES. Primers for all potential causative variants were designed using Primer3 version 0.4.0 (http://primer3.ut.ee/). Amplified PCR products were cleaned up using EXO-SAP-IT and sequenced using the BigDye Terminator kit v3.1 (Applied Biosystems, USA) on a the ABI 3130xl Genetic Analyzer (Applied Biosystems, USA). DNA chromatograms were inspected and analyzed based on cDNA sequence in accordance with the GenBank entry NM_004933.2 for CDH15 and NM_020732.3 for ARID1B using the Sequencing Analysis® 5.3 software (Applied Biosystems) and ClustalW2 algorithms (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
RESULTS
Clinical Data
The parents are second cousins Emirati nationals of Baloushi origin (Fig. 1A). They have six children, three of them are affected. The parent’s appearances are normal and they have a normal IQ. There is no family history of similar problem.
FIG. 1.
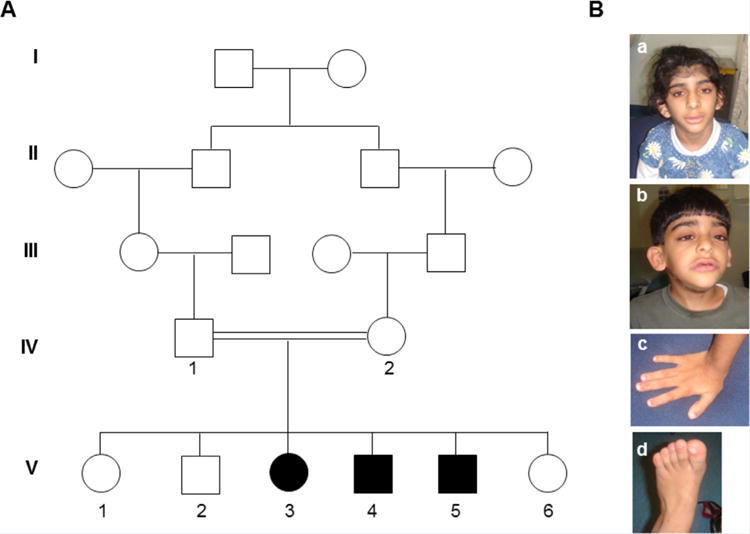
Pedigree of consanguineous family with three intellectual disable children. A: Pedigree of consanguineous family showing three affected children with intellectually disable and dysmorphic features. Circles and squares denote females and males respectively, filled symbols represent affected members, double lines denote consanguineous marriage, roman numbers indicate the first generation until their offspring, arabic numbers depict all participants in this study. B: Pictures of affected children showing the typical feature of ARID1B-related ID. B.a,b: showing abnormal head shape and low-set, posteriorly rotated, and abnormally shaped ears, low anterior hair line with thick eyebrows, broad nasal tip, large mouth with thick lower lip and hypertrichosis. B.c: shows shows short fingers. B:d: shows broad big toe. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/ajmga]
Patient 1
A 14 year-old female (Fig. 1A_V-3), product of normal pregnancy and term normal vaginal delivery. Her birth weight was 2,420 g (3rd centile) and head circumference 34 cm (25th centile). During the first few weeks of life, parents noted that she was hypoactive with feeding difficulties. She was seen in the Paediatric clinic at the age of 8 months due to delayed developmental milestones. She weighed 6 kg (below 3rd centile) and had a head circumference of 43 cm (50th centile). She has dysmorphic features including brachycephaly, narrow forehead, low hair line, long eyelashes, and low set ears. There was alternating squint but detailed eye examination was normal. She was unable to sit or role over, there was severe head lag, axial hypotonia with exaggerated reflexes. There was feeding and sucking problems. Chromosome analysis was normal. MRI brain showed complete agenesis of corpus callosum. Renal ultrasound was normal. She was evaluated in the genetic clinic at the age of 10 years. She was noticed to have global developmental delay with delayed speech (says few words only) and autistic behavior. Parents reported that she did not like loud sound and tends to put her hands on her ears when there is any loud noise. She was dysmorphic with narrow forehead and low anterior hair line, thick eyebrows with medial flare, long eyelashes, broad nasal tip, wide mouth with thick lower lip, low set prominent ears, and wide big toes. She had hypertrichosis over her entire body. She had autistic behavior with aggressive tendency.
Patient 2
A 10-year-old boy is the product of normal pregnancy and term delivery (Fig. 1A_V-4). No birth measurements are available. He was kept in NICU for 13 days due to irregular heart beat and echocardiography revealed a small VSD. He was assessed by a neurologist at the age of 6.5 months because of developmental delay. He had head lag with axial hypotonia and hyperreflexia and reasonable social interaction. There was a soft systolic murmur at the left sternal edge. His developmental milestones as following: he started to sit alone at 14 months of age, stand at 2 years of age, and walk at 27 months of age. Brain CT scan showed corpus callosum dysgenesis, hearing test and vision assessments were normal. Karyotype, amino acids chromatography, echocardiography, and skeletal surveys were all normal. He was evaluated in genetic clinic at the age of 6 years. His weight was 22 kg (>50th centile), height 110 cm (25th centile) and head circumference 50 cm (25th centile). He had dysmorphic features including brachycephaly, narrow forehead with low anterior hair line, thick eyebrows with medial flare, broad nasal tip, prominent philtrum, wide mouth with thick lower lip, and low set prominent ears. He had short fingers, broad big toes, and hypertrichosis over his entire body. He had moderate intellectual disability with delayed speech (says 2–3 word sentences). Microarray and fragile X mutation analysis were normal.
Patient 3
A 9-year-old boy the product of normal pregnancy and delivery (Fig. 1A_V-5). His birth weight was 2,430 g (>3rd centile) and head circumference 33 cm (10th centile). He was assessed in the pediatric clinic at the age of 2 months because of slow sucking, and failure to gain weight. He was found to have facial dysmorphic features with low anterior hair line, low set ears, high arched palate, hypotonia with head lag, and hyperreflexia. Echocardiography showed bicuspid aortic valve. Amino acid chromatography, urine organic acid chromatography, MRI brain, ultrasound kidney, and karyotyping were all normal. Ophthalmology examination and hearing assessment were normal. He was evaluated in genetic clinic at the age of 5 years. His weight was 18 kg (50th centile), height 105 cm (>10th centile) and head circumference 50 cm (40th centile). He had developmental delay and delayed speech (no words) with dysmorphic features including brachycepaly, narrow forehead with low anterior hairline, medial flare of the eyebrows, wide nasal tip, prominent philtrum, thick lips, low set ears, short fingers, broad big toes, and hypertrichosis over his entire body.
Whole-exome Sequencing Revealed a Novel Heterozygous Variant in ARID1B With Gonadal Mosaicism Segregation
Due to consanguinity and absence of phenotype in the parents, we assumed an autosomal recessive mode of inheritance in this family. However, filtering the whole-exome sequencing variants data failed to identify any homozygous or compound heterozygous variant that is in agreement with a recessive inheritance model. By accepting a dominant inheritance model, we were able to identify two heterozygous variants in known disease-causing genes, CDH15 and ARID1B. The variant in CDH15 is a known SNP (rs201368808) in exon 14 leading to the substitution of an arginine residue with glutamine at position 775 (c.2324G>A/p.R775Q). This variant has a minor allele frequency of A = 0.0018/9 in the 1000 Genomes Project database and 0.001154 in the ExAC database (http://exac.broadinstitute.org). In addition, Sanger sequencing showed that this variant is heterozygous in all three affected children, their mother (IV-2) and one unaffected sibling (V-2). SIFT, PROVEN, and Mutation Taster predicted this variant to be neutral while PolyPhen2 predicted it to be possibly damaging. Collectively, these findings strongly suggest that this variant (rs201368808) is benign without any clinical significance. The second variant is a novel single nucleotide substitution (c.4318C>T) detected in exon 18 of the ARID1B gene. Sanger sequencing validated the segregation of this deletion in the studied family showing that both parents and all unaffected siblings were homozygous for the wild-type allele while all three affected children were heterozygous (Fig. 2A). This variant results in a nonsense mutation with introduction of premature termination codon at amino acid position 1440 (p.Q1440*) of this 2249 amino acid protein. This mutation is not described in the exome variant server (http://evs.gs.washington.edu/EVS/), dbSNP database, 1000 Genomes project database, or in the ExAC database. In addition, p.Q1440* was absent in 200 ethnically matched normal control chromosomes which confirmed the novelty of this variant.
FIG. 2.
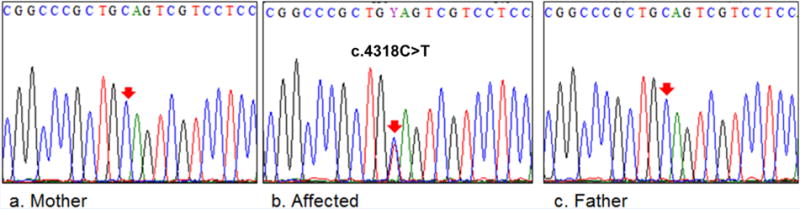
Molecular analysis of patients with ARID1B mutation. A: DNA chromatograms of ARID1B mutation compared to wild-type sequence. A.b: shows the heterozygous nonsense mutation (c.4318C>T) detected in the affected patients. A.a,c: show normal chromatograms in the parents. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/ajmga]
DISCUSSION
Heterozygous pathogenic variants and cytogenetic abnormalities involving ARID1B are considered the leading cause of Coffin–Siris syndrome (CSS) being found in 68–83% of cases [Santen et al., 2013; Wieczorek et al., 2013; Santen et al., 2014; Tsurusaki et al., 2014]. These abnormalities are also found in 0.9% of unselected intellectual disability (ID) patients [Hoyer et al., 2012]. All the reported disease-producing variants in ARID1B are truncating and predicted to result in haploinsufficiency of the protein product [Santen et al., 2014]. The phenotypic features of the syndromic ARID1B haploinsufficiency are variable ranging from mild to severe ID associated with speech delay and facial features that include low anterior hair line, coarse face, thick eyebrow, broad nasal tip, large mouth with thick lower lip. There is usually hypertrichosis, corpus callosum abnormalities, and hypoplasia of the nails of fifth fingers/toes. A recent review of 60 patients with ARID1B mutations from several CSS cohort studies in the literature concluded that there is a broad variation in the phenotype and speculated on the possibility of unidentified genetic factors that may modify the phenotype and determine the severity of phenotype in affected patients [Santen et al., 2014]. This study also concluded that the major features of the ARID1B phenotype, present in almost all patients with prior CSS diagnosis are: intellectual disability (100%), speech delay (100%), coarse features (90%) and hypertrichosis (95%) while minor features present in a smaller but significant fraction of patients such as small 5th finger or toe nails (81%), short 5th finger (73%), feeding difficulties (65%), agenesis of corpus callosum (35%), seizures (23%), and growth delay (19%). The authors suggested that patients with 2/3 major features and at least two minor features should be tested for ARID1B gene mutations [Santen et al., 2014]. Mari et al. [2015] suggested that other hand and feet anomalies, including broadening of distal phalanges and broad hallux, might be present in this syndrome [Mari et al., 2015]. All reported cases so far have been sporadic suggesting high rates of new mutations in this gene [Santen et al., 2014].
The family we describe here is consanguineous with three of six children affected with moderate degree of ID and dysmorphic features. All three children had low anterior hair line with thick eyebrows, broad nasal tip, large mouth with thick lower lip and hypertrichosis. In addition, two of them had hypoplastic corpus callosum. There was no hypoplasia of the nails of 5th fingers or toes but there were broad halluces in all of them. The parents were completely phenotypically normal. The presence of consanguinity with three affected children of both sexes and phenotypically normal parents suggests autosomal recessive inheritance in this family. However, WES failed to identify a homozygous or compound heterozygous variant that could explain the etiology of the phenotype in this family. We then considered autosomal dominant inheritance with incomplete penetrance or highly variable expressivity [Beratis et al., 1971; Russell et al., 1995]. However, careful examination of the parents and absence of the variant in their constitutional DNA ruled this possibility out. Therefore, we conclude the most likely explanation is that one of the parents is a germline mosaic. Germline mosaicism has been reported in several autosomal dominant conditions [Ng et al., 2004; Chiang et al., 2010; Barbaro et al., 2012; Danda et al., 2014; Hufnagel et al., 2014; Armaroli et al., 2015] and gives the impression of autosomal recessive inheritance when there is recurrence of an autosomal dominant trait among offspring of phenotypically normal parents. This possibility should be considered when analyzing exome sequencing data.
Acknowledgments
UAEU; Grant number: 31R017; Grant sponsor: National Human Genome Research Institute; Grant number: 1U54HG006542.
We are indebted the family members for their participation in this study. We are grateful to UAEU for their financial support (grant number 31R017). A grant from the National Human Genome Research Institute (1U54HG006542) also provided support for this work.
Footnotes
Conflict of interest: All authors have declared that no competing interests exist.
References
- Armaroli A, Trabanelli C, Scotton C, Venturoli A, Selvatici R, Brisca G, Merlini L, Bruno C, Ferlini A, Gualandi F. Paternal germline mosaicism in collagen VI related myopathies. Eur J Paediatr Neurol. 2015;19:533–536. doi: 10.1016/j.ejpn.2015.04.002. [DOI] [PubMed] [Google Scholar]
- Barbaro V, Nardiello P, Castaldo G, Willoughby CE, Ferrari S, Ponzin D, Amato F, Bonifazi E, Parekh M, Calistri A, Parolin C, Di Iorio E. A novel de novo missense mutation in TP63 underlying germline mosaicism in AEC syndrome: Implications for recurrence risk and prenatal diagnosis. Am J Med Genet A. 2012;158A:1957–1961. doi: 10.1002/ajmg.a.35414. [DOI] [PubMed] [Google Scholar]
- Beratis NG, Hsu LY, Hirschhorn K. Familial de Lange syndrome. Report of three cases in a sibship. Clin Genet. 1971;2:170–176. [PubMed] [Google Scholar]
- Biegel JA, Busse TM, Weissman BE. SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet. 2014;166C:350–366. doi: 10.1002/ajmg.c.31410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JM, Chen HW, Tang RP, Chen JS, Changchien CR, Hsieh PS, Wang JY. Mutation analysis of the APC gene in Taiwanese FAP families: Low incidence of APC germline mutation in a distinct subgroup of FAP families. Fam Cancer. 2010;9:117–124. doi: 10.1007/s10689-009-9292-2. [DOI] [PubMed] [Google Scholar]
- Danda S, van Rahden VA, John D, Paul P, Raju R, Koshy S, Kutsche K. Evidence of Germline Mosaicism for a Novel BCOR Mutation in Two Indian Sisters with Oculo-Facio-Cardio-Dental Syndrome. Mol Syndromol. 2014;5:251–256. doi: 10.1159/000365768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maquire JR, Hartington C, Philippakis AA, del Angel C, Rivas MA, Hanna M, McKenna A, Fennel TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Alcantar A, Gonzalez-Sandoval A, Escalante-Alcalde D, Lomelí H. Dynamics of expression of ARID1A and ARID1B subunits in mouse embryos and in cells during the cell cycle. Cell Tissue Res. 2011;345:137–148. doi: 10.1007/s00441-011-1182-x. [DOI] [PubMed] [Google Scholar]
- Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: Genetics, genomics and mechanisms. Cell Res. 2011;21:396–420. doi: 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, Wiesener A, Wohlleber E, Dufke A, Rossier E, Petsch C, Zweier M, Göhring I, Zink AM, Rappold G, Schröck E, Wieczorek D, Riess O, Engels H, Rauch A, Reis A. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet. 2012;90:565–572. doi: 10.1016/j.ajhg.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufnagel SB, Weaver KN, Hufnagel RB, Bader PI, Schorry EK, Hopkin RJ. A novel dominant COL11A1 mutation resulting in a severe skeletal dysplasia. Am J Med Genet A. 2014;164A:2607–2612. doi: 10.1002/ajmg.a.36688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlstone AF, Olave IA, Barker N, van Noort M, Clevers H. Cloning and characterization of hELD/OSA1, a novel BRG1 interacting protein. Biochem J. 2002;364:255–264. doi: 10.1042/bj3640255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Crabtree GR. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell. 2013;153:71–85. doi: 10.1016/j.cell.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron. 2007;55:201–215. doi: 10.1016/j.neuron.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari F, Marozza A, Mencarelli MA, Lo Rizzo C, Fallerini C, Dosa L, Di Marco C, Carignani G, Baldassarri M, Cianci P, Vivarelli R, Vascotto M, Grosso S, Rubegni P, Caffarelli C, Pretegiani E, Fimiani M, Garavelli L, Cristofoli F, Vermeesch JR, Nuti R, Dotti MT, Balestri P, Hayek J, Selicorni A, Renieri A. Coffin-Siris and Nicolaides-Baraitser syndromes are a common well recognizable cause of intellectual disability. Brain Dev. 2015;37:527–536. doi: 10.1016/j.braindev.2014.08.009. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng D, Thakker N, Corcoran CM, Donnai D, Perveen R, Schneider A, Hadley DW, Tifft C, Zhang L, Wilkie AO, van der Smagt JJ, Gorlin RJ, Burgess SM, Bardwell VJ, Black GC, Biesecker LG. Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat Genet. 2004;36:411–416. doi: 10.1038/ng1321. [DOI] [PubMed] [Google Scholar]
- Nie Z, Yan Z, Chen EH, Sechi S, Ling C, Zhou S, Xue Y, Yang D, Murray D, Kanakubo E, Cleary ML, Wang W. Novel SWI/SNF chromatinremodeling complexes contain a mixed-lineage leukemia chromosomal translocation partner. Mol Cell Biol. 2003;23:2942–2952. doi: 10.1128/MCB.23.8.2942-2952.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronan JL, Wu W, Crabtree GR. From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet. 2013;14:347–359. doi: 10.1038/nrg3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell MB, Andersson PG, Thomsen LL, Iselius L. Cluster headache is an autosomal dominantly inherited disorder in some families: A complex segregation analysis. J Med Genet. 1995;32:954–956. doi: 10.1136/jmg.32.12.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EA, Hilhorst-Hofstee Y, Wessels MW, den Hollander NS, Ruivenkamp CA, van Ommen GJ, Breuning MH, den Dunnen JT, van Haeringen A, Kriek M. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet. 2012;44:379–380. doi: 10.1038/ng.2217. [DOI] [PubMed] [Google Scholar]
- Santen GW, Aten E, Vulto-van Silfhout AT, Pottinger C, van Bon BW, van Minderhout IJ, Snowdowne R, van der Lans CA, Boogaard M, Linssen MM, Vijfhuizen L, van der Wielen MJ, Vollebregt MJ, Breuning MH, Kriek M, van Haeringen A, den Dunnen JT, Hoischen A, Clayton-Smith J, de Vries BB, Hennekam RC, van Belzen MJ, consortium CS Coffin-Siris syndrome and the BAF complex: Genotype-phenotype study in 63 patients. Hum Mutat. 2013;34:1519–1528. doi: 10.1002/humu.22394. [DOI] [PubMed] [Google Scholar]
- Santen GW, Clayton-Smith J, consortium ABC The ARID1B phenotype: What we have learned so far. Am J Med Genet C Semin Med Genet. 2014;166C:276–289. doi: 10.1002/ajmg.c.31414. [DOI] [PubMed] [Google Scholar]
- Singhal N, Graumann J, Wu G, Araúzo-Bravo MJ, Han DW, Greber B, Gentile L, Mann M, Schöler HR. Chromatin-Remodeling Components of the BAF Complex Facilitate Reprogramming. Cell. 2010;141:943–955. doi: 10.1016/j.cell.2010.04.037. [DOI] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Boehm C, Valle D, Hamosh A. New tools for Mendelian disease gene identification: PhenoDB variant analysis module; and GeneMatcher, a web-based tool for linking investigators with an interest in the same gene. Hum Mutat. 2015;36:425–431. doi: 10.1002/humu.22769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurusaki Y, Okamoto N, Ohashi H, Mizuno S, Matsumoto N, Makita Y, Fukuda M, Isidor B, Perrier J, Aggarwal S, Dalal AB, Al-Kindy A, Liebelt J, Mowat D, Nakashima M, Saitsu H, Miyake N. Coffin-Siris syndrome is a SWI/SNF complex disorder. Clin Genet. 2014;85:548–554. doi: 10.1111/cge.12225. [DOI] [PubMed] [Google Scholar]
- Wieczorek D, Bögershausen N, Beleggia F, Steiner-Haldenstätt S, Pohl E, Li Y, Milz E, Martin M, Thiele H, Altmüller J, Alanay Y, Kayserili H, Klein-Hitpass L, Böhringer S, Wollstein A, Albrecht B, Boduroglu K, Caliebe A, Chrzanowska K, Cogulu O, Cristofoli F, Czeschik JC, Devriendt K, Dotti MT, Elcioglu N, Gener B, Goecke TO, Krajewska-Walasek M, Guillén-Navarro E, Hayek J, Houge G, Kilic E, Simsek-Kiper P, López-González V, Kuechler A, Lyonnet S, Mari F, Marozza A, Mathieu Dramard M, Mikat B, Morin G, Morice-Picard F, Ozkinay F, Rauch A, Renieri A, Tinschert S, Utine GE, Vilain C, Vivarelli R, Zweier C, Nürnberg P, Rahmann S, Vermeesch J, Lüdecke HJ, Zeschnigk M, Wollnik B. A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum Mol Genet. 2013;22:5121–5135. doi: 10.1093/hmg/ddt366. [DOI] [PubMed] [Google Scholar]
- Wu JN, Roberts CW. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013;3:35–43. doi: 10.1158/2159-8290.CD-12-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]