

Abstract
BACKGROUND
Traumatic brain injury (TBI) confers a high risk of venous thrombosis, but early prevention with heparinoids is often withheld, fearing cerebral hematoma expansion. Yet, studies have shown heparinoids not only to be safe but also to limit brain edema and contusion size after TBI. Human TBI data also suggest faster radiologic and clinical neurologic recovery with earlier heparinoid administration. We hypothesized that enoxaparin (ENX) after TBI blunts in vivo leukocyte (LEU) mobilization to injured brain and cerebral edema, while improving neurologic recovery without increasing the size of the cerebral hemorrhagic contusion.
METHODS
CD1 male mice underwent either TBI by controlled cortical impact (CCI, 1-mm depth, 6 m/s) or sham craniotomy. ENX (1 mg/kg) or vehicle (VEH, 0.9% saline, 1 mL/kg) was administered at 2, 8, 14, 23, and 32 hours after TBI. At 48 hours, intravital microscopy was used to visualize live LEUs interacting with endothelium and microvascular leakage of fluorescein isothiocyanate–albumin. Neurologic function (Neurological Severity Score, NSS), activated clotting time, hemorrhagic contusion size, as well as brain and lung wet-to-dry ratios were evaluated post mortem. Analysis of variance with Bonferroni correction was used for statistical comparisons between groups.
RESULTS
Compared with VEH, ENX significantly reduced in vivo LEU rolling on endothelium (72.7 ± 28.3 LEU/100 μm/min vs. 30.6 ± 18.3 LEU/100 μm/min, p = 0.02) and cerebrovascular albumin leakage (34.5% ± 8.1% vs. 23.8% ± 5.5%, p = 0.047). CCI significantly increased ipsilateral cerebral hemisphere edema, but ENX treatment reduced post-CCI edema to near control levels (81.5% ± 1.5% vs. 77.6% ± 0.6%, p < 0.01). Compared with VEH, ENX reduced body weight loss at 24 hours (8.7% ± 1.2% vs. 5.8% ± 1.1%, p < 0.01) and improved NSS at 24 hours (14.5 ± 0.5 vs. 16.2 ± 0.4, p < 0.01) and 48 hours (15.1 ± 0.4 vs. 16.7 ± 0.5, p < 0.01) after injury. There were no significant differences in activated clotting time, hemorrhagic contusion size, and lung water content between the groups.
CONCLUSION
ENX reduces LEU recruitment to injured brain, diminishing visible microvascular permeability and edema. ENX may also accelerate neurologic recovery without increasing cerebral contusion size. Further study in humans is necessary to determine safety, appropriate dosage, and timing of ENX administration early after TBI.
Keywords: Enoxaparin, traumatic brain injury, leukocyte, blood-brain barrier, mice
Traumatic brain injury (TBI) is the leading cause of death and disability in young Americans, with 1.7 million cases occurring yearly. It is characterized by a primary mechanical insult followed by a multifaceted secondary response, which further exacerbates brain swelling and functional impairments. Inflammatory processes initiated by the innate host immune response, in particular circulating leukocytes (LEUs) and their intense communication with cerebrovascular endothelium are believed to be one of the major etiologic contributors to secondary brain injury. After the initial injury, circulating LEU interact with an activated blood-brain barrier (BBB) and progress through conformational changes, ending in the migration into the cerebral parenchyma.2,3 In conditions of severe tissue injury, this sequence may become overexuberant and lead to the unbridled activation of proteases, release of reactive oxygen species, and production of proinflammatory cytokines, and ultimately lead to BBB impairment and permeability.4–8
Enoxaparin (ENX), a low-molecular-weight heparin, has a long established history of reducing injury-related venous thrombotic complications by blocking two key coagulation factors, factor Xa and factor II. Compared with unfractionated heparin, ENX may have superior bioavailability, higher anti-Xa/anti-IIa ratio, and a longer half-life, making it the agent of choice following multiple injuries and TBI.9–11 Intriguingly, growing evidence indicates that irrespective of its antithrombotic effect, ENX may cross the BBB12,13 and reduce cerebral edema and cerebral lesion size after experimental TBI and stroke.13–17 Some human TBI data also suggest faster radiologic resolution and neurologic recovery with earlier heparinoid administration after TBI.18 In different animal tissue injury models, ENX was found to blunt tissue inflammation through inhibition of LEU activation and adhesion to endothelial cells (ECs),19 thereby diminishing LEU invasion,14 markedly suppressing neutrophil elastase release,20 and reducing tumor necrosis factor α (TNF-α) levels.21 Nevertheless, it remains unknown whether ENX treatment, after TBI interferes with cerebrovascular LEU-ECs interactions and whether this results in diminished brain edema and ultimately improved neurologic recovery. To address these questions, we sought to observe the live cerebral microcirculation following treatment of TBI with ENX and hypothesized that ENX would reduce pericontusional cerebrovascular permeability and cerebral edema as well as improve cognitive function without increasing intracranial bleeding.
MATERIALS AND METHODS
Experimental Design and Study Groups
All experimental procedures were conducted with approval of the Institutional Animal Care and Use Committee of the University of Pennsylvania. Adult male CD1 mice weighing 25 g to 30 g were housed in standard facilities for 5 days to 7 days before study with access to water and food ad libitum. Thirty-eight animals were randomly assigned to one of four groups as follows: (1) sham craniotomy and vehicle (VEH, 0.9% normal saline) without CCI (no CCI + VEH, n = 9); (2) sham craniotomy and ENX (no CCI + ENX, n = 9); (3) TBI and vehicle (0.9% normal saline) (CCI + VEH, n = 11); (4) TBI and ENX (CCI + ENX, n = 9). ENX (1 mg/kg, Winthrop, Sanofiaventis, NJ) or an equal volume of 0.9% saline (1 mL/kg, Baxter, Deerfield, IL) were injected subcutaneously at 2, 8, 14, 23, and 32 hours after CCI (Fig. 1). The dose and frequency of administration were based on previous reports, indicating clinical relevance with an associated neuroprotective effect and minimal risk of hemorrhage.15,22
Figure 1.

Experimental procedures. A, Timeline of experimental procedures. B, Schematic representation of the localization of both craniotomies and CCI-induced cortical contusion surrounded by penumbral area. IVM, intravital microscopy; s.c., subcutaneously.
Severe TBI Model Preparation
CCI was used to create brain injury through a widely used and well-validated model of TBI in rodents as explained elsewhere.23–25 In brief, on Day 1, mice were anesthetized with intraperitoneal ketamine, xylazine, and acepromazine (KXA: 100, 10, 1 mg/kg, respectively) and placed prone in a stereotactic device. After exposure of the skull through an apical scalp incision, a left-sided, 4-mm craniotomy was created between bregma and lambda sutures using a dental drill (Henry Schein, Melville, NY). Special attention was given to leave uninjured the underlying dura mater. The left parietotemporal cortex was then injured via a controlled cortical impactor (AMS201, AmScien Instruments, Richmond, VA), which resulted in a reproducible injury (3-mm-diameter impactor, tip, impact velocity of 6 m/s, and cortical deformation of 1 mm).
Pial Intravital Microscopy Preparation
In vivo assessment of the cerebral microcirculation was conducted using pial intravital microscopy 48 hours after CCI on the live animal as described previously.24,26 After intraperitoneal anesthesia using KXA as mentioned earlier, the right jugular vein was cannulated for the administration of fluorescent dyes. A second 2.5-mm craniotomy was then created immediately anterior to and, using the same procedure as in the first craniotomy, and covered with a 5-mm coverslip (Fisher Scientific, Waltham, MA). To maintain physiologic conditions, warm normal saline was used to continuously superfuse the dura mater beneath the window. Animals were again placed in the stereotactic device and transferred to an intravital microscope (ECLIPSE FN1, Nikon Instruments, Melville, NY). Fifty microliters of 0.3% rhodamine 6G (Sigma-Aldrich, St. Louis, MO) was injected in travenously to label LEU. Using a 40× water immersion objective, the cerebral microcirculation was visualized and nonbranching venules (diameter, 25–50 μm) were randomly selected for video capture. One-minute long footage was recorded under a 590-nm epiillumination emission filter using a digital camera (QuantEM, Photometrics, Tucson, AR) and saved as an .avi file. Thereafter, 100-mg/kg fluorescein isothiocyanate (FITC)–labeled albumin (Sigma-Aldrich) were administrated intravenously for in vivo analysis of transendothelial macromolecular leakage. Footage of the same pial sites was captured digitally for 10 seconds through a 488-nm emission filter. All video clips were subsequently analyzed offline by a blinded observer.
Offline Quantification of LEU/EC Interactions and Albumin Leakage
Microscopy video recordings were analyzed frame-by-frame by a blinded observer who quantified LEU/EC cell interactions in 100-μm venular segments using NIS-Elements software (Nikon Instruments, Melville, NY). Fluorescently labeled intravascular cells 7 μm to 12 μm in diameter were counted as LEU, and their interactions with endothelium were documented as follows: (1) rolling leukocytes, travelling slower than the central blood flow velocity; (2) adherent leukocytes, remaining at the same location for at least 30 seconds. Leukocyte-endothelial interactions were quantified and reported as number of cells per 100 μm per minute. Microvascular permeability was determined by calculating the ratio of FITC-labeled albumin fluorescence intensity (grays) in three separate locations immediately outside the vessel wall (Iout) to that in the vessel for three separate locations immediately within the venule (Iin) (Fig. 2B).
Figure 2.

In vivo leukocyte/endothelial interactions and microvascular permeability. A, Representative image showing LEU interacting with endothelium. White arrows indicate live circulating LEUs labeled with Rhodamine 6G. B, Representative image showing FITC-albumin leakage in the cerebral microcirculation. BBB permeability index was calculated as the ratio of mean fluorescence of three separate locations outside the vessel wall (Iout) to mean fluorescence within the venule (Iin). C, Bar graphs indicate the number of rolling LEUs and FITC-albumin leakage in the pial penumbral microcirculation 48 hours after injury. The increased number of rolling LEUs induced by CCI was significantly reduced after ENX treatment. Concurrently, the elevated FITC-albumin leakage following injury was reduced by ENX. *p < 0.05 and **p < 0.01 versus CCI + VEH group.
Physiologic Parameters
Animal weight loss following cerebral injury is common and illustrates diminished ability for the animal to conduct activities of daily living. Animal body weight was thus obtained at 0, 24, and 48 hours after CCI and results were expressed as body weight loss ratio [(W0h − W24h or 48h)/W0h]. In some animals, activated clotting time (ACT) (25) was obtained from blood drawn from the jugular line 48 hours after CCI using an i-STAT blood analyzer (Abbot Laboratories, Abbott Park, IL).
Gross Assessment of Hemorrhagic Contusion Area
Animals were sacrificed with ketamine overdose and cervical dislocation and systemically perfused with 1% phosphate-buffered solution (Life Technologies, Carlsbad, CA) and 10% formalin (Sigma-Aldrich) through the right cardiac ventricle. Brains were procured, and the dorsal surface of the injured hemisphere was evaluated for size of the hemorrhagic contusion using Adobe Photoshop CS6 Extended software (Adobe Systems, San Jose, CA). The surface area encompassed by the hemorrhagic contusion was reported as a percentage of the total surface ipsilateral hemisphere area.
Brain and Lung Water Content
After procurement, the brain was divided along the midline into hemispheres. The animal lungs were then obtained following sternotomy. Both organs were weighed immediately (wet weight, WW) and dried at 70°C for 72 hours and weighed again to obtain dry weight (DW). Water content was calculated as a percentage of WW (% water content = [WW − DW]/WW × 100%).
Functional Neurologic Recovery
Neurologic function was assessed 24 and 48 hours after CCI using the validated modified Neurological Severity Scale (NSS),27 which scores motor, sensory, reflex, and balance ability to a maximum sum score of 18 points.
Statistical Analysis
All data are expressed as mean ± SD. Statistical analyses were performed using SPSS software (version 19, SPSS, Chicago, IL). Comparisons between multiple groups were conducted using analysis of variance using post hoc Bonferroni correction. p < 0.05 was considered statistically significant.
RESULTS
In Vivo Leukocyte Rolling and Microvascular Permeability
Forty-eight hours after CCI, LEU rolling was greatly increased in injured animals, but this was notably reduced to uninjured control levels by ENX treatment (72.7 ± 28.3 LEUs/100 μm/min vs. 30.6 ± 18.3 LEUs/100 μm/min, p = 0.021, Fig. 2C). Leukocyte adhesion was rare and did not differ between the groups (overall mean ± SD, 0.29 ± 0.49 LEU/100 μm/min). Concurrently, 48 hours after CCI, in vivo microvascular penumbral permeability as indicated by FITC-labeled albumin leakage was significantly elevated in injured animals (CCI + VEH group) as compared with the uninjured counterparts (34.5% ± 8.1% vs. 10.8% ± 7.5%, p < 0.01, Fig. 2C). Again, ENX treatment following CCI partially restored BBB integrity, reducing albumin leakage that was found in the CCI + VEH group (23.8% ± 5.5% vs. 34.5% ± 8.1%, p = 0.047).
Brain and Lung Tissue Edema 48 Hours After TBI
Mice in the CCI + VEH group demonstrated a significantly greater brain water content in both cerebral hemispheres 48 hours after TBI as compared with the no CCI + VEH group (ipsilateral, 81.5% ± 1.5% vs. 75.6% ± 1.6%, p < 0.01; contralateral, 77.5% ± 0.8% vs. 75.2% ± 0.9%, p < 0.01; Fig. 3A). Compared with the CCI + VEH group, cerebral hemisphere water content ipsilateral to the injury was markedly attenuated by ENX administration (81.5% ± 1.5% vs. 77.6% ± 0.6%, p < 0.01). Brain water content contralateral to the injury was similar in the CCI + VEH and the CCI + ENX groups (77.5% ± 0.8% vs. 76.7% ± 0.4%). No significant differences were found in lung water content among the different groups (Fig. 3B).
Figure 3.

Animal outcomes. A, Brain water content in the hemisphere ipsilateral to the injury was greatly increased by CCI (CCI + VEH [n = 8]) as compared with both uninjured controls (no CCI + VEH [n = 6] and no CCI + ENX [n = 6]) 48 hours after injury. ENX treatment (CCI + ENX [n = 6]) markedly decreased brain water content in the ipsilateral hemisphere without altering that found in the hemisphere contralateral to the injury. Ipsilateral, **p < 0.01 versus CCI + VEH group; contralateral, ##p < 0.01 versus CCI + VEH. B, Lung water content was similar in all groups (n = 22) 48 hours after CCI. C, Posttrauma body weight loss was greatest in the CCI + VEH group (n = 8). ENX treatment blunted animal weight loss to near control levels 24 hours after CCI. **p < 0.01 versus CCI + VEH; 48 hours: #p < 0.05 versus CCI + VEH. D, Neurologic performance as judged by the NSS was uniformly normal in uninjured animals throughout. Injured animals demonstrated impaired neurologic function both 24 hours and 48 hours after CCI. ENX treatment significantly restored NSS scores at both time intervals following injury. 24 hours, **p < 0.01 versus CCI + VEH; 48 hours, ##p < 0.01 versus CCI + VEH.
Body Weight Loss, ACT, and Size of Hemorrhagic Contusion
As shown in Figure 3C, body weight loss was greatest 24 hours after TBI, but ENX treatment reduced weight loss significantly (8.7% ± 1.2% vs. 5.8% ± 1.1%, p < 0.01). Body weight loss ratios were similar in all groups at 48 hours. No significant differences were found in ACT among the different groups. The proportion of the surface hemisphere that was contused was similar in both injured groups without change after ENX administration (CCI + VEH group, 31.5% ± 3.4% vs. CCI + ENX group, 27.0% ± 6.0%; p > 0.05; Fig. 4).
Figure 4.
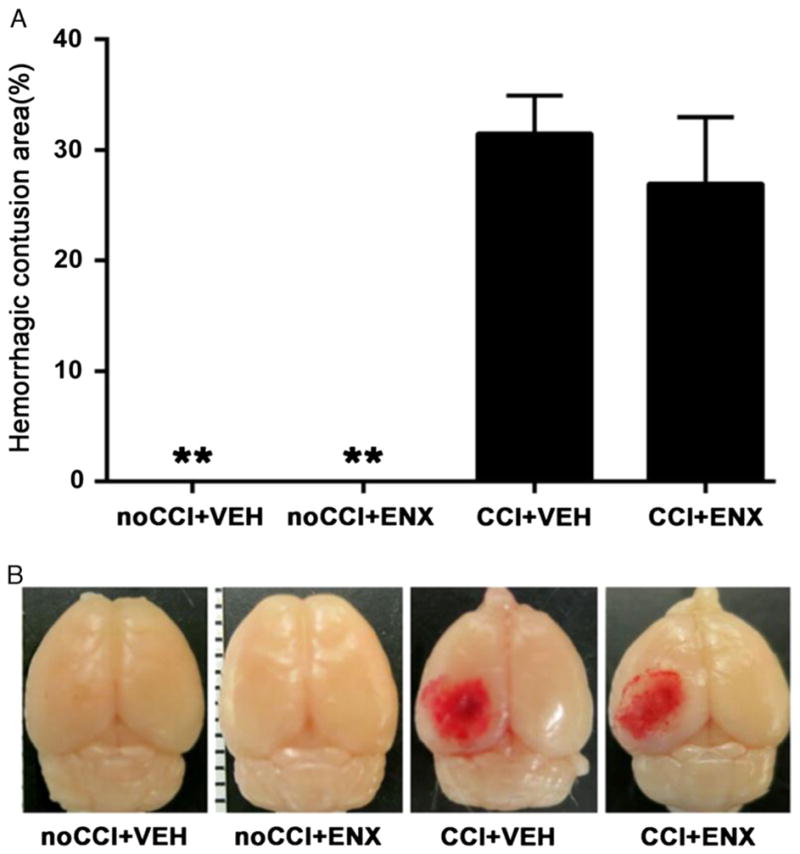
Pial surface contusion. A, Injury resulted in a mean dorsal surface hematoma proportion of the hemisphere of (29.2% ± 5.0%). ENX administration did not cause further expansion of CCI-induced hemorrhagic contusion. **p < 0.01 versus CCI + VEH. B, Representative images showing gross pathologic specimens for each group 48 hours after CCI.
Neurologic Recovery During Post-TBI Period
Compared with either uninjured group, neurologic functional impairment was evident both at 24 hours and 48 hours after CCI in the CCI + VEH animals (24 hours, 18.0 ± 0 vs. 14.5 ± 0.5, p < 0.01; 48 hours, 18 ± 0 vs. 15.1 ± 0.4, p < 0.01; Fig. 3D). In injured animals, ENX administration improved functional neurologic activity at both time intervals after CCI as compared with vehicle (24 hours, 16.2 ± 0.4 vs. 14.5 ± 0.5, p < 0.01; 48 hours, 16.7 ± 0.5 vs. 15.1 ± 0.4, p < 0.01).
DISCUSSION
In the present study, we provide the first in vivo evidence that a low molecular weight heparin blocks cerebrovascular interactions between activated neutrophils and endothelium and that this leads to a visible reduction in microvascular permeability. This effect is subsequently confirmed by ex vivo findings, revealing edema in the injured cerebral hemisphere without expansion of the surface cerebral hematoma despite the known anticoagulant effect of ENX. Furthermore, ENX-associated reductions in edema were associated with improved clinical recovery as evidenced by less animal weight loss and improved cognitive recovery in the early postinjury period. Taken together, these findings suggest that a commonly used low molecular weight heparin administered after TBI may not only be safe but, in this controlled animal model, also lead to improved neurologic recovery through blunting of LEU-mediated BBB permeability and progression of brain swelling.
It is well established that systemic injury greatly promotes venous thrombotic events and that TBI doubles this risk.28,29 Nonetheless, specific recommendations on when to use heparinoids and in what dose or frequency have remained uncertain because of safety concerns that they may exacerbate intracranial bleeding.11,30,31
Excessive posttraumatic neuroinflammation has been characterized as a major contributor to secondary brain injury in different models of cerebral insult. In particular, peripheral LEU infiltration into brain tissue with subsequent activation of resident immune cells such as microglia and astrocytes has been found to occur through an imbalance between local concentrations of proinflammatory and anti-inflammatory cytokines.32,33 In this process, activation of circulating LEU and their sequential interaction with activated endothelium has been proposed as one of the earliest initiators of the microvascular leakiness and permeability that leads to local tissue edema.34 To emigrate into the brain parenchyma, LEUs follow a sequence of steps, that is, margination and rolling on endothelium followed by firm adhesion and transmigration through the BBB into the tissue phase.7,35 In conditions of severe tissue injury, this process may become unbridled, driven by excessive up-regulation of surface adhesion molecules on both LEU and endothelium. 36 It has been shown in a variety of models how activated LEUs release cytotoxic substances, including matrix metalloproteinases, reactive oxygen species, and other proteolytic enzymes, in part through greater secretion of local proinflammatory cytokines (TNF-α, interleukin 1β). 37,38 At the BBB, these processes facilitate disproportionate activation of resident astrocytes and microglia, leading to further tissue injury and pervasive disruption of BBB integrity.39 Injured cerebrovascular endothelium may also promote microvascular LEU trafficking through up-regulation of endothelial adhesion molecules, including E-selectin, VCAM-1, and ICAM-1, which specifically promote LEU rolling and adhesion to endothelium and further stimulate LEU capture and sequestration.40 Taken together, this evidence advocates an intimate relationship between the magnitude of post-traumatic LEU accumulation and the extent of brain edema and brain tissue destruction in secondary brain injury.41–43
In the last decade, increasing evidence has surfaced, indicating that independent of their anticoagulant effect, heparinoids also act as pleiotropic modulators of tissue inflammation particularly through blunting of LEU activation.15,19,44 ENX was found to block LEU infiltration in myocardial ischemia and lung injury 14,45 and was further observed to inhibit surface expression of EC adhesion receptors (ICAM-1, VCAM-1, E-selectin) responsible for interactions with LEU.19,46 In experimental endotoxemia, Iba et al. used intravital microscopy in rats and found reductions in LEU/EC interactions with better perfusion of tissues and, similar to our study, found no increased bleeding complications.47,48 In central nervous system (CNS) diseases, previous studies focused on potential neuroprotective effects of ENX in various conditions, without investigating the effect of ENX on LEU-mediated cerebral inflammatory responses or correlating this to reduced edema and improved cognitive function. In a rodent stroke model, ENX was found to reduce infarct size, resulting in improved cognitive function.13,49 In a separate study using a rodent moderate-TBI model, ENX was found to reduce oxidative damage, inflammation, and astrocytosis.22 In a different TBI rat model, repeated ENX treatment resulted in reduced cerebral edema in multiple regions of the brain and reduced cognitive impairment as measured with a Lashley maze task assessment.15 To our knowledge, the current study is the first reported observation of in vivo modulation by ENX on LEU/ECs interactions in the cerebral microcirculation. In agreement with previous reports, our study linked live reductions in LEU/EC interactions and reduced microvascular leakiness. While ENX effects on LEU may not have been causative of improved cognitive function, the undeniable association between both could be a window into the neuroprotective mechanisms of ENX.
Interestingly, our group recently reviewed 75 sequential severe TBI patients and found faster neuroclinical recovery and less progression of cerebral hemorrhage and edema on serial postinjury head computed tomographic scans if heparin was started within 3 days of injury.18
While this study may present novel evidence of ENX neuroprotective effects, it is a small animal study and has a number of limitations. First, we did not examine the dose-dependent effect of ENX on LEU-ECs interactions, and doses used (1 mg/kg) mirrored therapeutic human doses, not prophylactic low molecular weight heparin. Nonetheless, the dose used reflected previously published models of CNS rodent studies15,22 and, despite the higher doses used, did not translate into greater hematoma expansion. Second, we only assessed bleeding complications by evaluating the relative surface size of the hemorrhagic contusions. Additional histologic measurement of hematoma depth and immunohistochemical analysis confirming reduced LEU brain sequestration would have been optimal. Finally, ACT was used to determine ENX coagulopathic activity instead of the more traditional measurement of anti-Xa levels, but good evidence exists suggesting that ACT may be as sensitive as anti-Xa levels for gauging ENX activity.50
Taken together, previous findings have demonstrated how ENX blunts activation of LEU and endothelium and separately how it also reduces cerebral edema and improves cognitive recovery after CNS insults. In our opinion, the current study adds novel evidence linking how ENX blunts visible LEU/EC interactions concurrently with reducing microvascular permeability, in vivo, in the same pericontusional pial region. It further confirms improved cognitive recovery following ENX administration in the same animals without evidence of increased contusional bleeding. While counterintuitive, these results may help elucidate the mechanism behind the observed neuroprotective effects of heparinoids and lead to clinical studies that will confirm or refute this effect in humans.
Acknowledgments
We thank Ms. Robin Armstrong for her technical and organizational assistance. We also thank Isabel Xu for her assistance in the analysis of the in vivo video footage.
Footnotes
This study was presented at the 28th Annual Scientific Assembly of the Eastern Association for the Surgery of Trauma, January 13–17, 2015, in Lake Buena Vista, Florida.
AUTHORSHIP
Each author has contributed significantly to and is willing to take public responsibility for one or more aspects of the study, its design, data acquisition, as well as analysis and interpretation of data.
DISCLOSURE
The authors declare no conflicts of interest.
References
- 1.Rosenfeld JV, Maas AI, Bragge P, Morganti-Kossmann MC, Manley GT, Gruen RL. Early management of severe traumatic brain injury. Lancet. 2012;380(9847):1088–1098. doi: 10.1016/S0140-6736(12)60864-2. [DOI] [PubMed] [Google Scholar]
- 2.Lukaszewicz AC, Soyer B, Payen D. Water, water, everywhere: sodium and water balance and the injured brain. Curr Opin Anaesthesiol. 2011;24(2):138–143. doi: 10.1097/ACO.0b013e32834458af. [DOI] [PubMed] [Google Scholar]
- 3.Soares HD, Hicks RR, Smith D, McIntosh TK. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J Neurosci. 1995;15(12):8223–8233. doi: 10.1523/JNEUROSCI.15-12-08223.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shojo H, Kaneko Y, Mabuchi T, Kibayashi K, Adachi N, Borlongan CV. Genetic and histologic evidence implicates role of inflammation in traumatic brain injury-induced apoptosis in the rat cerebral cortex following moderate fluid percussion injury. Neuroscience. 2010;171(4):1273–1282. doi: 10.1016/j.neuroscience.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 5.Morganti-Kossmann MC, Rancan M, Otto VI, Stahel PF, Kossmann T. Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock. 2001;16(3):165–177. doi: 10.1097/00024382-200116030-00001. [DOI] [PubMed] [Google Scholar]
- 6.Hunt RF, Boychuk JA, Smith BN. Neural circuit mechanisms of post-traumatic epilepsy. Front Cell Neurosci. 2013;7:89. doi: 10.3389/fncel.2013.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Buul JD, Hordijk PL. Signaling in leukocyte transendothelial migration. Arterioscler Thromb Vasc Biol. 2004;24(5):824–833. doi: 10.1161/01.ATV.0000122854.76267.5c. [DOI] [PubMed] [Google Scholar]
- 8.McKeating EG, Andrews PJ, Mascia L. Leukocyte adhesion molecule profiles and outcome after traumatic brain injury. Acta Neurochir Suppl. 1998;71:200–202. doi: 10.1007/978-3-7091-6475-4_57. [DOI] [PubMed] [Google Scholar]
- 9.Gould MK, Garcia DA, Wren SM, Karanicolas PJ, Arcelus JI, Heit JA, Samama CM. Prevention of VTE in nonorthopedic surgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(Suppl 2):e227S–e277S. doi: 10.1378/chest.11-2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knudson MM, Morabito D, Paiement GD, Shackleford S. Use of low molecular weight heparin in preventing thromboembolism in trauma patients. J Trauma. 1996;41(3):446–459. doi: 10.1097/00005373-199609000-00010. [DOI] [PubMed] [Google Scholar]
- 11.Dudley RR, Aziz I, Bonnici A, Saluja RS, Lamoureux J, Kalmovitch B, Gursahaney A, Razek T, Maleki M, Marcoux J. Early venous thromboembolic event prophylaxis in traumatic brain injury with low-molecular-weight heparin: risks and benefits. J Neurotrauma. 2010;27(12):2165–2172. doi: 10.1089/neu.2010.1366. [DOI] [PubMed] [Google Scholar]
- 12.Mary V, Wahl F, Uzan A, Stutzmann JM. Enoxaparin in experimental stroke: neuroprotection and therapeutic window of opportunity. Stroke. 2001;32(4):993–999. doi: 10.1161/01.str.32.4.993. [DOI] [PubMed] [Google Scholar]
- 13.Quartermain D, Li YS, Jonas S. The low molecular weight heparin enoxaparin reduces infarct size in a rat model of temporary focal ischemia. Cerebrovasc Dis. 2003;16(4):346–355. doi: 10.1159/000072556. [DOI] [PubMed] [Google Scholar]
- 14.Libersan D, Khalil A, Dagenais P, Quan E, Delorme F, Uzan A, Latour JG. The low molecular weight heparin, enoxaparin, limits infarct size at re-perfusion in the dog. Cardiovasc Res. 1998;37(3):656–666. doi: 10.1016/s0008-6363(97)00292-7. [DOI] [PubMed] [Google Scholar]
- 15.Wahl F, Grosjean-Piot O, Bareyre F, Uzan A, Stutzmann JM. Enoxaparin reduces brain edema, cerebral lesions, and improves motor and cognitive impairments induced by a traumatic brain injury in rats. J Neurotrauma. 2000;17(11):1055–1065. doi: 10.1089/neu.2000.17.1055. [DOI] [PubMed] [Google Scholar]
- 16.Stutzmann JM, Mary V, Wahl F, Grosjean-Piot O, Uzan A, Pratt J. Neuroprotective profile of enoxaparin, a low molecular weight heparin, in in vivo models of cerebral ischemia or traumatic brain injury in rats: a review. CNS Drug Rev. 2002;8(1):1–30. doi: 10.1111/j.1527-3458.2002.tb00213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pratt J, Boudeau P, Uzan A, Imperato A, Stutzmann J. Enoxaparin reduces cerebral edema after photothrombotic injury in the rat. Haemostasis. 1998;28(2):78–85. doi: 10.1159/000022416. [DOI] [PubMed] [Google Scholar]
- 18.Kim L, Schuster J, Holena DN, Sims CA, Levine J, Pascual JL. Early initiation of prophylactic heparin in severe traumatic brain injury is associated with accelerated improvement on brain imaging. J Emerg Trauma Shock. 2014;7(3):141–148. doi: 10.4103/0974-2700.136846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manduteanu I, Voinea M, Capraru M, Dragomir E, Simionescu M. A novel attribute of enoxaparin: inhibition of monocyte adhesion to endothelial cells by a mechanism involving cell adhesion molecules. Pharmacology. 2002;65(1):32–37. doi: 10.1159/000056183. [DOI] [PubMed] [Google Scholar]
- 20.Gikakis N, Khan MM, Hiramatsu Y, Gorman JH, 3rd, Hack CE, Sun L, Rao AK, Niewiarowski S, Colman RW, Edmunds LH., Jr Effect of factor Xa inhibitors on thrombin formation and complement and neutrophil activation during in vitro extracorporeal circulation. Circulation. 1996;94(9 Suppl):II341–II346. [PubMed] [Google Scholar]
- 21.Tanne D, Katzav A, Beilin O, Grigoriadis NC, Blank M, Pick CG, Landenberg P, Shoenfeld Y, Chapman J. Interaction of inflammation, thrombosis, aspirin and enoxaparin in CNS experimental antiphospholipid syndrome. Neurobiol Dis. 2008;30(1):56–64. doi: 10.1016/j.nbd.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 22.Zupan Z, Pilipovic K, Dangubic B, Frkovic V, Sustic A, Zupan G. Effects of enoxaparin in the rat hippocampus following traumatic brain injury. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(8):1846–1856. doi: 10.1016/j.pnpbp.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 23.Schwarzmaier SM, Kim SW, Trabold R, Plesnila N. Temporal profile of thrombogenesis in the cerebral microcirculation after traumatic brain injury in mice. J Neurotrauma. 2010;27(1):121–130. doi: 10.1089/neu.2009.1114. [DOI] [PubMed] [Google Scholar]
- 24.Pascual JL, Murcy MA, Li S, Gong W, Eisenstadt R, Kumasaka K, Sims C, Smith DH, Browne K, Allen S, et al. Neuroprotective effects of progesterone in traumatic brain injury: blunted in vivo neutrophil activation at the blood-brain barrier. Am J Surg. 2013;206(6):840–845. doi: 10.1016/j.amjsurg.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, McIntosh TK. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma. 1995;12(2):169–178. doi: 10.1089/neu.1995.12.169. [DOI] [PubMed] [Google Scholar]
- 26.Marks JA, Li S, Gong W, Sanati P, Eisenstadt R, Sims C, Smith DH, Reilly PM, Pascual JL. Similar effects of hypertonic saline and mannitol on the inflammation of the blood-brain barrier microcirculation after brain injury in a mouse model. J Trauma Acute Care Surg. 2012;73(2):351–357. doi: 10.1097/TA.0b013e3182592f76. discussion 7. [DOI] [PubMed] [Google Scholar]
- 27.Schaar KL, Brenneman MM, Savitz SI. Functional assessments in the rodent stroke model. Exp Transl Stroke Med. 2010;2(1):13. doi: 10.1186/2040-7378-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knudson MM, Ikossi DG. Venous thromboembolism after trauma. Curr Opin Crit Care. 2004;10(6):539–548. doi: 10.1097/01.ccx.0000144941.09650.9f. [DOI] [PubMed] [Google Scholar]
- 29.Kim J, Gearhart MM, Zurick A, Zuccarello M, James L, Luchette FA. Preliminary report on the safety of heparin for deep venous thrombosis prophylaxis after severe head injury. J Trauma. 2002;53(1):38–42. doi: 10.1097/00005373-200207000-00008. [DOI] [PubMed] [Google Scholar]
- 30.Bratton SL, Chestnut RM, Ghajar J, McConnell Hammond FF, Harris OA, Hartl R, Manley GT, Nemecek A, Newell DW, Rosenthal G, et al. Guidelines for the management of severe traumatic brain injury. V. Deep vein thrombosis prophylaxis. J Neurotrauma. 2007;24(Suppl 1):S32–S36. doi: 10.1089/neu.2007.9991. [DOI] [PubMed] [Google Scholar]
- 31.Farooqui A, Hiser B, Barnes SL, Litofsky NS. Safety and efficacy of early thromboembolism chemoprophylaxis after intracranial hemorrhage from traumatic brain injury. J Neurosurg. 2013;119(6):1576–1582. doi: 10.3171/2013.8.JNS13424. [DOI] [PubMed] [Google Scholar]
- 32.Kubes P, Ward PA. Leukocyte recruitment and the acute inflammatory response. Brain Pathol. 2000;10(1):127–135. doi: 10.1111/j.1750-3639.2000.tb00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147(Suppl 1):S232–S240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whalen MJ, Carlos TM, Kochanek PM, Heineman S. Blood-brain barrier permeability, neutrophil accumulation and vascular adhesion molecule expression after controlled cortical impact in rats: a preliminary study. Acta Neurochir Suppl. 1998;71:212–214. doi: 10.1007/978-3-7091-6475-4_61. [DOI] [PubMed] [Google Scholar]
- 35.Gavins F, Yilmaz G, Granger DN. The evolving paradigm for blood cell-endothelial cell interactions in the cerebral microcirculation. Microcirculation. 2007;14(7):667–681. doi: 10.1080/10739680701404903. [DOI] [PubMed] [Google Scholar]
- 36.Pascual JL, Khwaja KA, Chaudhury P, Christou NV. Hypertonic saline and the microcirculation. J Trauma. 2003;54(Suppl 5):S133–S140. doi: 10.1097/01.TA.0000064526.33647.63. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen HX, O’Barr TJ, Anderson AJ. Polymorphonuclear leukocytes promote neurotoxicity through release of matrix metalloproteinases, reactive oxygen species, and TNF-alpha. J Neurochem. 2007;102(3):900–912. doi: 10.1111/j.1471-4159.2007.04643.x. [DOI] [PubMed] [Google Scholar]
- 38.DiStasi MR, Ley K. Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability. Trends Immunol. 2009;30(11):547–556. doi: 10.1016/j.it.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moxon-Emre I, Schlichter LC. Neutrophil depletion reduces blood-brain barrier breakdown, axon injury, and inflammation after intracerebral hemorrhage. J Neuropathol Exp Neurol. 2011;70(3):218–235. doi: 10.1097/NEN.0b013e31820d94a5. [DOI] [PubMed] [Google Scholar]
- 40.Stanimirovic DB, Wong J, Shapiro A, Durkin JP. Increase in surface expression of ICAM-1, VCAM-1 and E-selectin in human cerebromicrovascular endothelial cells subjected to ischemia-like insults. Acta Neurochir Suppl. 1997;70:12–16. doi: 10.1007/978-3-7091-6837-0_4. [DOI] [PubMed] [Google Scholar]
- 41.Schoettle RJ, Kochanek PM, Magargee MJ, Uhl MW, Nemoto EM. Early polymorphonuclear leukocyte accumulation correlates with the development of posttraumatic cerebral edema in rats. J Neurotrauma. 1990;7(4):207–217. doi: 10.1089/neu.1990.7.207. [DOI] [PubMed] [Google Scholar]
- 42.Semple BD, Bye N, Ziebell JM, Morganti-Kossmann MC. Deficiency of the chemokine receptor CXCR2 attenuates neutrophil infiltration and cortical damage following closed head injury. Neurobiol Dis. 2010;40(2):394–403. doi: 10.1016/j.nbd.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 43.Semple BD, Bye N, Rancan M, Ziebell JM, Morganti-Kossmann MC. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2−/− mice. J Cereb Blood Flow Metab. 2010;30(4):769–782. doi: 10.1038/jcbfm.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simard JM, Tosun C, Ivanova S, Kurland DB, Hong C, Radecki L, Gisriel C, Mehta R, Schreibman D, Gerzanich V. Heparin reduces neuroinflammation and transsynaptic neuronal apoptosis in a model of subarachnoid hemorrhage. Transl Stroke Res. 2012;3(Suppl 1):155–165. doi: 10.1007/s12975-012-0166-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li LF, Huang CC, Lin HC, Tsai YH, Quinn DA, Liao SK. Unfractionated heparin and enoxaparin reduce high-stretch ventilation augmented lung injury: a prospective, controlled animal experiment. Crit Care. 2009;13(4):R108. doi: 10.1186/cc7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manduteanu I, Voinea M, Antohe F, Dragomir E, Capraru M, Radulescu L, Simionescu M. Effect of enoxaparin on high glucose-induced activation of endothelial cells. Eur J Pharmacol. 2003;477(3):269–276. doi: 10.1016/j.ejphar.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 47.Iba T, Takayama T. Enoxaparin attenuates endothelial damage with less bleeding compared with unfractionated heparin in endotoxemic rats. Shock. 2009;32(5):530–534. doi: 10.1097/SHK.0b013e3181a2e279. [DOI] [PubMed] [Google Scholar]
- 48.Yeh YC, Wang MJ, Lin CP, Fan SZ, Tsai JC, Sun WZ, Ko WJ. Enoxaparin sodium prevents intestinal microcirculatory dysfunction in endotoxemic rats. Crit Care. 2012;16(2):R59. doi: 10.1186/cc11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quartermain D, Li Y, Jonas S. Enoxaparin, a low molecular weight heparin decreases infarct size and improves sensorimotor function in a rat model of focal cerebral ischemia. Neurosci Lett. 2000;288(2):155–158. doi: 10.1016/s0304-3940(00)01223-4. [DOI] [PubMed] [Google Scholar]
- 50.Cavusoglu E, Lakhani M, Marmur JD. The activated clotting time (ACT) can be used to monitor enoxaparin and dalteparin after intravenous administration. J Invasive Cardiol. 2005;17(8):416–421. [PubMed] [Google Scholar]