

Significance
Prions (infectious proteins) pose a substantial risk to yeast, as they do for humans. Overproduction of the disaggregase, Hsp104, has long been known capable of curing [PSI+], a prion based on amyloid formation by the Sup35 protein. We find that most [PSI+] variants arising spontaneously in the absence of this Hsp104 overproduction curing activity are cured when that activity is restored at normal levels. This activity is thus an antiprion system, largely protecting the cells from prion formation by this protein.
Keywords: prion, antiprion system, [PSI+], Sup35p
Abstract
Overproduction or deficiency of many chaperones and other cellular components cure the yeast prions [PSI+] (formed by Sup35p) or [URE3] (based on Ure2p). However, at normal expression levels, Btn2p and Cur1p eliminate most newly arising [URE3] variants but do not cure [PSI+], even after overexpression. Deficiency or overproduction of Hsp104 cures the [PSI+] prion. Hsp104 deficiency curing is a result of failure to cleave the Sup35p amyloid filaments to make new seeds, whereas Hsp104 overproduction curing occurs by a different mechanism. Hsp104(T160M) can propagate [PSI+], but cannot cure it by overproduction, thus separating filament cleavage from curing activities. Here we show that most [PSI+] variants arising spontaneously in an hsp104(T160M) strain are cured by restoration of just normal levels of the WT Hsp104. Both strong and weak [PSI+] variants are among those cured by this process. This normal-level Hsp104 curing is promoted by Sti1p, Hsp90, and Sis1p, proteins previously implicated in the Hsp104 overproduction curing of [PSI+]. The [PSI+] prion arises in hsp104(T160M) cells at more than 10-fold the frequency in WT cells. The curing activity of Hsp104 thus constitutes an antiprion system, culling many variants of the [PSI+] prion at normal Hsp104 levels.
Most organisms devote a substantial part of their genome to opposing viral propagation and takeover of cellular processes. Humoral and cellular immune systems, RNAi-based systems, innate immunity, physical barriers to penetration, interferons, the SKI-based systems, and many others have been selected to keep virus propagation in check. Infectious proteins, prions, have not been studied as widely, but already antiprion systems have been recognized.
The translation termination subunit, Sup35p, can form an infectious protein, [PSI+] by conversion to a self-propagating amyloid form (1–6). Similarly, Ure2p, a mediator of nitrogen catabolite repression, can form the amyloid-based [URE3] prion (1, 7–9) and Rnq1p forms the [PIN+] prion (10, 11). Amyloid is a filamentous β-sheet–rich protein polymer, and the yeast prion amyloids have a folded, in-register, parallel β-sheet architecture (12–15). This architecture provides a mechanism by which proteins can template their conformation, much as DNA templates its sequence, and explains the rather stable propagation of many different prion variants (called “prion strains” in mammals) based on different conformations of a single prion protein (16, 17).
Chernoff’s seminal discovery that Hsp104 overproduction or deficiency could cure the [PSI+] prion (18, 19) led to detailed dissection of the mechanisms of these effects, and discovery of the involvement of many other chaperones and cochaperones. Hsp104 (20) is a disaggregating chaperone, which acts with Hsp70s and Hsp40s to solubilize proteins (21). Monomers are removed from the aggregate and fed through the central cavity of the Hsp104 hexamer, thereby denaturing them and allowing them a chance to properly refold (22–24). Millimolar guanidine HCl is a surprisingly specific inhibitor of Hsp104 (25–29), and has been used to show that the effect of Hsp104 inactivation on prion propagation is to block the generation of new seeds (also called propagons) (30–32). Hsp104’s prion-propagating activity, like its general disaggregating activity, also involves Hsp70s and nucleotide-exchange factors, as well as Hsp40s. Hsp70s, the cytoplasmic Ssas of Saccharomyces cerevisiae, are necessary for stable prion propagation (33–37), and can antagonize the curing of [PSI+] by overproduction of Hsp104 (38), an effect requiring Sgt2 (39). The Hsp40 role in prion propagation includes considerable prion-specificity of the various Hsp40s (40, 41). The need for collaboration between Hsp104, Hsp70s, Hsp40s, and nucleotide-exchange factors in prion propagation was shown by the ability of Escherichia coli homologs, ClpB, DnaK, and GrpE, to substitute for their yeast relatives only if they could interact with their E. coli partners (42).
The mechanism of Hsp104 overproduction curing of [PSI+] differs from that of its prion–propagation action. Mutation or complete deletion of the Hsp104 N-terminal domain has no effect on [PSI+] propagation, but eliminates the ability of the overproduced protein to cure [PSI+] (43). Sti1p and Cpr7 are cochaperones containing tetratricopeptide repeat sequences that determine their binding to conserved EEVD/DDLD sites at the C termini of Hsp70, Hsp90, and Hsp104 (44, 45). The sti1Δ mutation also prevents curing of [PSI+] by overproduced Hsp104 (46, 47), although this deletion does not affect propagation of the same [PSI+] variant in an otherwise WT strain (48). However, deletion of the Hsp104 C-terminal DDLD prevents its binding to Sti1p (45), but does not prevent overproduced Hsp104 from curing [PSI+] (47), suggesting Sti1p is needed for the Hsp104 overproduction curing via another interaction. The part of Sti1p most important for Hsp104 curing of [PSI+] is the tetratricopeptide repeat 2 domain involved in interaction with Hsp90s (47). Indeed, inhibition of Hsp90s with radicicol blocks the curing activity of overproduced Hsp104 (47). These results suggest a role of Hsp90s in the Hsp104 prion curing process.
The mechanism of Hsp104 overproduction curing of [PSI+] remains controversial. One proposal is that overproduced Hsp104 binds to a special site in the middle (M) domain of Sup35p (49) and so prevents Hsp70s from having access to the filaments, which access is believed necessary for the Hsp104-Hsp70-Hsp40 machine to extract a monomer from the filament and thereby cleave it (37). Another model proposes that overproduction curing represents removal of Sup35p monomers from the ends of filaments, thereby eventually solubilizing the filaments (50). A third group posits assymetric segregation of prion seeds that have been collected by the overproduced Hsp104, with some cells emerging from the division with no seeds (51). There is substantial evidence for each of these models.
Although extensive studies have probed the mechanism of Hsp104 overproduction curing, less is known about its biological role. During the usual heat-shock regime, little curing of [PSI+] occurs, perhaps because the higher levels of Hsp104 are largely occupied renaturing the wide array of denatured cellular proteins. Transient heat shock produces some curing of a weak [PSI+], a phenomenon attributed to a demonstrated temporary excess of Hsp104 over Hsp70s of the Ssa group (52). However, it is likely that other heat-shock proteins and other factors are also varying in amount or activity during this treatment.
The rare occurrence of the [PSI+] and [URE3] prions in wild yeast (53) is a function of their low frequency of generation, their spontaneous loss, their spread by mating, and their effects on their host, the latter varying from lethal to mild (54–57; reviewed in ref. 58). As organisms have an array of systems to deal with viral, bacterial, and parasite infections, it is not surprising that yeast has antiprion systems. The ribosome-associated Hsp70s, Ssb1 and Ssb2, at their normal levels, repress the formation of the [PSI+] prion and, if overproduced, can cure [PSI+] (59, 60). The Ssb proteins are involved in assuring correct folding of nascent proteins (61, 62). Most variants of [URE3] are quickly eliminated by the normal levels of Btn2p and Cur1p and overproduction of either protein cures all known [URE3] variants (63, 64). Btn2p acts by collecting Ure2p amyloid aggregates to one place in the cell so that following division, one of the daughter cells is cured (63, 64). Btn2p and Cur1p are each also able to collect nonprion protein aggregates (63, 65, 66), but neither cures [PSI+] (63).
The fact that Hsp104 overproduction cures [PSI+], and that this prion-curing activity appears to be distinct from Hsp104’s prion-propagation activity, led us to suspect that, as in the Btn2p/Cur1p effects on [URE3], the prion-curing activity of Hsp104 might be working without overexpression of the protein to eliminate many [PSI+] variants as they arise.
Results
Hsp104 at Its Normal Level Cures Many [PSI+] Variants.
Overproduction of Hsp104T160M fails to cure [PSI+], but this mutant is fully able to propagate [PSI+] (43). The hsp104T160M mutation does not simply lower amyloid fiber cutting activity, as such a change would make a strong [PSI+] appear to be weaker. In fact, the mutation makes a weak [PSI+] appear stronger (43). We isolated spontaneous [PSI+] variants in a [PIN+] hsp104T160M strain (Fig. S1A). We restored the WT Hsp104 (with the curing activity, but at normal levels) by cytoduction (cytoplasmic transfer) (Methods) of these prions into an isogenic WT host, or into another hsp104T160M strain as a control (Table 1). We also performed cytoductions of [PSI+]s generated in a WT strain as a control.
Fig. S1.

Comparison of WT and hsp104T160M strains. (A) hsp104T160M mutation does not result in the loss of the [PIN+] prion as assayed by florescence microscopy using Rnq1-GFP fusion protein as a reporter of the presence of the Rnq1p aggregates in the cell. 3.A1, 3.A2, 3.A3, and 3.A4 are meiotic segregants from a tetrad formed by the HSP104/hsp104T160M diploid strain AG457 × AG478. (Magnification, 1,000×.) (B) The range of phenotypes that spontaneously generated [PSI+]s isolated either in WT or hsp104T160M background had on 1/2 YPD (Table 1). [PSI+] cells of several different variants were spotted on 1/2 YPD and grown at 30 °C for 2 d followed by 3 d at room temperature. Differences in the intensity of the prion phenotype (weak/strong) as well in prion stability are evident.
Table 1.
Restoration of the WT Hsp104 (at normal levels) cures most of the spontaneously arising [PSI+] variants
| Donor HSP104 genotype and [PSI+] variant (phenotype on 1/2 YPD) | Recipient | Cytoductants | Ade+ cytoductants | % Ade+ | P value |
| WT [PSI+1] (ss) | WT | 12 | 11 | 92 | |
| WT [PSI+1] (ss) | hsp104T160M | 9 | 9 | 100 | |
| WT [PSI+2] (ss) | WT | 12 | 2 | 17 | 6 × 10−5 |
| WT [PSI+2] (ss) | hsp104T160M | 16 | 16 | 100 | |
| WT [PSI+3] (ss) | WT | 13 | 12 | 92 | |
| WT [PSI+3] (ss) | hsp104T160M | 14 | 14 | 100 | |
| hsp104T160M [PSI+4] (vwu) | WT | 32 | 13 | 41 | 4 × 10−4 |
| hsp104T160M [PSI+4] (vwu) | hsp104T160M | 22 | 21 | 95 | |
| hsp104T160M [PSI+5] (vwvu) | WT | 13 | 0 | 0 | |
| hsp104T160M [PSI+5] (vwvu) | hsp104T160M | 12 | 0 | 0 | |
| hsp104T160M [PSI+6] (vwu) | WT | 21 | 17 | 81 | |
| hsp104T160M [PSI+6] (vwu) | hsp104T160M | 15 | 15 | 100 | |
| hsp104T160M [PSI+7] (vwu) | WT | 23 | 6 | 26 | 1 × 10−4 |
| hsp104T160M [PSI+7] (vwu) | hsp104T160M | 15 | 12 | 80 | |
| hsp104T160M [PSI+8] (vwu) | WT | 3 | 0 | 0 | 2 × 10−3 |
| hsp104T160M [PSI+8] (vwu) | hsp104T160M | 19 | 19 | 100 | |
| hsp104T160M [PSI+9] (vwvu) | WT | 37 | 6 | 16 | 1 × 10−5 |
| hsp104T160M [PSI+9] (vwvu) | hsp104T160M | 18 | 15 | 83 | |
| hsp104T160M [PSI+10] (vwu) | WT | 28 | 18 | 64 | 1 × 10−5 |
| hsp104T160M [PSI+10] (vwu) | hsp104T160M | 21 | 20 | 95 | |
| hsp104T160M [PSI+11] (vwvu) | WT | 26 | 7 | 27 | 1 × 10−5 |
| hsp104T160M [PSI+11] (vwvu) | hsp104T160M | 14 | 14 | 100 |
The spontaneously arising prion variants isolated in either WT (AG666) or hsp104T160M (AG667) background were used as cytoduction donors to isogenic WT (AG686) or hsp104T160M (AG687) recipients. The cytoduction efficiency of the [PSI+2], [PSI+4], [PSI+7], [PSI+8], [PSI+9], [PSI+10], and [PSI+11] prion variants into hsp104T160M recipients was significantly higher than into WT recipients. ss, strong stable; vwu, very weak unstable; vwvu, very weak very unstable (Fig. S1B). Statistical tests were carried out as described in Methods.
Most [PSI+]s arising spontaneously in the hsp104T160M strain propagated poorly when cytoduced to WT, but not when cytoduced to hsp104T160M recipients. This finding supports the hypothesis that Hsp104, at its normal level, cures a significant fraction of the [PSI+]s appearing spontaneously in a yeast cell. We call such prions [PSI+hhs] for Hsp104 hypersensitive. To make reading the tables of data easier, we use “wt” for variants isolated in a WT host and “hsp” for those isolated in an hsp104T160M host. Variant numbers are “Ax” or “Bx” (isolated in two different experiments). In most of the tables, [PSI+hhs] variants are shown in orange, and those that are not hypersensitive are shown in green, and will be referred to by those colors herein.
We also performed the same experiment with [PSI+] cytoduction donors generated by Sup35NM overexpression in either WT or hsp104T160M background (Table 2). All of the donors were guanidine-curable (Fig. 1A). As expected, we found [PSI+] variants generated in the hsp104T160M background that cytoduced significantly better to hsp104T160M than to WT recipients (orange variants hspB5, hspB7, hspB20; hspA3, hspA4, hspA5, hspA6, hspA7, hspA9, hspA15, hspA19, hspA20). We also found a few such variants among those generated in the WT background (wtB19; wtA3, wtA13). Two of these were very unstable in the WT parent and were apparently stabilized by the hsp104T160M mutation. Most variants generated by Sup35NM overexpression in the WT background cytoduced well to both WT and hsp104T160M recipients (28 of 40, green variants). The fraction of orange variants among those generated in the hsp104T160M background was higher than in variants generated in the WT background [12 of 40 in hsp104T160M vs. 3 of 40 isolated in the WT host (P < 10−4)]. We also found variants that cytoduced to both recipients poorly, but with near equal efficiency [white variants (shown as white in the tables)]. Surprisingly, we found several variants generated in the hsp104T160M background that cytoduced significantly better to WT recipients than to hsp104T160M ones (variants hspB1, hspB3, hspB10, hspB13). Apparently, normal Hsp104 curing activity aids their propagation. These variants are reminiscent of a [PSI+] described by Borchsenius et al., which requires elevated Hsp104 for its propagation (67).
Table 2.
[PSI+] isolates induced by overproduction of Sup35NM
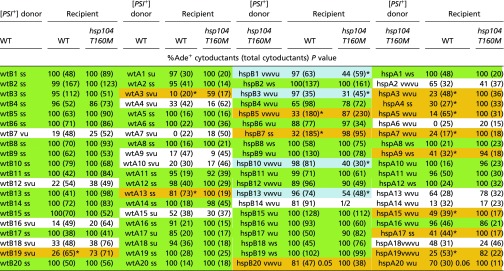 |
WT (wt, AG666) and hsp104T160M (hsp, AG667) [psi−] [PIN+] strains were induced to [PSI+] by transient overproduction of Sup35NM. Guanidine curable clones were each used as cytoduction donors to WT (wt, AG686) and hsp104T160M (hsp, AG687) [psi−] ρo recipients. The color-coding in this table is as follows: “green” variants cytoduce well to both WT or hsp104T160M recipients; “orange” variants cytoduce well to hsp104T160M but not to WT recipients; “blue” variants cytoduce well to WT but not to hsp104T160M recipients; and “white” variants cytoduce poorly, but with approximately equal efficiency to both recipients. *P < 10−5.
Fig. 1.

Characterization of the [PSI+] cytoduction donors generated by Sup35NM overexpression in either wt or hsp104T160M background (Table 2). [PSI+], positive control. [psi−], negative control. (A) [PSI+] prion variants were grown on 1/2 YPD medium with or without 3 mM guanidine HCl (Gdn). Strong [PSI+] variants are white and weak variants are pink reflecting the lower or higher amount of the soluble Sup35 in the cytoplasm. Red colonies are prion-free. Only guanidine-curable donors were used in further experiments. Phenotypes: ss, strong stable; su, strong unstable; svu, strong very unstable; ws, weak stable; wu, weak unstable; wvu, weak very unstable; vwvu, very weak very unstable. (B) The [PSI+] prion seed number determined for the indicated strains (Methods). LiebW and LiebS are weak and strong strains from S. Liebman, University of Nevada, Reno, NV.
Ade− HSP104 Cytoductants Are [psi−].
To confirm that Ade− cytoductants from an hsp104T160M [PSI+hhs] strain to a WT recipient were really [psi−], we performed back-cytoductions from Ade− cytoductants (resulting from cytoduction from hspB7 to the WT) (Table 2) to WT and hsp104T160M [psi−] recipients (Fig. 2). All of the cytoductants and diploids formed were Ade−, including the hsp104T160M cytoductants, confirming that [PSI+] was really lost upon cytoduction from hspB7 to the WT recipient. The phenotypic assay for [PSI+] measures translational read-through of the ade2-1 mutation that results from the deficiency of translation termination factor Sup35p, produced by its being largely tied up in the amyloid filaments. A phenotypic masking effect would be a direct effect of the hsp104T160M mutation on translational read-through. It has been previously shown that hsp104T160M has no such [PSI+]-independent effect (43).
Fig. 2.

Ade− phenotype of cytoductants is because of loss of [PSI+hhs]. Cytoduction of Hsp104 hypersensitive [PSI+] from hsp104T160M to a WT recipient produced mostly Ade− clones, but also a minority of Ade+ cytoductants (Tables 1 and 2). Back-cytoductions from the Ade− cytoductants into either WT or hsp104T160M recipients produced only Ade− cytoductants and diploids. (A) The scheme of the back-cytoduction experiment. The hspB7 orange prion variant was cytoduced into WT recipient resulting in 126 Ade− cytoductants and 59 Ade+ cytoductants. Two Ade− (see B) and several Ade+ cytoductants (Table 3) were used as donors and were back-cytoduced into WT and hsp104T160M recipients. (B) The results of the back-cytoduction of Ade− cytoductants. Cytoductant colonies grow on YPG medium (ρ+) but not on medium selective for diploids (Methods). The back-cytoduction of the Ade− cytoductants into their original hsp104T160M background did not result in any Ade+ cytoductants. This proves that their Ade− phenotype is a result of the loss of [PSI+] and is not because of phenotype masking.
We also did back-cytoductions from Ade+ cytoductants (Table 3). The orange variant converted to a green variant when transferred to a WT. This result is consistent with the “cloud of variants” model according to which a [PSI+] cell contains a mixture of prion variants, with one or another becoming stochastically dominant (68–70). By cytoducing this mixture from the curing-defective hsp104T160M host to one with a fully functional Hsp104, we applied selective pressure that eliminated the susceptible (orange) [PSI+hhs] variants leaving resistant (green) variants uncured. As expected, passing hspB7 through the hsp104T160M recipient maintained the orange character of the [PSI+] variant as no new selective pressure was applied by this process. Note that the green character of a [PSI+] is not changed in an hsp104T160M host.
Table 3.
Back-cytoductions from Ade+ guanidine-curable cytoductants
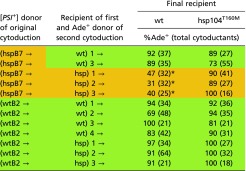 |
Some cytoductants of Hsp104 hypersensitive [PSI+]s (e.g., hspB7) to WT recipients remained Ade+. These cytoductants were used as donors to WT (AG679) and hsp104T160M (AG680) recipients. The results indicate that the [PSI+]s were no longer Hsp104 hypersensitive. Controls were Ade+ cytoductants originating in WT cells. *P < 10−5.
Loss of [PSI+hhs] Was Indeed Because of Hsp104.
To make sure that the T160M mutation in HSP104 and not some other accompanying mutation underlies the inability of our hsp104T160M recipient strain to cure a significant fraction of spontaneously appearing [PSI+] variants, we used the CRISPR-Cas technique to restore the WT allele of HSP104 in this particular strain, and performed the cytoductions as previously. The results confirm the primary role of the T160M mutation in making Hsp104 unable to cure some [PSI+] variants because transmission of orange prion variants into both WT-CRISPR strains was significantly less efficient compared with cytoduction into hsp104T160M recipients, whereas green variants were well propagated by the CRISPR-restored hosts (Table 4).
Table 4.
Cytoductions into CRISPR-CAS – corrected and uncorrected recipients
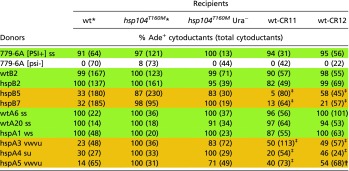 |
To confirm that the loss of the hypersensitive [PSI+] variants from WT recipients was because of their normal Hsp104, the hsp104T160M mutant recipient was converted to WT using CRISPR-CAS gene editing (SI Methods) and several [PSI+] variants were introduced by cytoduction into the mutant strain (hsp104T160M Ura−, AG730) and two corrected WT strains (C11, C12; AG780, AG781).
The data for the cytoductions from green and orange donors to these WT and hsp104T160M recipients are the same as in Table 2. The statistics relate the CRISPR-corrected recipient strains wt-CR11 and wt-CR12 to the mutant parent, hsp104T160M Ura− (strain AG730).
P < 10−5.
P < 10−4.
Strong/Weak, Seed Number, and Hsp104 Hypersensitivity.
We do not find a correlation between the strong/weak or stable/unstable nature of a variant and its ability/inability to propagate in the presence of normal levels of WT Hsp104. For example, hspA4 is a strong very stable [PSI+] (Fig. 1A), but is highly sensitive to curing by normal levels of Hsp104 (Table 2). Similarly, the weak very unstable variant hspA5 (Fig. 1) shows a similar sensitivity to normal levels of WT Hsp104 (Table 2). Furthermore, we did not observe any correlation between seeds/cell and the green vs. orange property of variants (Fig. 1C).
Mutability of Hsp104 Hypersensitive [PSI+].
We noted the gradual change of orange variants toward green on repeated passage in an hsp104T160M host on –Ade medium (Table S1). The difference in cytoduction efficiencies of these variants into WT and hsp104T160M recipients gradually decreased with growth. In contrast, we did not observe such changes in the case of green variants. This finding suggests that initially orange variants include a small minority of green variants (the prion cloud), and that the green variants have an advantage over orange variants, even in an hsp104T160M host. Note that even the strong, stable orange variant hspA4 gradually becomes green on repeated culturing (Table S1).
Table S1.
Repeated passage lessens the sensitivity of [PSI+] variants to normal levels of Hsp104
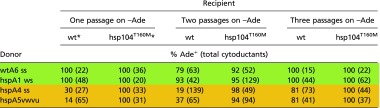 |
Each passage comprised about 10 generations.
The data for cytoductions from green and orange donors to wt and hsp104T160M recipients are the same as in Table 2.
Proteins Associated with Hsp104 and Hsp104(T160M).
We used coimmunoprecipitation (co-IP) to compare the binding partners of WT Hsp104 and Hsp104T160M. The binding of most interactors with the mutant Hsp104 was approximately as effective as with WT Hsp104 (Table S2). The only protein that consistently co-IP with Hsp104T160M less effectively was Ura2 (aspartate transcarbamylase), a known Hsp90 client (71). Both Hsp90 and its cochaperone, Cpr6, were shown to be able to independently bind Ura2 (71), and the binding of Cpr6 to the mutant Hsp104 was slightly reduced (Table S2), thus leaving the possibility that the decrease in Ura2 binding is a result of somewhat weaker interaction of Cpr6 with Hsp104T160M. As for three other Hsp90 cochaperones, the T160M mutation slightly impairs interaction with Sti1 (Table S2). Hsp104 is a hexamer and, as expected, we detected the binding of Hsp104 to itself that was not changed in the mutant strain (Table S2).
Table S2.
Co-IP of proteins with Hsp104 and Hsp104T160M
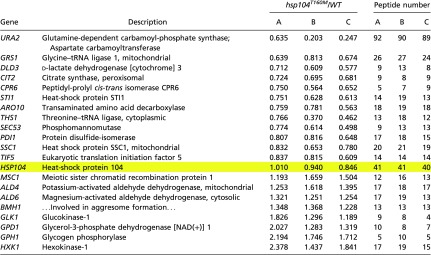 |
Anti-Hsp104 antiserum was used to isolate Hsp104 from extracts of WT and hsp104T160M cells, along with associated proteins. Proteins were methyl-labeled with deuterated (hsp104T160M) or protonated (WT) methyl groups, protease digested and peptides identified by mass spectrometry (see Methods). A, B, and C are biological replicates. The interaction of Hsp104 with itself is not changed in the mutant strain (yellow highlight).
Increased Frequency of Spontaneous [PSI+] in hsp104T160M Strains.
Finding that many variants generated in an hsp104T160M strain are curable by normal levels of Hsp104 predicts that the frequency of spontaneous [PSI+] generation may be increased in this mutant. We crossed a WT (779-6A [psi−] [PIN+]) strain with isogenic WT or hsp104T160M::hisG URA3 hisG [psi−] [pin−] strains and the spontaneous [PSI+] generation frequency was measured for each spore clone (Table 5). As anticipated, the average frequency of [PSI+] generation in hsp104T160M mutant strains was approximately 10 times higher than in WT (130 × 10−6 vs. 10 × 10−6, respectively) (Table 5 and Fig. S2). These differences are probably not a result of different variants of [PIN+] because each spore clone of a tetrad should get the same [PIN+] variant. The cosegregation of elevated [PSI+] frequency with hsp104T160M again indicates that it is this mutation that allows the orange variants to propagate, and not some adventitious change at another locus. The hsp104T160M meiotic segregants were [PIN+] (Fig. S1A), and [PSI+] generation in these strains was still [PIN+]-dependent, as no [PSI+] colonies were found when their [pin−] derivatives were tested (Table 5). Most [PSI+]s generated spontaneously in hsp104T160M strains were weak (dark pink) and unstable on 1/2 YPD, whereas most [PSI+]s generated spontaneously in WT strains were strong (white or slightly pink) and stable (Fig. S1B). However, in most cases [PSI+] colonies generated in a hsp104T160M mutant grew on –Ade medium significantly faster than those generated in WT strains, as previously shown for a standard [PSI+] variant (43).
Table 5.
Cosegregation of elevated spontaneous [PSI+] generation and hsp104T160M
| No. | Spore clone | MAT | HSP104 | [PSI+] per 106 cells* |
| 1 | 1.A1 | α | WT | 4.7 |
| 2 | 1.A2 | a | WT | 0.3 |
| 3 | 1.A3 | a | WT | 0.3 |
| 4 | 1.A4 | α | WT | 5.3 |
| 5 | 3.A1† | α | WT | 12 |
| 6 | 3.A2† | a | WT | 7 |
| 7 | 3.A3† | a | T160M | 500 |
| 8 | 3.A4† | α | T160M | 120 |
| 9 | 2A | α | WT | 1 |
| 10 | 2B | a | T160M | 140 |
| 11 | 2C | a | WT | 33 |
| 12 | 2D | α | T160M | 80 |
| 13 | 6A | α | WT | 2 |
| 14 | 6B | a | WT | 59 |
| 15 | 6C | α | T160M | 96 |
| 16 | 6D | a | T160M | 55 |
| 17 | 10A | a | WT | 2 |
| 18 | 10B | a | T160M | 91 |
| 19 | 10C | α | T160M | 89 |
| 20 | 10D | α | WT | 1 |
| 21 | 11A | α | WT | 31 |
| 22 | 11B | a | T160M | 53 |
| 23 | 11C | α | T160M | 120 |
| 24 | 11D | a | WT | 3 |
| 25 | 14A | a | WT | 5 |
| 26 | 14B | α | T160M | 95 |
| 27 | 14C | α | WT | 12 |
| 28 | 14D | a | T160M | 140 |
| 29 | 17A | α | WT | 5 |
| 30 | 17B | a | WT | 12 |
| 31 | 17C | a | T160M | 100 |
| 32 | 17D | α | T160M | 210 |
| 33 | 19A | α | WT | 2 |
| 34 | 19B | a | WT | 3 |
| 35 | 19C | a | T160M | 91 |
| 36 | 19D | α | T160M | 110 |
| 37 | 8–1 [pin-]† | a | T160M | 0 |
| 38 | 8–2 [pin-] | a | T160M | 0 |
Meiotic tetrads of strains AG457 × AG417 (nos. 1–4) and AG457 × AG478 (nos. 5–38) were plated for Ade+ colonies. The Ade+ clones were tested for guanidine-curability.
[PSI+] is guanidine-curable Ade+.
See Fig. S1A.
Fig. S2.

Spontaneous [PSI+] generation frequency of WT and hsp104T160M meiotic segregants (data from Table 5).
Chaperone Levels in HSP104 vs. hsp104T160M.
Potentially, the difference in curing ability of WT and hsp104T160M recipient strains could be a result of differences in protein levels of Hsp104 or other chaperones/cochaperones and related proteins between these strains. Western blot analysis (Fig. 3) showed that the levels of Hsp104, Ssa1-4, Sti1, Cpr7, and Ydj1 were not changed in the hsp104T160M mutant compared with the isogenic WT, whereas the level of Sis1 was 1.4 times increased, and the level of Sse1/2 was 1.3 times decreased. It is known that having two copies of the SIS1 gene in the cell does not influence [PSI+] propagation (72), so it is unlikely that this slight protein level increase is the cause of the phenotype. Similarly, deletion of SSE1 has generally detrimental effects on [PSI+] generation and propagation (73, 74), so it is unlikely that the slight decrease of Sse1 level leads to better propagation of orange [PSI+] variants in hsp104T160M than in WT. The antibody used in our work recognizes both Sse1 and Sse2, but without heat shock, SSE2 transcripts are nearly undetectable and Sse2 is known not to support [PSI+] propagation (74). We also find the levels of Sup35 and Hsp104 are approximately the same in the mutant as in the CRISPR-corrected WT (Fig. 3).
Fig. 3.

Western blot analysis of the protein levels in the WT and hsp104T160M strains. (A) The comparison of the protein levels in the original WT and hsp104T160M strains. (B) The comparison of the protein levels in the strains in which WT allele of HSP104 was restored using the CRISPR-Cas technique (wt-CR11 and wt-CR12) and their parent strain (hsp104T160M Ura−) (Methods).
Proteins Modulating the Curing of [PSI+]s by Normal Levels of Hsp104.
To determine whether any of the factors affecting [PSI+] curing by overexpressed Hsp104 can modulate [PSI+] curing by normal levels of Hsp104, we generated a set of mutant recipients and performed cytoductions into them from green and orange [PSI+] donors, as well as from standard [PSI+] and [psi−] strains (Table 6). Depletion of Ssa1p (in favor of Ssa2p) is known to be detrimental to [PSI+] propagation (75). Moreover, overproduction of Ssa1p counteracts curing of [PSI+] by overproduction of Hsp104 (38). Consistent with these findings, we also observed that the cytoduction efficiency of some variants (e.g., orange hspB5, hspB7, hspA9; white wtA4, wtA10, hspA2) into ssa1Δ was impaired compared with that into a WT recipient. Sgt2p is known to help overproduced Ssa1p antagonize Hsp104 overproduction curing of [PSI+], indicating a pro-[PSI+] activity for Sgt2 (39). We found that most orange variants (e.g., wtA3, hspA4, hspA5, hspA15, hspA19) cytoduced into sgt2Δ nearly as efficiently as into the hsp104T160M recipient, and generally better than into the WT. Several variants (e.g., white wtA10 and hspA14; orange hspA3, hspA4, hspA5, hspA6 and hspA7) cytoduced into sti1Δ significantly better than into the WT recipient. These results suggest that Sgt2 and Sti1 are helping, whereas Ssa1 is counteracting the curing of some [PSI+] variants by the normal level of Hsp104. Most orange variants (hspB5, wtA3, wtA7, hspA3, hspA4, hspA5, hspA6, hspA7, hspA9, hspA17, hspA19) cytoduced into sis1Δ338–352 recipients nearly as efficiently as into hsp104T160M (Table 6), consistent with this deletion’s known total abrogation of Hsp104 overproduction curing (72). Thus, Sis1 helps the normal level of Hsp104 to cure many orange [PSI+] variants.
Table 6.
Effects of other chaperones and cochaperones on Hsp104 normal level curing of [PSI+]
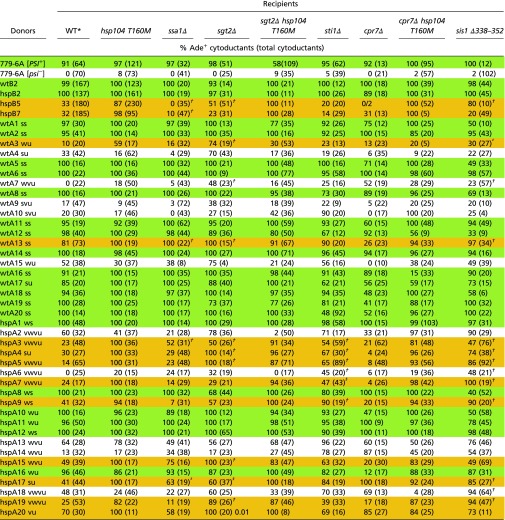 |
Because sti1Δ, cpr7Δ and sis1Δ3338–352 each are reported to prevent curing of [PSI+] by overproduction of Hsp104, we tested whether these mutations affect transmission of [PSI+] variants hypersensitive to Hsp104. Some probabilities shown as, for example, “(81%).05,” where 0.05 is the P value.
The data for cytoductions from green and orange donors to WT and hsp104T160M recipients are the same as in Table 2.
P < 10−5.
P < 10−4.
P < 10−3.
We could not find evidence for the involvement of Btn2 and Cur1 in curing of [PSI+] by the normal level of Hsp104 because both green and orange variants that we’ve tested cytoduced with similar efficiency into 74-D694 WT and 74-D694 btn2Δ cur1Δ recipients (Table S3). Deletion of HSP42 does not substantially affect the propagation of any variants tested (Table 7), consistent with our earlier findings (64).
Table S3.
Btn2p and Cur1p are not needed for the Hsp104 elimination of hypersensitive [PSI+] variants
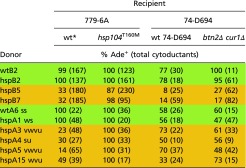 |
The btn2Δ cur1Δ strain AG563 and the isogenic 74-D694 were used as recipients to ascertain whether Btn2p or Cur1p, both involved in curing of [URE3], affect curing by normal levels of Hsp104.
Table 7.
Hsp90 is necessary for efficient elimination of [PSI+hhs] by normal levels of Hsp104
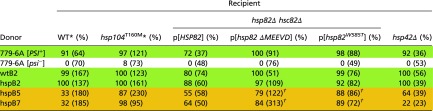 |
The data for cytoductions from green and orange donors to WT and hsp104T160M recipients are the same as in Table 2; the data for cytoductions from 779-6A [PSI+] and [psi−] to WT and hsp104T160M recipients are the same as in Table 6.
P < 10−5. Comparisons are shown between the hsp82Δ hsc82Δ strain complemented by WT HSP82 and mutant hsp82 plasmids.
Yeast cells need at least one of the highly similar isoforms of Hsp90 (HSP82 and HSC82) to be viable (76), so we used the strains in which the hsp82Δ hsc82Δ double deletion was compensated by either a WT or mutant copy of HSP82 expressed under the control of its native promoter from a centromere plasmid. We used either hsp82ΔMEEVD or hsp82W585T mutants in our experiments. The C-terminal MEEVD motif of Hsp82 is critical for interaction with tetratricopeptide repeat domain-containing cochaperones such as Cpr7, Sti1, Cpr6, and Cns1, so its deletion disturbs these interactions (45). Hsp82W585T protein is defective in client binding and chaperone activity (77). Mutant Hsp82 results in increased cytoduction efficiency of orange [PSI+] variants, suggesting that Hsp82 is involved in the Hsp104 normal-level curing (Table 7), as it has previously been shown to be involved in Hsp104 overexpression curing of [PSI+] (47).
Discussion
The lethal potential of yeast prion infections (54) suggested that cellular defense mechanisms should have evolved. Because overproduction of Hsp104 cures [PSI+] (18, 19), and the mechanism of this curing appears to be distinct from that of its prion propagation-promoting activity (43) (see Introduction), we suspected that normal levels of Hsp104 might be curing some [PSI+] variants as they arise.
We isolated [PSI+] variants in an hsp104T160M mutant shown by Hung and Masison (43) to lack the [PSI+]-curing activity, and tested each for ability to infect isogenic WT or mutant hosts. We found that most [PSI+] variants arising spontaneously in this mutant could not propagate in a WT host. These variants would arise in a WT strain but most would be quickly eliminated. Nonetheless, we did find a few [PSI+hhs] variants arising in the WT host. Like the elevated frequency of [URE3] in btn2Δ cur1Δ strains, we found that the frequency of [PSI+] arising spontaneously is about 10-fold higher in hsp104T160M strains than in isogenic WT, a phenotype that cosegregates with hsp104T160M and is eliminated by correcting the mutation. However, although Btn2p and Cur1p selectively cure [URE3] variants having low seed number, there is no such correlation for Hsp104 curing, with both high and low seed variants cured and not cured.
Because the same hsp104T160M mutant lacks both the overproduction curing activity and the ability to cure [PSI+hhs] variants, it seemed likely that the same Hsp104 activity was carrying out each of these processes. We further tested this notion by examining the influence on the latter process of other factors known to influence the former. Sti1p, a cochaperone of Hsp90s, Hsp70s, and Hsp104, has been shown to promote the curing of [PSI+] by overproduction of Hsp104 (46, 47), but not to be necessary for propagation of [PSI+] (46, 47, 78). We find that Sti1p is required for efficient elimination of most [PSI+hhs] variants by normal levels of WT Hsp104 (Table 6). Hsp90 is involved in Hsp104 overproduction curing of [PSI+] (47) and two mutations in Hsp90 also diminish the sensitivity to normal levels of Hsp104 of [PSI+hhs] variants (Table 7). Furthermore, the sis1Δ338–352 mutation that eliminates Hsp104 overproduction curing (72) also reduces the ability of cells with normal levels of WT Hsp104 to cure [PSI+hhs]. These results argue that the Hsp104 activity that cures [PSI+hhs] is the same as that which cures all [PSI+] variants on overproduction of Hsp104.
Although our work does not specifically address the mechanism, the fact that [PSI+hhs] curing occurs without Hsp104 overproduction suggests that overproduction is not an inherent component of the mechanism. The hsp104T160M mutation was originally isolated by its suppressing the [PSI+]-destabilizing dominant SSA1-21 mutation, and sti1, hsp90, and sis1 mutations had a similar effect (43, 47, 72). This earlier work also suggested that Hsp104 is part of an antiprion system that does not require overproduction to be active. Likewise, our [PSI+hss] variants are distinct from the nonsense-suppression observed by Salnikova et al. on sustained overexpression of Sup35p or Sup35NM (79).
Prion variants are central to the prion phenomenon. Properties tested for a single [PSI+] variant, for example, may not be representative of the whole range of [PSI+] variants. The commonly studied [URE3-1], originally isolated by Francois Lacroute (80), is evidently not cured by normal levels of Btn2p or Cur1p, but most [URE3] variants arising in the absence of these two proteins are cured by simply restoring their normal levels (64). Transmission of [URE3] across a species barrier also varies substantially with the prion variant (81). Similarly, transmission of [PSI+] from one Sup35p sequence polymorph to another is a rather low frequency event for one [PSI+] prion variant, but other [PSI+] variants are transmitted at high efficiency (82). One [PSI+] variant is very unstable in WT cells but is stabilized by Hsp104 overproduction (83). In our study, we find that most spontaneously arising [PSI+] isolates in the hsp104T160M mutant are cured by normal levels of WT Hsp104. These Hsp104 hypersensitive variants include both strong and weak [PSI+] variants and those with high or low seed number. It is not yet clear what structural aspects determine sensitivity to this antiprion system. Similarly, each of the four [PSI+] transmission variant types included strong and weak variants (68). By culling most spontaneous [PSI+] variants as they arise, Hsp104 evidently limits the potential damage to the cell.
We find that the Hsp104 hypersensitivity trait is not as stable as some other [PSI+] characteristics, apparently because even in the hsp104T160M mutant, there is selection against such variants. Conditional-lethal [PSI+] variants are gradually either lost or convert to a less pathogenic form, even under the “permissive condition” (54). It is likely that the permissive condition does not completely avoid the prion’s toxicity, so that there is continuing selection for loss or milder variants. Similarly, toxic [URE3] variants rapidly change or are lost as mitotic segregants with an altered variant are selected for (54). Similar instability has been noted for many other prion traits, including the transmission phenotype of [PSI+] (68) and Btn2/Cur1-sensitivity of [URE3] (64).
Sse1p is a chaperone and nucleotide exchange factor that has an interesting parallel with Hsp104. Sse1p is both necessary for [URE3] propagation and cures [URE3] when overproduced (84). SseI is necessary for the propagation of weak [PSI+], but not of strong variants (73, 84). However, overproduction of Sse1p, far from curing [PSI+], greatly stimulates the generation of [PSI+] (73). Completing this parallel, we note that overproduction of Hsp104 increases [URE3] generation (85). This is thus another example of a chaperone shaping the spectrum of prion variants.
Although there are now many means of curing yeast prions, few have been shown to be naturally acting curing mechanisms, in contrast to artificial imbalance of components involved in or affecting the prion propagation process. These antiprion systems, like DNA repair systems, are not completely effective, but certainly lower the burden of prions on the host, and shape the array of prion variants that will succeed in arising. The existence of at least two anti-[PSI+] systems argues that the cell does not view [PSI+] as an unalloyed blessing.
Methods
Yeast Culture and Genetic Manipulation.
Strains of S. cerevisiae are listed in Table S4 and growth media are as described by Sherman (86). Yeast were grown at 30 °C. Knockout mutations (87) were transferred by PCR amplification, transformation of the target strain, and confirmation, using PCR, of the absence of the normal allele and the presence, in the correct location, of the mutant allele.
Table S4.
Strains of Saccharomyces cerevisiae
| Description | Strain no. | Genotype |
| Parent of most strains | 779–6A | MATa ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 [psi−] [PIN+] (33) |
| WT donor parent strain | AG664 | MATa ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 [psi−] [PIN+] pRS314(CEN TRP1) |
| hsp104T160M donor parent strain | AG663 | 779-6A MATa hsp104T160M::hisG-URA3-hisG ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 [psi−] [PIN+] pRS314(CEN TRP1) |
| WT recipient parent strain | AG686 | 779-6A MATα ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 [psi−] [PIN+] |
| hsp104T160M recipient parent strain | AG687 | 779-6A MATα hsp104T160M::hisG-URA3-hisG ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 [psi−] [PIN+] |
| hsp104T160M Ura− recipient parent strain | AG730 | 779-6A MATα hsp104T160M ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 [psi−] [PIN+] (URA3 was lost on FOA) |
| wt-CR11 recipient parent strain | AG780 | 779-6A MATα HSP104 (reverted to WT, recoded) ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 [psi−] [PIN+] (see SI Methods) |
| wt-CR12 recipient parent strain | AG781 | 779-6A MATα HSP104 (reverted to wt, recoded) ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 [psi−] [PIN+] (see SI Methods) |
| hsp104T160M [pin−] 8–1 recipient parent strain | AG652 | 779-6A MATα hsp104T160M::hisG-URA3-hisG ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 [psi−] [pin−] |
| hsp104T160M [pin−] 8–2 recipient parent strain | AG653 | 779-6A MATα hsp104T160M::hisG-URA3-hisG ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 [psi−] [pin−] |
| HSP104/HSP104 mitotic segregants | AG457 (779-7A MATα ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 trp1 [psi−] [PIN+] ρo pRS314) X AG417 (779-7A MATa ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 trp1 [psi−] [pin−] ρ+ | |
| Table 5, nos. 1–4. | ||
| HSP104/hsp104T160M mitotic segregants: | AG457 (MATα ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 trp1 [psi−] [PIN+] ρo pRS314) X AG478 (779-6A MATa ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 trp1 hsp104T160M::hisG-URA3-hisG [psi−] [pin−] ρ+ | |
| Table 5, nos. 5–36. (Fig. S1A) | ||
| 3.A1 (Fig. S1A) | MATα WT | |
| 3.A2 (Fig. S1A) | MATa WT | |
| 3.A3 (Fig. S1A) | MATa hsp104T160M::hisG-URA3-hisG | |
| 3.A4 (Fig. S1A) | MATα hsp104T160M::hisG-URA3-hisG | |
| Cytoduction donors | ||
| Laboratory [psi−] | AG666 | 779-6A MATa ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 [psi−] [PIN+] pRS314(CEN TRP1) pHK006(CEN LEU2 PGal1-SUP35NM) |
| Laboratory [PSI+] | AG667 | 779-6A MATa hsp104T160M::hisG-URA3-hisG ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 [PSI+] [PIN+] pRS314(CEN TRP1) pHK006(CEN LEU2 PGal1-SUP35NM) |
| Cytoduction recipients | ||
| WT | AG571 | 779-6A MATα [psi−] ρ0 pRS313(CEN HIS3) |
| hsp104T160M | AG537 | 779-6A MATα [psi−] ρ0 hsp104T160M::hisG-URA3-hisG pRS313(CEN HIS3) |
| Δsgt2 | AG539 | 779-6A MATα [psi−] ρ0 Δsgt2::KanMX pRS313(CEN HIS3) |
| hsp104T160M Δsgt2 | AG541 | 779-6A MATα [psi−] ρ0 hsp104T160M::hisG-URA3-hisG Δsgt2::KanMX pRS313(CEN HIS3) |
| Δssa1 | AG548 | 779-6A MATα [psi−] ρ0 Δssa1::KanMX pRS313(CEN HIS3) |
| Δcpr7 | AG790 | 779-6A MATα [psi−] ρ0 Δcpr7::KanMX pRS313(CEN HIS3) |
| hsp104T160M Δcpr7 | AG552 | 779-6A MATα [psi−] ρ0 hsp104T160M::hisG-URA3-hisG Δcpr7::KanMX pRS313(CEN HIS3) |
| Δsti1 | AG560 | 779-6A MATα [psi−] ρ0 Δsti1::KanMX pRS313(CEN HIS3) |
| sis1Δ338–352 | AG561 | 779-6A MATα [psi−] ρ0 Δsis1::KanMX pRS314sis1Δ339–352 pRS313(CEN HIS3) |
| 4788 WT | AG562 | 74D-694 MATα [psi−] ρ0 pRS313(CEN HIS3) |
| 4788 Δbtn2 Δcur1 | AG563 | 74D-694 MATα [psi−] ρ0 Δbtn2::TRP1 Δcur1::KanMX pRS313(CEN HIS3) |
| Δhsp42 | AG678 | 779-6A MATα [psi−] ρ0 Δhsp42::KanMX pRS313(CEN HIS3) |
| Δhsc82 Δhsp82 p[HSP82] | AG732 | 779-6A MATα [psi−] ρ0 Δhsc82::KanMX Δhsp82::KanMX pMR62(CEN URA3 HSP82) pRS313(CEN HIS3) |
| Δhsc82 Δhsp82 p[hsp82ΔMEEVD] | AG733 | 779-6A MATα [psi−] ρ0 Δhsc82::KanMX Δhsp82::KanMX pMR63L(CEN LEU2 hsp82ΔMEEVD) pRS313(CEN HIS3) |
| Δhsc82 Δhsp82 p[hsp82W585T] | AG734 | 779-6A MATα [psi−] ρ0 Δhsc82::KanMX Δhsp82::KanMX pRS315-hsp82W585T(CEN LEU2 hsp82W585T) pRS313(CEN HIS3) |
| wt-CR11 | AG783 | 779-6A MATα [psi−] ρ0 HSP104 (reverted to WT, recoded) pRS313(CEN HIS3) |
| wt-CR12 | AG784 | 779-6A MATα [psi−] ρ0 HSP104 (reverted to WT, recoded) pRS313(CEN HIS3) |
| hsp104T160M Ura− | AG785 | 779-6A MATα [psi−] ρ0 hsp104T160M (hisG-URA3-hisG was lost on FOA) pRS313(CEN HIS3) |
| hsp104T160M [pin−] 8–1 | AG670 | 779-6A MATα [psi−] [pin−] ρ0 hsp104T160M::hisG-URA3-hisG ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 pRS313(CEN HIS3) |
| hsp104T160M [pin−] 8–2 | AG671 | 779-6A MATα [psi−] [pin−] ρ0 hsp104T160M::hisG-URA3-hisG ade2-1 SUQ5 trp1 kar1-1 his3 leu2 ura3 pRS313(CEN HIS3) |
| Back-cytoduction recipients | ||
| WT | AG679 | 779-6A MATa [psi−] ρ0 pRS314(CEN TRP1) |
| hsp104T160M | AG680 | 779-6A MATa [psi−] ρ0 hsp104T160M::hisG-URA3-hisG pRS314(CEN TRP1) |
In all cases (except where indicated specifically), the background of the strains was 779-6A.
Cytoduction Donor Isolation.
All [PSI+] variant strains used as cytoduction donors were isolated in either WT strain AG664 or hsp104T160M strain AG663 (both [psi−] [PIN+] and isogenic to 779-6A) (Table S4). To isolate spontaneously arising [PSI+] variants, or to measure its frequency, 105, 106, and 107 [psi−] cells were plated on –Ade (SD + HLU) medium. To raise the frequency of [PSI+] generation, AG664 and AG663 were transformed with pHK006 (LEU2 PGal1-SUP35NM), grown in YPAGal (1% yeast extract, 2% peptone, 0.004% adenine sulfate, 2% galactose) overnight, and plated on –Ade medium. After 5–6 d at 30 °C when most Ade+ colonies were at least 2–3 mm in diameter, clones were transferred into separate wells of 96-well plates containing 15% glycerol. Using a 48-pin replicator tool Ade+ isolates were stamped onto –Ade to double-check the Ade+ phenotype, 1/2 YPD to estimate the strength of the (presumed) [PSI+], 1/2 YPD with 3 mM guanidine hydrochloride to find the guanidine-curable (=[PSI+]) isolates, and YPG to confirm an intact mitochondrial genome that would be needed for cytoduction. The 96-well plates containing the Ade+ isolates were stored at −80 °C. To minimize the growth of the yeast cells, which could potentially change the [PSI+] variant, before each set of cytoduction experiments we thawed the 96-well plates at room temperature and “stamped” the isolates to –Ade plates, grew them for 4–5 d, suspended the cells in water, and used as cytoduction donors.
Cytoduction.
Donors and recipients were isogenic (to 779-6A in most cases) (Table S4). The donors of spontaneous [PSI+] variants carried the TRP1 vector pRS314. Donors of induced [PSI+] variants also carried the LEU2 plasmid pHK006 (bearing PGal1-SUP35NM which is not expressed on glucose-containing media) (Table S5). The recipient carried the HIS3 plasmid pRS313 to allow selection against donors and distinction of diploids and recipients. Recipients were made ρ0 by growth on media containing 25 µg/mL ethidium bromide, and transfer of mitochondrial DNA, as assayed by growth on glycerol, was used as an indicator that cytoplasm had been transferred from donor cells. Donor and recipients were mixed in water with a modest excess of donor cells, and spotted on a YPAD plate. After 6- to 8-h incubation at 30 °C, the mixture was streaked for single colonies on media selecting against the donor. Colonies were replica-plated to YPG, media selective for diploids (two types: one was –Leu and the other was –Trp) and –Ade. Clones growing on YPG but not on the media selective for diploids were cytoductants. Colonies that grew on –Ade media propagated [PSI+]. The back-cytoduction recipients carried the TRP1 vector pRS314 as a marker and, like the forward-cytoduction recipients, were ρ0.
Table S5.
Plasmids of Saccharomyces cerevisiae
Statistics.
We used the binomial distribution to assess the significance of differences of transmission frequency between strains or of frequency differences of a yes/no type between groups. For example, whether an orange variant's transmission to a WT recipient was significantly different from transmission to an isogenic hsp104 T160M recipient usually used this method. The mean transmission frequency (p), of the total population (adding cytoductants to both mutant and WT), the total number of cytoductants (N), and the binomial distribution allows calculation of the SD = sqrt[p(1 − p)N]. If the sample sizes are sufficiently large [i.e., if p(1 − p)N > ∼10], the actual differences of the mean transmission frequency to WT and mutant would show a normal distribution, with the SD calculated above and a mean of 0 if such differences were a result of chance. Using an online normal distribution calculator, this allows calculation of the likelihood that the observed difference is a result of chance.
Seed Number Measurement.
The [PSI+] strains were streaked to single colonies on 1/2 YPD medium supplemented with 3 mM guanidine hydrochloride. Individual colonies, with the underlying agar cube, were cut out of the plates, and the cells were suspended in water and plated on –Ade medium. The number of Ade+ colonies estimates the number of [PSI+] prion seeds contained by the cell which gave rise to the colony. For each strain, at least 10 individual colonies were tested (88).
Western Blot Analysis.
Yeast were grown overnight at 30 °C in YPAD, washed with water, and suspended in Disruption buffer [25 mM Tris⋅Cl pH 7.5, 150 mM NaCl, 1 mM DTT, 10 mM MgCl2, 1.2 mg ATP/mL, 1× Halt Protease Inhibitor Mixture (Thermo), one tablet Complete Mini Protease Inhibitor EDTA free (Roche)/8 mL, and 10 mM Prefabloc]. The cell suspension was placed, with glass beads, in screw-cap 2-mL tubes, and disrupted using a Bead Beater homogenizer (3 min, 4 °C). Protein concentrations were determined using the BCA assay and equalized between the samples using Disruption buffer. The samples were analyzed on a polyacrylamide gel and transferred to a PVDF membrane. The primary and the secondary antibodies used to perform the membrane staining are listed in Table S6.
Table S6.
Antibodies used in Fig. 3
| Antibody | Raised in | Poly/monoclonal? | Product ID/Source | Dilution for WB |
| Primary antibody | ||||
| Anti-Hsp104 | Rabbit | pAb | Polyclonal, present work | 1:5,000 |
| Anti-Sup35 | Mouse | mAb | (96) | 1:10,000 |
| Anti-Ssa1-4 | Rabbit | pAb | SPA757 (Enzo Lifesciences) | 1:1,000 |
| Anti-Cpr7 | Rabbit | pAb | (48) | 1:1,000 |
| Anti-Sti1 | Rabbit | pAb | (48) | 1:2,000 |
| Anti-Sse1/2 | Rabbit | pAb | (97) | 1:2,000 |
| Anti-Sis1 | Rabbit | pAb | (98) | 1:40,000 |
| Anti-Ydj1 | Rabbit | pAb | (98) | 1:10,000 |
| Secondary antibody | ||||
| Anti-Mouse IgG, HRP | Goat | pAb | 12-349 (Millipore) | 1:1,000 |
| Anti-Rabbit IgG, HRP | Goat | pAb | ab97080 (Abcam) | 1:10,000 |
Proteins Co-IP with Hsp104 and Hsp104T160M.
Cells of strain AG686 (WT) and AG687 (hsp104T160M) were grown and extracts made as described above for Western blots. Forty microliters of extract was mixed with 1 μL of anti-Hsp104 antibody and 460 μL of Disruption buffer (see Western blot method described above) in a 2-mL screw-cap tube and incubated overnight at 4 °C with slow mixing. Magnetic beads from the Pierce Classic Magnetic IP/co-IP kit were washed two times with 1 mL of Washing buffer (Disruption buffer without the protease inhibitors or detergents), the extract–antibody mixture was added to these beads, and the suspension was incubated for 1 h at room temperature with rotation. Beads were then washed three times with the Washing buffer, and proteins were eluted with 8 M urea in 100 mM Tris⋅Cl pH 7.5. Protein components were then subjected to proteolysis, on-column postdigestion reductive di-methylation, and quantitative proteomics essentially using approaches described by others (89–91), with details provided in SI Methods.
SI Methods
Using CRISPR-Cas to Revert hsp104T160M Back to WT.
The CRISPR-Cas procedure was performed as described elsewhere (92, 93) with some modifications. Strain AG687 (MATα hsp104T160M::hisG-URA3-hisG) (Table S4) was plated on FOA to select for loss of the URA3 marker cassette. The resulting Ura− strain AG730 was transformed with the p414-TEF1p-Cas9-CYC1t plasmid (a gift from George Church, Harvard Medical School, Cambridge, MA; Addgene plasmid # 43802) (92) to allow constitutive expression of Cas9. The linear backbone of the URA3 plasmid that will express the genome-targeting CRISPR guide RNA (gRNA) cassette was prepared by PCR using p426-SNR52p-gRNA.CAN1.Y-SUP4t (a gift from George Church; Addgene plasmid # 43803) (1) as a template and primers DRPE109 (GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGC) and DRPE110 (CGATCATTTATCTTTCACTGCGGAG). The gRNA cassette has ends homologous to the ends of the linear backbone (see above), namely the SNR52 promoter and the SUP4 terminator sequences, to allow homologous recombination to construct the gRNA-expressing plasmid in vivo (as in ref. 92). The gRNA was changed to target Cas9 to position 89093 on the chromosome XII which is inside the HSP104 gene and is right next to a protospacer-associated motif, a genomically encoded NGG nucleotide sequence directly 3′ of the 20-bp genome target known to be a crucial design constraint to the site specificity.
The HSP104-targeted gRNA cassette sequence was as follows (synthesized by Eurofins Genomics):
TGCGGTGTGAAATACCGCACAGATGCGTAAGGAGAAAATACCGCATCAGGAAATTGTAAGCGTTAATATTTTGTTAAAATTCGCGTTAAATTTTTGTTAAATCAGCTCATTTTTTAACCAATAGGCCGAAATCGGCAAAATCCCTTATAAATCAAAAGAATAGACCGAGATAGGGTTGAGTGTTGTTCCAGTTTGGAACAAGAGTCCACTATTAAAGAACGTGGACTCCAACGTCAAAGGGCGAAAAACCGTCTATCAGGGCGATGGCCCACTACGTGAACCATCACCCTAATCAAGTTTTTTGGGGTCGAGGTGCCGTAAAGCACTAAATCGGAACCCTAAAGGGAGCCCCCGATTTAGAGCTTGACGGGGAAAGCCTCTTTGAAAAGATAATGTATGATTATGCTTTCACTCATATTTATACAGAAACTTGATGTTTTCTTTCGAGTATATACAAGGTGATTACATGTACGTTTGAAGTACAACTCTAGATTTTGTAGTGCCCTCTTGGGCTAGCGGTAAAGGTGCGCATTTTTTCACACCCTACAATGTTCTGTTCAAAAGATTTTGGTCAAACGCTGTAGAAGTGAAAGTTGGTGCGCATGTTTCGGCGTTCGAAACTTCTCCGCAGTGAAAGATAAATGATCGGCTGATACGAACACACCTTGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGGTGCTTTTTTTGTTTTTTATGTCTCAGCTTTTGTTCCCTTTAGTGAGGGTTAATTGCGCGCTTGGCGTAATCATGGTCATAGCTGTTTCCTGTGTGAAATTGTTATCCGCTCACAATTCCACACAACATAGGAGCCGGAAGCATAAAGTGTAAAGCCTGGGGTGCCTAATGAGTGAGGTAACTCACATTAATTGCGTTGCGCTCACTGCCCGCTTTCCAGTCGGGAAACCTGTCGTGCCAGCTGCATTAATGAATCGGCCAACGCGCGGGGAGAGGCGGTTTGCGTATTGGGCGCTCTTCCGCTTCCTCGCTCACTGACTCGCTGCGCTCGGTCGTTCGGCTGCGGCGAGCGGTATCAGCTCACTCAAAGGCGGTAATACGGTTATCCACAGAATCAGGGGATAACGCAGGAAAGAACATGTGAGC
The double-stranded donor DNA to revert the mutant sequence was the 1,100 bp of the 5′ end of the HSP104 gene, with changes in the third bases of several codons around codon 160 so that once reverted, the gRNA will no longer recognize, and Cas9 will no longer cleave that site. Its sequence was as follows:
GlyAlaAspMetAsnThrProLeuGlu, T160M mutant amino acid sequence;
TGGCGCTGACATGAATACTCCATTGGAATATTT, mutant DNA sequence;
TGGCGCTGACACTAATACTCCATTGGAATATTT, WT HSP104 DNA sequence;
TGGCGCTGATACGAACACACCTTTGGAATATTT, CRISPR-Cas reverted DNA sequence;
GlyAlaAspThrAsnThrProLeuGlu, CRISPR-Cas reverted amino acid sequence.
We cotransformed the linearized gRNA cassette expressing plasmid, the HSP104-targeted gRNA cassette, and the double-stranded donor DNA into the cells constitutively expressing Cas9 and selected for Ura+ transformats (i.e., the ones in which the linear fragments of the gRNA expressing plasmid recombined because of homologous recombination and, presumably, took up the ds donor DNA). These candidates were screened for the presence of the heterology block in the chromosome using colony PCR with primers F4 (TTACAGAAAGGGCTCTAACGATTTT) and HB_R (TTCCAATGGAGTATTAGTgTC). The primer pair F4/R4 (TTTCCACAATAGATCTATATTCGTT) was used as a control for the WT sequence. In case of the candidates for which we observed the heterology block-containing PCR product, we performed the sequencing of the 5′ part of HSP104 gene using primers F4 and R4 to confirm the mutation. We grew the confirmed isolates on nonselective medium to allow loss of all of the plasmids. The plasmid-free strains were used to generate the cytoduction recipients (Table S4).
Mass Spectrometry.
Protein components present on the magnetic beads (Methods) were solubilized in 200 μL of 100 mM Tris (pH 8), 8 M urea for 30 min. The same volume of the liquid portion was then carefully removed to a fresh vial, an aliquot of hen ovalbumin in 100 mM Tris 8 M urea was added, and 20 μL of 100 mM DTT, 100 mM Tris (pH 8), 8 M urea was added and the sample incubated at 35 °C for 20 min. Next, the samples were placed in room temperature water for 5 min before the addition of 30 μL of 200 mM chloroacetamide, 100 mM Tris (pH 8), 8 M urea, and then incubated for 1 h in the dark. Next 30 μL, 200 mM β-mercaptoethanol, 100 mM Tris (pH 8), 8 M urea was added and the sample incubated after mixing for 15 min. Two micrograms of Lys-C was then added and the samples digested overnight. The next day each sample was diluted with three volumes of 50 mM ammonium acetate and 10 μg of trypsin was added. After 4 h the samples were made, 2% formic acid, an aliquot of predigested hen lysozyme peptides was added, and the sample was loaded in several applications on to a C8 Stage tip containing four layers of Empore C8 material (1.8-mM diameter) using a centrifuge with a spinning bucket (application force was 200 G). The flow through from this column was transferred using a pippettor to a similar column made with Empore C18. After application of the sample, wash solutions were similarly applied in a serial fashion. First, 200 μL of 1.6% formic acid was applied and next 100 μL of 0.16% formic acid. STAGE tips for each pair of subsamples were prematched using a test spin with water. Loading was performed at 4 °C.
After loading and washing were complete, the columns were allowed to warm up to room temperature for 15 min. Each STAGE tip now received 400 μL of either “light” or “intermediate” labeling solution (90) and this solution was allowed to slowly flow through the column at 100 G as an adaptation of protocol C from the same reference. After 30 min the centrifugal force was raised to 400 and the bed cleared. The columns were now placed at 4 °C while the flow-through reaction liquid from each C8/C18 pair of columns was combined and acidified and placed in ice water. After the columns and flow-through solution were ice cold, the reactant flow-through was applied in the same way that the sample was loaded. Subsequently, the columns were washed with 200 μL of 1.6% formic acid 100 mM ammonium acetate and then 100 μL of 0.4% formic acid, 20 mM ammonium acetate. Next, the peptides on each of the columns were eluted with 100 μL 0.4% formic acid 40% acetonitrile and then 100 μL 0.4% formic acid 80% acetonitrile. The solutions from the C8/C18 pairs were pooled and then the L/M pairs were positioned near a shared STAGE tip containing Empore SCX material. Next, matching portions of each subsample were mixed thoroughly in a fresh vial and applied to the SCX stage tip which was loaded at 300 G. After loading, each SCX column was washed with 400 μL of 0.4% formic acid 40% acetonitrile and then 400 μL 0.4% formic acid 80% acetonitrile and then 50 μL of 0.4% formic acid 20% acetonitrile. The six SCX fractions were created from this column using the SCX step solutions described by others (89).
LC/MS/MS analysis was performed using a vent configured NanoAcquity LC system(Trap; Symmetry C18 180 μm × 20 mm, analytical; 1.7 μm BEH130 C18 75 μm × 250 mm) interfaced to a Thermo-Fisher Q-Exactive mass spectrometer. The initial (A) solution for the LC system was 0.1% formic acid in water and the terminal (B) solution was 0.1% formic acid 99.9% acetonitrile. One-third of each fraction was injected onto the trap for 7 min at 7 μL/min at 99% A. Next, the vent was closed and the flow rate set to 0.1 μL/min. The LC system linearly raised the concentration to 23% B by minute 264, to 80% B in the subsequent 20 min, holding for 5 min at 80%, and then retuning to 99% A in 10 min. A contact closure signal was sent to the Q-Exactive to start data collection at minute 2. The Q-Exactive collected a parent spectrum at resolution 140K setting with a target of 3E6 (450–1,800, 100-ms injection) followed by up to 30 fragment spectra collected from candidate peaks, which were filtered to have charge states 2–4, apparent intensities above 20,000, and accumulation target of 2E5 (300-ms maximum injection).
All data were processed using MaxQuant 1.5.3.28 using default settings, except that multiplicity was set to two and light and heavy labels were set to appropriate dimethyl isotopamers, requantify was enabled, a minimum peptide length of 6 was used because of the size of the search space, second peptides were disallowed, and match between runs was enabled with a match time window of 3.5 min and an alignment window of 30 min. Protein sequences searched were the implied proteome of yeast (S288C and isoforms downloaded 2/17/16), selected proteins from the domestic chicken (which are internal controls), and a list of common contaminants. Direct linear ratios (without normalization) were used in evaluating the results of the experiment with internal controls present near unity ratios indicating the successful application of the dimethyl labeling approach.
Acknowledgments
We thank Eric Anderson (National Institute of Diabetes and Digestive and Kidney Diseases) for mass spectometry analysis; Harold Smith (National Institute of Diabetes and Digestive and Kidney Diseases) for next-generation sequencing; Sue Liebman for strains and antibodies; and Elizabeth Craig (University of Wisconsin) and Jeff Brodsky (Pittsburgh University) for antibodies. This work was supported by the Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1704016114/-/DCSupplemental.
References
- 1.Wickner RB. [URE3] as an altered URE2 protein: Evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 2.King C-Y, et al. Prion-inducing domain 2-114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc Natl Acad Sci USA. 1997;94:6618–6622. doi: 10.1073/pnas.94.13.6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. In vitro propagation of the prion-like state of yeast Sup35 protein. Science. 1997;277:381–383. doi: 10.1126/science.277.5324.381. [DOI] [PubMed] [Google Scholar]
- 4.Glover JR, et al. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell. 1997;89:811–819. doi: 10.1016/s0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 5.King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature. 2004;428:319–323. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 7.Edskes HK, Gray VT, Wickner RB. The [URE3] prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc Natl Acad Sci USA. 1999;96:1498–1503. doi: 10.1073/pnas.96.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masison DC, Wickner RB. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science. 1995;270:93–95. doi: 10.1126/science.270.5233.93. [DOI] [PubMed] [Google Scholar]
- 9.Brachmann A, Baxa U, Wickner RB. Prion generation in vitro: Amyloid of Ure2p is infectious. EMBO J. 2005;24:3082–3092. doi: 10.1038/sj.emboj.7600772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: The story of [PIN(+)] Cell. 2001;106:171–182. doi: 10.1016/s0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 11.Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics. 1997;147:507–519. doi: 10.1093/genetics/147.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shewmaker F, Wickner RB, Tycko R. Amyloid of the prion domain of Sup35p has an in-register parallel β-sheet structure. Proc Natl Acad Sci USA. 2006;103:19754–19759. doi: 10.1073/pnas.0609638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baxa U, et al. Characterization of β-sheet structure in Ure2p1-89 yeast prion fibrils by solid-state nuclear magnetic resonance. Biochemistry. 2007;46:13149–13162. doi: 10.1021/bi700826b. [DOI] [PubMed] [Google Scholar]
- 14.Wickner RB, Dyda F, Tycko R. Amyloid of Rnq1p, the basis of the [PIN+] prion, has a parallel in-register β-sheet structure. Proc Natl Acad Sci USA. 2008;105:2403–2408. doi: 10.1073/pnas.0712032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorkovskiy A, Thurber KR, Tycko R, Wickner RB. Locating folds of the in-register parallel β-sheet of the Sup35p prion domain infectious amyloid. Proc Natl Acad Sci USA. 2014;111:E4615–E4622. doi: 10.1073/pnas.1417974111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wickner RB, Shewmaker F, Kryndushkin D, Edskes HK. Protein inheritance (prions) based on parallel in-register β-sheet amyloid structures. BioEssays. 2008;30:955–964. doi: 10.1002/bies.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wickner RB, et al. Amyloids and yeast prion biology. Biochemistry. 2013;52:1514–1527. doi: 10.1021/bi301686a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chernoff YO, Ono B-I. Dosage-dependent modifiers of PSI-dependent omnipotent suppression in yeast. In: Brown AJP, Tuite MF, McCarthy JEG, editors. Protein Synthesis and Targeting in Yeast. Springer; Berlin: 1992. pp. 101–107. [Google Scholar]
- 19.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez Y, Lindquist SL. HSP104 required for induced thermotolerance. Science. 1990;248:1112–1115. doi: 10.1126/science.2188365. [DOI] [PubMed] [Google Scholar]
- 21.Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 22.Schlieker C, Tews I, Bukau B, Mogk A. Solubilization of aggregated proteins by ClpB/DnaK relies on the continuous extraction of unfolded polypeptides. FEBS Lett. 2004;578:351–356. doi: 10.1016/j.febslet.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 23.Lum R, Niggemann M, Glover JR. Peptide and protein binding in the axial channel of Hsp104. Insights into the mechanism of protein unfolding. J Biol Chem. 2008;283:30139–30150. doi: 10.1074/jbc.M804849200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lum R, Tkach JM, Vierling E, Glover JR. Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J Biol Chem. 2004;279:29139–29146. doi: 10.1074/jbc.M403777200. [DOI] [PubMed] [Google Scholar]
- 25.Tuite MF, Mundy CR, Cox BS. Agents that cause a high frequency of genetic change from [psi+] to [psi-] in Saccharomyces cerevisiae. Genetics. 1981;98:691–711. doi: 10.1093/genetics/98.4.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung G, Masison DC. Guanidine hydrochloride inhibits Hsp104 activity in vivo: A possible explanation for its effect in curing yeast prions. Curr Microbiol. 2001;43:7–10. doi: 10.1007/s002840010251. [DOI] [PubMed] [Google Scholar]
- 27.Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol. 2001;40:1357–1369. doi: 10.1046/j.1365-2958.2001.02478.x. [DOI] [PubMed] [Google Scholar]
- 28.Jung G, Jones G, Masison DC. Amino acid residue 184 of yeast Hsp104 chaperone is critical for prion-curing by guanidine, prion propagation, and thermotolerance. Proc Natl Acad Sci USA. 2002;99:9936–9941. doi: 10.1073/pnas.152333299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grimminger V, Richter K, Imhof A, Buchner J, Walter S. The prion curing agent guanidinium chloride specifically inhibits ATP hydrolysis by Hsp104. J Biol Chem. 2004;279:7378–7383. doi: 10.1074/jbc.M312403200. [DOI] [PubMed] [Google Scholar]
- 30.Wegrzyn RD, Bapat K, Newnam GP, Zink AD, Chernoff YO. Mechanism of prion loss after Hsp104 inactivation in yeast. Mol Cell Biol. 2001;21:4656–4669. doi: 10.1128/MCB.21.14.4656-4669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ness F, Ferreira P, Cox BS, Tuite MF. Guanidine hydrochloride inhibits the generation of prion “seeds” but not prion protein aggregation in yeast. Mol Cell Biol. 2002;22:5593–5605. doi: 10.1128/MCB.22.15.5593-5605.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem. 2003;278:49636–49643. doi: 10.1074/jbc.M307996200. [DOI] [PubMed] [Google Scholar]
- 33.Jung G, Jones G, Wegrzyn RD, Masison DC. A role for cytosolic hsp70 in yeast [PSI(+)] prion propagation and [PSI(+)] as a cellular stress. Genetics. 2000;156:559–570. doi: 10.1093/genetics/156.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones GW, Masison DC. Saccharomyces cerevisiae Hsp70 mutations affect [PSI+] prion propagation and cell growth differently and implicate Hsp40 and tetratricopeptide repeat cochaperones in impairment of [PSI+] Genetics. 2003;163:495–506. doi: 10.1093/genetics/163.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts BT, Moriyama H, Wickner RB. [URE3] prion propagation is abolished by a mutation of the primary cytosolic Hsp70 of budding yeast. Yeast. 2004;21:107–117. doi: 10.1002/yea.1062. [DOI] [PubMed] [Google Scholar]
- 36.Hines JK, et al. [SWI], the prion formed by the chromatin remodeling factor Swi1, is highly sensitive to alterations in Hsp70 chaperone system activity. PLoS Genet. 2011;7:e1001309. doi: 10.1371/journal.pgen.1001309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winkler J, Tyedmers J, Bukau B, Mogk A. Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J Cell Biol. 2012;198:387–404. doi: 10.1083/jcb.201201074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newnam GP, Wegrzyn RD, Lindquist SL, Chernoff YO. Antagonistic interactions between yeast chaperones Hsp104 and Hsp70 in prion curing. Mol Cell Biol. 1999;19:1325–1333. doi: 10.1128/mcb.19.2.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiktev DA, et al. Regulation of chaperone effects on a yeast prion by cochaperone Sgt2. Mol Cell Biol. 2012;32:4960–4970. doi: 10.1128/MCB.00875-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Higurashi T, Hines JK, Sahi C, Aron R, Craig EA. Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Proc Natl Acad Sci USA. 2008;105:16596–16601. doi: 10.1073/pnas.0808934105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Troisi EM, Rockman ME, Nguyen PP, Oliver EE, Hines JK. Swa2, the yeast homolog of mammalian auxilin, is specifically required for the propagation of the prion variant [URE3-1] Mol Microbiol. 2015;97:926–941. doi: 10.1111/mmi.13076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reidy M, Miot M, Masison DC. Prokaryotic chaperones support yeast prions and thermotolerance and define disaggregation machinery interactions. Genetics. 2012;192:185–193. doi: 10.1534/genetics.112.142307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hung GC, Masison DC. N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics. 2006;173:611–620. doi: 10.1534/genetics.106.056820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheufler C, et al. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 45.Abbas-Terki T, Donzé O, Briand P-A, Picard D. Hsp104 interacts with Hsp90 cochaperones in respiring yeast. Mol Cell Biol. 2001;21:7569–7575. doi: 10.1128/MCB.21.22.7569-7575.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moosavi B, Wongwigkarn J, Tuite MF. Hsp70/Hsp90 co-chaperones are required for efficient Hsp104-mediated elimination of the yeast [PSI(+)] prion but not for prion propagation. Yeast. 2010;27:167–179. doi: 10.1002/yea.1742. [DOI] [PubMed] [Google Scholar]
- 47.Reidy M, Masison DC. Sti1 regulation of Hsp70 and Hsp90 is critical for curing of Saccharomyces cerevisiae [PSI+] prions by Hsp104. Mol Cell Biol. 2010;30:3542–3552. doi: 10.1128/MCB.01292-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones G, Song Y, Chung S, Masison DC. Propagation of Saccharomyces cerevisiae [PSI+] prion is impaired by factors that regulate Hsp70 substrate binding. Mol Cell Biol. 2004;24:3928–3937. doi: 10.1128/MCB.24.9.3928-3937.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Helsen CW, Glover JR. Insight into molecular basis of curing of [PSI+] prion by overexpression of 104-kDa heat shock protein (Hsp104) J Biol Chem. 2012;287:542–556. doi: 10.1074/jbc.M111.302869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park YN, et al. Hsp104 overexpression cures Saccharomyces cerevisiae [PSI+] by causing dissolution of the prion seeds. Eukaryot Cell. 2014;13:635–647. doi: 10.1128/EC.00300-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ness F, Cox BS, Wongwigkarn J, Naeimi WR, Tuite MF. Over-expression of the molecular chaperone Hsp104 in Saccharomyces cerevisiae results in the malpartition of [PSI(+) ] propagons. Mol Microbiol. 2017;104:125–143. doi: 10.1111/mmi.13617. [DOI] [PubMed] [Google Scholar]
- 52.Newnam GP, Birchmore JL, Chernoff YO. Destabilization and recovery of a yeast prion after mild heat shock. J Mol Biol. 2011;408:432–448. doi: 10.1016/j.jmb.2011.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakayashiki T, Kurtzman CP, Edskes HK, Wickner RB. Yeast prions [URE3] and [PSI+] are diseases. Proc Natl Acad Sci USA. 2005;102:10575–10580. doi: 10.1073/pnas.0504882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McGlinchey RP, Kryndushkin D, Wickner RB. Suicidal [PSI+] is a lethal yeast prion. Proc Natl Acad Sci USA. 2011;108:5337–5341. doi: 10.1073/pnas.1102762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelly AC, Shewmaker FP, Kryndushkin D, Wickner RB. Sex, prions, and plasmids in yeast. Proc Natl Acad Sci USA. 2012;109:E2683–E2690. doi: 10.1073/pnas.1213449109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Halfmann R, et al. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature. 2012;482:363–368. doi: 10.1038/nature10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kelly AC, Busby B, Wickner RB. Effect of domestication on the spread of the [PIN+] prion in Saccharomyces cerevisiae. Genetics. 2014;197:1007–1024. doi: 10.1534/genetics.114.165670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wickner RB, Kelly AC. Prions are affected by evolution at two levels. Cell Mol Life Sci. 2016;73:1131–1144. doi: 10.1007/s00018-015-2109-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chernoff YO, Newnam GP, Kumar J, Allen K, Zink AD. Evidence for a protein mutator in yeast: Role of the Hsp70-related chaperone ssb in formation, stability, and toxicity of the [PSI] prion. Mol Cell Biol. 1999;19:8103–8112. doi: 10.1128/mcb.19.12.8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chacinska A, et al. Ssb1 chaperone is a [PSI+] prion-curing factor. Curr Genet. 2001;39:62–67. doi: 10.1007/s002940000180. [DOI] [PubMed] [Google Scholar]
- 61.Nelson RJ, Ziegelhoffer T, Nicolet C, Werner-Washburne M, Craig EA. The translation machinery and 70 kd heat shock protein cooperate in protein synthesis. Cell. 1992;71:97–105. doi: 10.1016/0092-8674(92)90269-i. [DOI] [PubMed] [Google Scholar]
- 62.Pfund C, et al. The molecular chaperone Ssb from Saccharomyces cerevisiae is a component of the ribosome-nascent chain complex. EMBO J. 1998;17:3981–3989. doi: 10.1093/emboj/17.14.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kryndushkin DS, Shewmaker F, Wickner RB. Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. EMBO J. 2008;27:2725–2735. doi: 10.1038/emboj.2008.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wickner RB, Bezsonov E, Bateman DA. Normal levels of the antiprion proteins Btn2 and Cur1 cure most newly formed [URE3] prion variants. Proc Natl Acad Sci USA. 2014;111:E2711–E2720. doi: 10.1073/pnas.1409582111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Malinovska L, Kroschwald S, Munder MC, Richter D, Alberti S. Molecular chaperones and stress-inducible protein-sorting factors coordinate the spatiotemporal distribution of protein aggregates. Mol Biol Cell. 2012;23:3041–3056. doi: 10.1091/mbc.E12-03-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kryndushkin D, Ihrke G, Piermartiri TC, Shewmaker F. A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity. Mol Microbiol. 2012;86:1531–1547. doi: 10.1111/mmi.12075. [DOI] [PubMed] [Google Scholar]
- 67.Borchsenius AS, Wegrzyn RD, Newnam GP, Inge-Vechtomov SG, Chernoff YO. Yeast prion protein derivative defective in aggregate shearing and production of new ‘seeds’. EMBO J. 2001;20:6683–6691. doi: 10.1093/emboj/20.23.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bateman DA, Wickner RB. The [PSI+] prion exists as a dynamic cloud of variants. PLoS Genet. 2013;9:e1003257. doi: 10.1371/journal.pgen.1003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kimberlin RH, Cole S, Walker CA. Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J Gen Virol. 1987;68:1875–1881. doi: 10.1099/0022-1317-68-7-1875. [DOI] [PubMed] [Google Scholar]
- 70.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 71.Zuehlke AD, Wren N, Tenge V, Johnson JL. Interaction of heat shock protein 90 and the co-chaperone Cpr6 with Ura2, a bifunctional enzyme required for pyrimidine biosynthesis. J Biol Chem. 2013;288:27406–27414. doi: 10.1074/jbc.M113.504142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kirkland PA, Reidy M, Masison DC. Functions of yeast Hsp40 chaperone Sis1p dispensable for prion propagation but important for prion curing and protection from prion toxicity. Genetics. 2011;188:565–577. doi: 10.1534/genetics.111.129460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fan Q, Park K-W, Du Z, Morano KA, Li L. The role of Sse1 in the de novo formation and variant determination of the [PSI+] prion. Genetics. 2007;177:1583–1593. doi: 10.1534/genetics.107.077982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moran C, Kinsella GK, Zhang ZR, Perrett S, Jones GW. Mutational analysis of Sse1 (Hsp110) suggests an integral role for this chaperone in yeast prion propagation in vivo. G3 (Bethesda) 2013;3:1409–1418. doi: 10.1534/g3.113.007112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sharma D, Masison DC. Functionally redundant isoforms of a yeast Hsp70 chaperone subfamily have different antiprion effects. Genetics. 2008;179:1301–1311. doi: 10.1534/genetics.108.089458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Borkovich KA, Farrelly FW, Finkelstein DB, Taulien J, Lindquist S. hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol Cell Biol. 1989;9:3919–3930. doi: 10.1128/mcb.9.9.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Genest O, et al. Uncovering a region of heat shock protein 90 important for client binding in E. coli and chaperone function in yeast. Mol Cell. 2013;49:464–473. doi: 10.1016/j.molcel.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kumar N, Gaur D, Gupta A, Puri A, Sharma D. Hsp90-associated immunophilin homolog Cpr7 is required for the mitotic stability of [URE3] prion in Saccharomyces cerevisiae. PLoS Genet. 2015;11:e1005567. doi: 10.1371/journal.pgen.1005567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Salnikova AB, Kryndushkin DS, Smirnov VN, Kushnirov VV, Ter-Avanesyan MD. Nonsense suppression in yeast cells overproducing Sup35 (eRF3) is caused by its non-heritable amyloids. J Biol Chem. 2005;280:8808–8812. doi: 10.1074/jbc.M410150200. [DOI] [PubMed] [Google Scholar]
- 80.Lacroute F. Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. J Bacteriol. 1971;106:519–522. doi: 10.1128/jb.106.2.519-522.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Edskes HK, McCann LM, Hebert AM, Wickner RB. Prion variants and species barriers among Saccharomyces Ure2 proteins. Genetics. 2009;181:1159–1167. doi: 10.1534/genetics.108.099929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bateman DA, Wickner RB. [PSI+] Prion transmission barriers protect Saccharomyces cerevisiae from infection: Intraspecies ‘species barriers’. Genetics. 2012;190:569–579. doi: 10.1534/genetics.111.136655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Borchsenius AS, Müller S, Newnam GP, Inge-Vechtomov SG, Chernoff YO. Prion variant maintained only at high levels of the Hsp104 disaggregase. Curr Genet. 2006;49:21–29. doi: 10.1007/s00294-005-0035-0. [DOI] [PubMed] [Google Scholar]
- 84.Kryndushkin D, Wickner RB. Nucleotide exchange factors for Hsp70s are required for [URE3] prion propagation in Saccharomyces cerevisiae. Mol Biol Cell. 2007;18:2149–2154. doi: 10.1091/mbc.E07-02-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kryndushkin DS, Engel A, Edskes H, Wickner RB. Molecular chaperone Hsp104 2can promote yeast prion generation. Genetics. 2011;188:339–348. doi: 10.1534/genetics.111.127779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sherman F. Getting started with yeast. In: Guthrie C, Fink GR, editors. Guide to Yeast Genetics and Molecular Biology. Academic; San Diego: 1991. pp. 3–21. [Google Scholar]
- 87.Brachmann CB, et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 88.Cox B, Ness F, Tuite M. Analysis of the generation and segregation of propagons: Entities that propagate the [PSI+] prion in yeast. Genetics. 2003;165:23–33. doi: 10.1093/genetics/165.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11:319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- 90.Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJ. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc. 2009;4:484–494. doi: 10.1038/nprot.2009.21. [DOI] [PubMed] [Google Scholar]
- 91.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 92.DiCarlo JE, et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013;41:4336–4343. doi: 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Horwitz AA, et al. Efficient multiplexed integration of synergistic alleles and metabolic pathways in yeasts via CRISPR-Cas. Cell Syst. 2015;1:88–96. doi: 10.1016/j.cels.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 94.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Edskes HK, et al. Sporadic distribution of prion-forming ability of Sup35p from yeasts and fungi. Genetics. 2014;198:605–616. doi: 10.1534/genetics.114.166538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bagriantsev SN, Kushnirov VV, Liebman SW. Analysis of amyloid aggregates using agarose gel electrophoresis. Methods Enzymol. 2006;412:33–48. doi: 10.1016/S0076-6879(06)12003-0. [DOI] [PubMed] [Google Scholar]
- 97.Goeckeler JL, Stephens A, Lee P, Caplan AJ, Brodsky JL. Overexpression of yeast Hsp110 homolog Sse1p suppresses ydj1-151 thermosensitivity and restores Hsp90-dependent activity. Mol Biol Cell. 2002;13:2760–2770. doi: 10.1091/mbc.02-04-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yan W, Craig EA. The glycine-phenylalanine-rich region determines the specificity of the yeast Hsp40 Sis1. Mol Cell Biol. 1999;19:7751–7758. doi: 10.1128/mcb.19.11.7751. [DOI] [PMC free article] [PubMed] [Google Scholar]