

Abstract
Pemphigus and pemphigoid are the prototypical immunobullous diseases. Although it has been well established that they are caused by deposition of autoreactive antibodies directed against adherence proteins within the skin, the specific genetic and environmental factors leading to development of these diseases continue to be an area of investigation. Herein, we discuss several of the potential environmental triggers that may induce patients to develop immunobullous diseases including medications, viral infections, UV exposure or other radiation injury and dietary factors. In addition, the potential genetic and immunologic mechanisms contributing to the pathogenesis of pemphigus and pemphigoid will be reviewed. The multifactorial nature of these diseases contributes to their complexity and highlights the importance of a detailed personal and family history when caring for these patients.
Keywords: Autoimmune blistering diseases, desmoglein, pemphigoid, pemphigus
What was known?
Autoimmune bullous diseases such as pemphigus and pemphigoid are caused by a variety of autoreactive antibodies directed against different adherence proteins within the skin.
Introduction
Pemphigus and pemphigoid result from deposition of autoreactive antibodies directed against various intraepithelial and subepidermal proteins, resulting in the formation of bullae or erosions.[1] Pemphigus is derived from the Greek word for blister, pemphix, and has several major variants: Pemphigus foliaceus, paraneoplastic pemphigus, pemphigus vegetans, pemphigus erythematosus, IgA pemphigus, drug-induced pemphigus, pemphigus fogo selvage and pemphigus vulgaris (PV).[2,3] PV is the most common and well-characterized variant.
In pemphigoid, the dermal-epidermal junction is the autoimmune target resulting in a complete separation of the epidermis from the dermis. Bullous pemphigoid (BP) is the most common variant from the pemphigoid class of blistering diseases. Related diseases include cicatricial pemphigoid, linear IgA bullous dermatosis (LAD), epidermolysis bullosa acquisita (EBA), lichen-planus pemphigoides, and anti-p200 pemphigoid.[1,4]
Although it has been well established that autoantibodies directed against adherence proteins within the skin is the underlying cause of all autoimmune bullous diseases, it is still unclear what factors lead to their occurrence. We plan to review the pathophysiology of the two most common autoimmune bullous diseases: PV and BP. Specifically, we will discuss the roles that genetic predisposition and environmental triggers play in the onset of these diseases.
Epidemiology, Presentation and Physical Exam
Incidence of pemphigus is estimated to be between 1 and 10 per million and individuals of Ashkenazi Jewish descent or of Mediterranean origin are at an increased risk. The peak incidence is between 30 and 60 years of age and the mean age of onset is between 50 and 60 years.[5] The prevalence in men and women is roughly equal.
In PV, autoantibodies are directed against desmoglein proteins within the epidermis, which leads to formation of characteristic flaccid blisters on the patient's cutaneous and mucosal surfaces [Figure 1]. The hallmark physical exam findings are the direct and indirect Nikolskiy signs. A positive direct Nikolskiy sign is noted when digital pressure on intact skin results in sloughing off of the outermost layer. A positive indirect Nikolskiy sign (Asboe–Hansen sign) is when digital pressure to an intact bullous lesion leads to enlargement of the lesion to involve adjacent clear skin.[6,7] In 66% of cases, disease onset occurs within the oral mucosa[8] and disease may be limited to the oral cavity for months before progressing to the keratinized surfaces of the patient.[9] Approximately 80% of patients with PV will have some oral involvement.[10] In addition to skin and oral involvement, 50% of patients will have additional sites affected by PV including their nails, presenting as tender periungual erythema and edema; pharynx and larynx, resulting in dysphagia and hoarseness; and nasal cavity, presenting as nasal congestion with bloody mucus.[11,12] In all cases, blisters may be associated with severe pain, pruritus, a burning sensation, or paresthesias. Prior to the discovery of an effective therapy, PV was fatal in approximately 70% of cases as a result of fluid/electrolyte-associated abnormalities and infection.[13]
Figure 1.
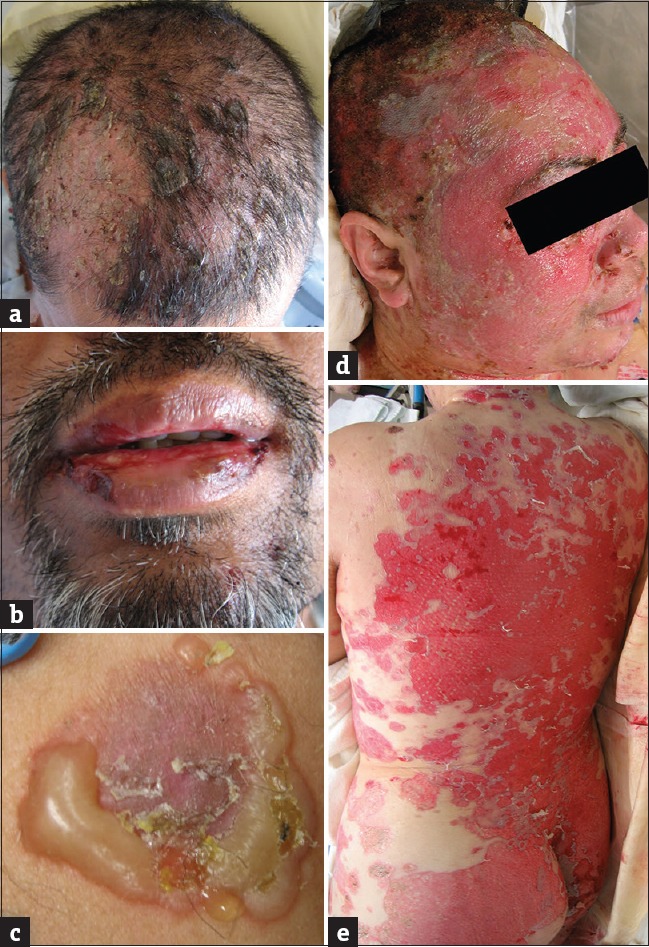
Clinical photos of pemphigus lesions. (a) Flaccid bullae involving the scalp. (b) Erosions of oral cavity following blister rupture. (c) Characteristic flaccid blister with previously ruptured areas. (d and e) Widespread cutaneous erosions following rupture of bullae
Pemphigoid [Figure 2] presents as tense bullae rather than flaccid ones and mucosal involvement is not as common. In contrast to pemphigus, only 33-50% of BP patients will develop oral manifestations of their disease and only 10% will present with oral findings.[8] The disease clinically manifests most commonly in the elderly (above 65 years of age) with urticarial lesions, tense blisters, and erosions that may involve the mucous membrane. The incidence of pemphigoid is higher in males and increases significantly over the age of 70 with patients over 90 years of age having a 300-fold higher relative risk than those under the age of 60.[14]
Figure 2.
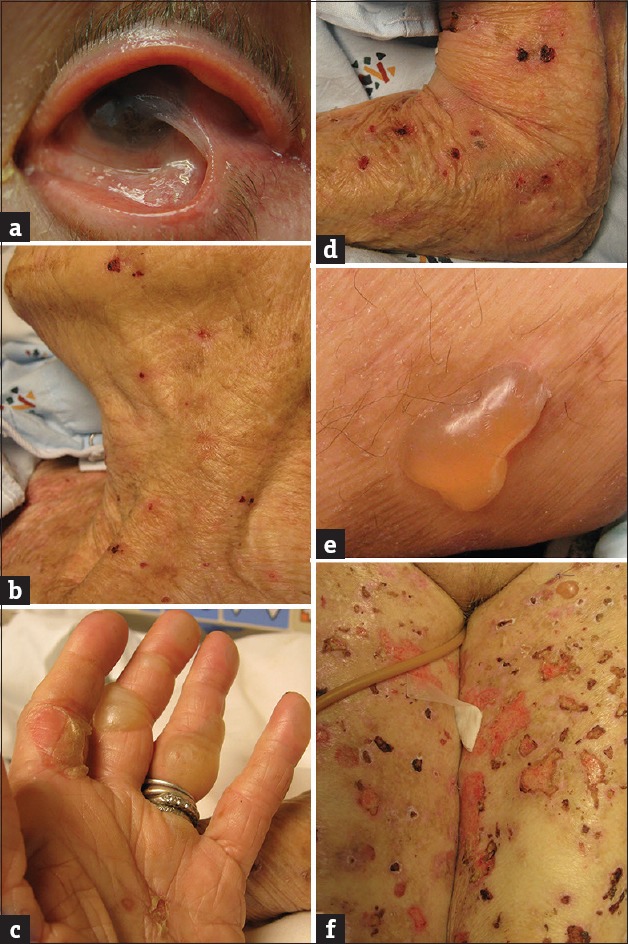
Clinical photographs of pemphigoid lesions. (a) Ocular involvement by cicatricial pemphigoid with formation of symblepharon (adhesion between bulbar and palpebral conjunctivae). (b) Superficial erosions following rupture of cutaneous bullae. (c) Ruptured bullae on thumb and second digit, intact tense bullae on third digit. (d) Erosions following rupture of bullae. (e) Tense bullous lesion on medial thigh. (f) Bullous lesions in various stages, including intact as well as ruptured bullae and resulting erosions
Pathophysiology
The pathogenesis of PV was established by Beutner and Jordon in 1964 when it was noted that pemphigus patients possessed autoantibodies to intercellular antigens found within the malphighian epithelia.[15] Through in vitro studies on human skin and disease-associated IgG passive transfer experiments in mice, autoreactive IgG was identified as the reason for the acantholysis and bullae formation in PV.[16,17] The most well-understood and accepted target antigens have been the calcium-dependent intercellular adhesion proteins that form the desmosomes, desmoglein (DSG) 1 and 3. The desmogleins are members of the cadherin protein family and are responsible for anchoring keratin intermediate filaments to the cell membrane of epidermal cells. The type of desmoglein affected has clinical significance, as the location of PV lesions depends on the specific desmoglein targeted by the autoreactive immune response. DSG1 is most readily found within superficial layers of the epidermis. In contrast, DSG3 is more predominant within non-keratinized epithelia such as mucosal surfaces.[9] The “compensation theory” established by Stanley and colleagues details how this distribution of target antigens leads to the different clinical presentations of PV.[18] In areas where DSG 1 and DSG 3 are equally present, one desmoglein can compensate for the other's loss of function.[19] For example, a patient with auto-IgG4 to DSG1 will present with superficial cutaneous blisters because DSG3 is not present in sufficient quantities in the upper epidermis to compensate for the autoantibody-mediated functional loss of DSG1. These patients are unlikely to have mucosal involvement due to the compensatory action of DSG3 at those sites.
Although the role of DSG1 and 3 has been well established in pemphigus, the presence of other non-desmoglein antigens has also been suggested. For instance, anti-E-cadherin antibodies have been detected in PV patients at significantly higher levels than healthy controls.[20] Keratinocyte acetylcholine receptors (AChRs) also play a role in cell–cell adhesion by regulating the intraepithelial expression of desmoglein and may act as potential non-desmoglein antigens.[21] Supporting this theory is the association between PV and other autoimmune disorders involving AChRs including myasthenia gravis and thymoma-associated autoimmunity.[9,21] Furthermore, in keratinocyte monolayers, addition of anti-α9 AChR IgG induced acantholysis that could be reversed with addition of the cholinergic agonist, carbachol.[21]
The pemphigoid class of diseases is caused by autoantibodies against various components of hemidesmosomes, which are the cellular adhesion proteins linking individual basal keratinocytes to the underlying extracellular matrix of the epidermis. Specifically, autoreactive IgG1 and IgG4 antibodies target BP antigen 1 (BP230) and BP antigen 2 (BP180, also known as collagen XVII). BP180 is a type II transmembrane hemidesmosome protein, while BP230 is a cytoplasmic protein. Because BP230 is intercellular, there is some controversy regarding whether anti-BP230 IgG alone can cause BP.[22,23,24] The majority of BP patients possess IgGs that specifically target the non-collagenous region, 16A (NC16A) of the BP180 ectodomain, and there is evidence that these patients also develop tissue resident memory B cells.[23,24]
Although autoimmunity is the underlying cause of BP, it is still unknown how the loss of self-tolerance actually occurs. Some believe that autoimmunity results from a deficient interaction between Treg and autoreactive Th cells. In such a scenario, presentation of the BP180 and BP230 autoantigens by an MHC class II-expressing APC to self-reactive T cells will lead to T-cell activation, release of pro-inflammatory cytokines, up-regulation of co-stimulatory molecules on the surface of the APC, and activation of autoreactive BP180-specific B cells.[25] Since characteristically BP involves recruitment of eosinophils and neutrophils, IL-17-secreting Th17 cells appear to be involved, which is similar to that seen in other inflammatory conditions such as psoriasis, systemic lupus erythematous, and rheumatoid arthritis. Another hypothesized mechanism involves the binding of B-cell receptors and toll-like receptors (TLR) to self-antigens that possess an endogenous TLR ligand,[25,26] resulting in B-cell activation and autoantibody secretion.
Recently, the existence of anti-BP180 IgE in BP patients has been reported, which is thought to synergize with autoreactive IgG1 and IgG4 to elicit complement activation, degranulation of mast cells, and release of leukotrienes, platelet-activating factor, TNF, and other cytokines. Recruited neutrophils and eosinophils also participate in the pathophysiology by releasing proteolytic enzymes which disrupt adhesion molecules, thereby contributing to the subepidermal blister formation.[22] IgE involvement in BP is further supported by the finding that when a BP180 ectodomain (LABD97)-specific IgE-secreting hybridoma was injected into a SCID mouse, a basement membrane blistering disease developed that was histologically identical to BP.[27]
Genetic Predisposition
The role of nature versus nurture is an ongoing debate in autoimmunity, as many diseases are multifactorial with both genetics and environment factors playing a role in their pathogenesis. The autoimmune blistering diseases PV and BP are no exception. Almost all patients with autoimmunity will first have a genetic predisposition for the disease such as disease-associated HLA gene or a mutation in a key regulatory protein of the immune system. But not all patients with a genetic predisposition will develop autoimmunity, suggesting that genetics alone is insufficient to cause autoimmunity. This argument has been supported by reports of PV occurring in only one monozygotic twin, and accounts of only two of three siblings with identical haplotypes developing disease.[28,29]
PV has the strongest association with HLA class II genes, particularly DR4.[30] Molecular gene subtyping showed the prevalence of either DRB1*0402 or DQB1*0503 in over 95% of patients.[31] These two alleles are often associated with one another and their prevalence is increased in the Ashkenazi Jewish population. Other PV-associated DRB1 polymorphisms include DRB1*1401, DRB1*1404, and DRB1*1454. Further molecular analysis of DQB1 polymorphisms showed an increase in DQB1*0302 (in association with DRB1*04). The PV-associated MHC class II molecules have similarities within their peptide-binding groove, which supports the notion that PV is initiated by the activation of MHC class II-restricted desmoglein-specific CD4+ T cells. These activated T cells then drive B-cell activity and autoantibody production.[31,32,33] In addition to MHC class II associations, some research has revealed the involvement of HLA class IA molecules (A3, A10, A26) and HLA class IB molecules (B15, B35, B38, B44, and B60).[31] The latter group has been linked with an increased frequency of a deletion variant of HLA-G. HLA-E*0103 has also been reported to be associated with PV in some patients.[31]
Patients with pemphigoid tend to have an increased frequency of the allele DQB1*0301,[34] and some studies have demonstrated that BP180-specific Th1 and Th2 cells are restricted to HLA-DQB1*0301.[35] Even DQB1*0301 individuals without clinical evidence of BP have been found to have BP180-specific T cells, predominantly of the Th1 phenotype.[35] Some studies have found that patients with BP have identical amino acid residues at positions 71–77 of their DQB1 gene.[36]
Nurture: Impact of Environmental Factors on Pemphigus and Pemphigoid
As mentioned above, genetic predisposition alone is often not sufficient to promote development of pemphigus or pemphigoid. Environmental factors play a role in triggering autoimmunity in individuals with an underlying genetic predisposition. Medications, viral infections, allergens, radiation therapy, diet and emotional stress have all been reported to induce immune dysregulation, which may lead to a flare of pemphigus or pemphigoid in susceptible individuals.[9]
With respect to medications, thiol-containing drugs such as penicillin, cephalosporins and captopril can bind to cysteine molecules in keratinocytes and interrupt cell–cell cohesion, causing a non-antibody-mediated pemphigus-like disease.[37] These medications may also bind to desmogleins and other adhesion proteins causing them to adapt an altered conformation, which in turn precipitates the immune system to recognize them as “foreign,” eliciting production of DSG-specific antibodies.[38,39] In drug-induced BP, thiols may behave as haptens by binding to proteins within the basement membrane, stimulating production of anti-basement membrane antibodies.[40,41,42,43] Foods high in thiols (garlic, leeks, onions) and polyphenols (black pepper, red chili pepper, cherry, red wine) are also thought to induce PV by a similar mechanism. BP, in comparison, has not been reported to have any dietary associations.[4]
Like thiols, it has been reported that phenols may induce autoimmune blistering diseases.[9] After phenol exposure, keratinocytes release IL-1 and tumor necrosis factor (TNF), which may promote keratinocyte acantholysis.[39] Common phenol-containing medications associated with development of PV include aspirin, rifampin, levodopa, heroin and some cephalosporins (which can also contain thiol groups). Aspirin can also induce BP via a mechanism similar to that of the thiol-based drugs. Specifically, it might bind to basement membrane proteins and act as a hapten to elicit an antigenic response.[44] BP induced by TNF-blocking agents such as etanercept and adalimumab has also been reported but the exact mechanism for this effect remains unclear.[45,46,47]
Various non-thiol or phenol-containing medications such as angiotensin-converting enzyme (ACE) inhibitors (other than captopril), vaccines, interferons, and nonsteroidal anti-inflammatory drugs (NSAIDs) have all been associated with the development of antibody-negative drug-induced PV.[9] ACE inhibitors may promote acantholysis by inhibiting tissue transglutaminase, an enzyme involved in keratinocyte aggregation.[48] In the case of vaccines and interferons, activation of the immune system is thought to result in autoantibody production and release of plasminogen activators by keratinocytes leading to acantholysis.[9]
Viral infections, especially herpesvirus, cause inflammation that is marked by up-regulation of IFN-γ and the B-cell-supporting cytokines IL-4 and IL-10, along with other pro-inflammatory cytokines and antibodies, which have the potential to cause epithelial damage. In a study of 20 pemphigus patients, DNA sequencing identified the presence of herpes simplex virus, Epstein Barr virus, and human herpes virus-6 in the patient's lymphocytes and skin lesions.[49] However, it is unclear if these viral infections are responsible for the induction of PV through a molecular mimicry mechanism or if their presence in the skin lesions is an incidental finding.[50,51] Viral infections may likewise play a role in pemphigoid, particularly the herpesviruses.[52] Although molecular mimicry is a conceivable mechanism linking infection with the onset of PV, the process of autoimmunity is often multifactorial, and as a result, such observations are difficult to interpret.[53]
Traumatic injuries to the skin caused by excessive sun exposure or radiation therapy may lead to blister and bullae formation at the sites of injury due to localized exposure of self-antigens. The up-regulated local inflammatory response at sites of cutaneous injury may further contribute to an environment favoring autoantibody production.[54,55] Radiation therapy has been noted to commonly cause BP in female breast cancer patients, by potentially modifying the basal membrane to expose BP antigens, resulting in antibody formation, complement activation, and a proinflammatory state.[56,57] Muramatsu and colleagues postulated that UVB exposure directly causes conformational changes in BP antigens.[58] During the radiation-induced inflammatory event, leukocytes migrate to the lamina lucida, which may cause proteolytic cleavage and eventually, subepidermal bullae. Inhibition of Tregs may also contribute to the formation of an unregulated proinflammatory state and antibody production in both diseases.[59,60]
Pesticide exposure has also been associated with PV onset. In an international survey of 126 pemphigus patients, 23.9% of them reported prior exposure to pesticides.[61] The mechanism behind this association may be related to the activity of organophosphates on acetylcholinesterase. Organophosphate pesticides act by inhibiting the enzyme, acetylcholinesterase, which increases the concentration of acetylcholine.[62] With prolonged exposure, the high concentration of the neurotransmitter will naturally induce a down-regulation of nicotinic and muscarinic receptors.[63] As mentioned earlier, AChRs in the skin help regulate expression of desmogleins and thus a down-regulation of AChRs can lead to disruption of cell–cell adhesion.[64] This mechanism also helps explain why cigarette smoking has been reported to have an inverse relationship with PV occurrence. Nicotine in tobacco activates nicotinic cholinergic receptors and thus may increase desmoglein expression.[65]
Conclusions
A number of potentially causative factors, both genetic and environmental, have been proposed to influence the development of the autoimmune blistering diseases. The variety of inciting events and agents identified in the pathogenesis of these diseases is continually expanding. A detailed clinical history, especially with regard to medications, is important not only for identifying cases of drug-induced pemphigus but also for expanding our knowledge of environmental factors that may trigger the disease.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
What is new?
The pathophysiology of autoimmune bullous diseases may actually be multifactorial where both, genetics and various different environmental factors, may be contributing to disease onset.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
References
- 1.Baum S, Sakka N, Artsi O, Trau H, Barzilai A. Diagnosis and classification of autoimmune blistering diseases. Autoimmun Rev. 2014;13:482–9. doi: 10.1016/j.autrev.2014.01.047. [DOI] [PubMed] [Google Scholar]
- 2.Maverakis E, Goodarzi H, Wehrli LN, Ono Y, Garcia MS. The etiology of paraneoplastic autoimmunity. Clin Rev Allergy Immunol. 2012;42:135–44. doi: 10.1007/s12016-010-8248-5. [DOI] [PubMed] [Google Scholar]
- 3.Kershenovich R, Hodak E, Mimouni D3. Diagnosis and classification of pemphigus and bullous pemphigoid. Autoimmun Rev. 2014;13:477–81. doi: 10.1016/j.autrev.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 4.Lo Schiavo A, Ruocco E, Brancaccio G, Caccavale S, Ruocco V, Wolf R. Bullous pemphigoid: Etiology, pathogenesis, and inducing factors: Facts and controversies. Clin Dermatol. 2013;31:391–9. doi: 10.1016/j.clindermatol.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis) Clin Dermatol. 2011;29:432–6. doi: 10.1016/j.clindermatol.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 6.Asboe-Hansen G. Blister-spread induced by finger-pressure, a diagnostic sign in pemphigus. J Invest Dermatol. 1960;34:5–9. doi: 10.1038/jid.1960.3. [DOI] [PubMed] [Google Scholar]
- 7.Grando SA, Grando AA, Glukhenky BT, Doguzov V, Nguyen VT, Holubar K. History and clinical significance of mechanical symptoms in blistering dermatoses: A reappraisal. J Am Acad Dermatol. 2003;48:86–92. doi: 10.1067/mjd.2003.39. [DOI] [PubMed] [Google Scholar]
- 8.Bean SF, Alt TH, Katz HI. Oral pemphigus and bullous pemphigold. Immunofluorescent studies of two patients. JAMA. 1971;216:673–4. [PubMed] [Google Scholar]
- 9.Ruocco V, Ruocco E, Lo Schiavo A, Brunetti G, Guerrera LP, Wolf R. Pemphigus: Etiology, pathogenesis, and inducing or triggering factors: Facts and controversies. Clin Dermatol. 2013;31:374–81. doi: 10.1016/j.clindermatol.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Shephard M, Hodgson T, Hegarty AM. Vesiculobullous disorders affecting the oral cavity. Br J Hosp Med (Lond) 2014;75:502–8. doi: 10.12968/hmed.2014.75.9.502. [DOI] [PubMed] [Google Scholar]
- 11.Tosti A, André M, Murrell DF. Nail involvement in autoimmune bullous disorders. Dermatol Clin. 2011;29:511–3. doi: 10.1016/j.det.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 12.Robati RM, Rahmati-Roodsari M, Dabir-Moghaddam P, Farnaghi A, Mahboobi-rad F, Rahimi H, et al. Mucosal manifestations of pemphigus vulgaris in ear, nose, and throat; before and after treatment. J Am Acad Dermatol. 2012;67:e249–52. doi: 10.1016/j.jaad.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 13.Tichy M, Urbanek J, Sternbersky J, Ditrichova D, Hercogova J. Life-threatening course of pemphigus vulgaris complicated by sepsis caused by azathioprine-induced bone marrow suppression, successfully managed with combination therapy. Dermatol Ther. 2014;27:183–6. doi: 10.1111/dth.12114. [DOI] [PubMed] [Google Scholar]
- 14.Jung M, Kippes W, Messer G, Zillikens D, Rzany B. Increased risk of bullous pemphigoid in male and very old patients: A population-based study on incidence. J Am Acad Dermatol. 1999;41(2 Pt 1):266–8. doi: 10.1016/s0190-9622(99)70061-7. [DOI] [PubMed] [Google Scholar]
- 15.Beutner EH, Jordon RE. Demonstration of Skin Antibodies in Sera of Pemphigus Vulgaris Patients by Indirect Immunofluorescent Staining. Proc Soc Exp Biol Med. 1964;117:505–10. doi: 10.3181/00379727-117-29622. [DOI] [PubMed] [Google Scholar]
- 16.Hu CH, Michel B, Schiltz JR. Epidermal acantholysis induced in vitro by pemphigus autoantibody. An ultrastructural study. Am J Pathol. 1978;90:345–62. [PMC free article] [PubMed] [Google Scholar]
- 17.Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA. Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med. 1982;306:1189–96. doi: 10.1056/NEJM198205203062001. [DOI] [PubMed] [Google Scholar]
- 18.Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999;103:461–8. doi: 10.1172/JCI5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hertl M, Zillikens D. Clinical and molecular characterization of autoimmune bullous diseases. J Invest Dermatol. 2008;128:E19–21. doi: 10.1038/skinbio.2008.3. [DOI] [PubMed] [Google Scholar]
- 20.Oliveira ME, Culton DA, Prisayanh P, Qaqish BF, Diaz LA. E-cadherin autoantibody profile in patients with pemphigus vulgaris. Br J Dermatol. 2013;169:812–8. doi: 10.1111/bjd.12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen VT, Ndoye A, Grando SA. Novel human alpha9 acetylcholine receptor regulating keratinocyte adhesion is targeted by Pemphigus vulgaris autoimmunity. Am J Pathol. 2000;157:1377–91. doi: 10.1016/s0002-9440(10)64651-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasperkiewicz M, Zillikens D. The pathophysiology of bullous pemphigoid. Clin Rev Allergy Immunol. 2007;33:67–77. doi: 10.1007/s12016-007-0030-y. [DOI] [PubMed] [Google Scholar]
- 23.Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: Pathogenesis, diagnosis, and treatment. Autoimmunity. 2012;45:55–70. doi: 10.3109/08916934.2011.606447. [DOI] [PubMed] [Google Scholar]
- 24.Di Zenzo G, Marazza G, Borradori L. Borradori, Bullous pemphigoid: Physiopathology, clinical features and management. Adv Dermatol. 2007;23:257–88. doi: 10.1016/j.yadr.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 25.Grando SA. Pemphigus autoimmunity: Hypotheses and realities. Autoimmunity. 2012;45:7–35. doi: 10.3109/08916934.2011.606444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shlomchik MJ. Activating systemic autoimmunity: B's, T's, and tolls. Curr Opin Immunol. 2009;21:626–33. doi: 10.1016/j.coi.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zone JJ, Taylor T, Hull C, Schmidt L, Meyer L. IgE basement membrane zone antibodies induce eosinophil infiltration and histological blisters in engrafted human skin on SCID mice. J Invest Dermatol. 2007;127:1167–74. doi: 10.1038/sj.jid.5700681. [DOI] [PubMed] [Google Scholar]
- 28.Ruocco V, Peluso G, Pisani M. Pemphigus vulgaris in only one of two monozygotic twins. J Am Acad Dermatol. 1985;12:587–9. doi: 10.1016/s0190-9622(85)80104-3. [DOI] [PubMed] [Google Scholar]
- 29.Revenga-Arranz F, Martínez-Lasso J, Vanaclocha-Sebastián F. Pemphigus vulgaris in two MHC-haploidentical brothers. Dermatology. 1996;193:71–2. doi: 10.1159/000246212. [DOI] [PubMed] [Google Scholar]
- 30.Lombardi ML, Mercuro O, Ruocco V, Lo Schiavo A, Lombari V, Guerrera V, et al. Common human leukocyte antigen alleles in pemphigus vulgaris and pemphigus foliaceus Italian patients. J Invest Dermatol. 1999;113:107–10. doi: 10.1046/j.1523-1747.1999.00626.x. [DOI] [PubMed] [Google Scholar]
- 31.Sinha AA. The genetics of pemphigus. Dermatol Clin. 2011;29:381–91. doi: 10.1016/j.det.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 32.Sercarz EE, Maverakis E. Mhc-guided processing: Binding of large antigen fragments. Nat Rev Immunol. 2003;3:621–9. doi: 10.1038/nri1149. [DOI] [PubMed] [Google Scholar]
- 33.Maverakis E. Sercarzian immunology-In memoriam. Eli E. Sercarz, 1934-2009. Cell Immunol. 2012;273:99–108. doi: 10.1016/j.cellimm.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 34.Hertl M, Eming R, Veldman C. T cell control in autoimmune bullous skin disorders. J Clin Invest. 2006;116:1159–66. doi: 10.1172/JCI28547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haase C, Büdinger L, Borradori L, Yee C, Merk HF, Yancey K, et al. Detection of IgG autoantibodies in the sera of patients with bullous and gestational pemphigoid: ELISA studies utilizing a baculovirus-encoded form of bullous pemphigoid antigen 2. J Invest Dermatol. 1998;110:282–6. doi: 10.1038/sj.jid.5602955. [DOI] [PubMed] [Google Scholar]
- 36.Delgado JC, Turbay D, Yunis EJ, Yunis JJ, Morton ED, Bhol K, et al. A common major histocompatibility complex class II allele HLA-DQB1* 0301 is present in clinical variants of pemphigoid. Proc Natl Acad Sci U S A. 1996;93:8569–71. doi: 10.1073/pnas.93.16.8569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruocco V, Pisani M, de Angelis E, Lombardi ML. Biochemical acantholysis provoked by thiol drugs. Arch Dermatol. 1990;126:965–6. [PubMed] [Google Scholar]
- 38.Ruocco V, De Angelis E, Lombardi ML. Drug-induced pemphigus. II. Pathomechanisms and experimental investigations. Clin Dermatol. 1993;11:507–13. doi: 10.1016/0738-081x(93)90158-9. [DOI] [PubMed] [Google Scholar]
- 39.Ruocco V, Sacerdoti G. Pemphigus and bullous pemphigoid due to drugs. Int J Dermatol. 1991;30:307–12. doi: 10.1111/j.1365-4362.1991.tb03867.x. [DOI] [PubMed] [Google Scholar]
- 40.Walsh SR, Hogg D, Mydlarski PR. Mydlarski, Bullous pemphigoid: From bench to bedside. Drugs. 2005;65:905–26. doi: 10.2165/00003495-200565070-00002. [DOI] [PubMed] [Google Scholar]
- 41.Mallet L, Cooper JW, Thomas J. Bullous pemphigoid associated with captopril. DICP. 1989;23:63. doi: 10.1177/106002808902300115. [DOI] [PubMed] [Google Scholar]
- 42.Wozniak K, Kowalewski C, Hashimoto T, Ishii N, Glinska-Wielochowska M, Schwartz RA. Penicillin-induced anti-p200 pemphigoid: An unusual morphology. Acta Derm Venereol. 2006;86:443–6. doi: 10.2340/00015555-0117. [DOI] [PubMed] [Google Scholar]
- 43.Hodak E, Ben-Shetrit A, Ingber A, Sandbank M. Bullous pemphigoid-an adverse effect of ampicillin. Clin Exp Dermatol. 1990;15:50–2. doi: 10.1111/j.1365-2230.1990.tb02020.x. [DOI] [PubMed] [Google Scholar]
- 44.Durdu M, Baba M, Seçkin D. A case of bullous pemphigoid induced by aspirin. J Am Acad Dermatol. 2011;65:443–4. doi: 10.1016/j.jaad.2010.02.032. [DOI] [PubMed] [Google Scholar]
- 45.Kluk J, Goulding JM, Bhat J, Finch TM. Drug-induced bullous pemphigoid: Cases triggered by intravenous iodine and etanercept. Clin Exp Dermatol. 2011;36:871–3. doi: 10.1111/j.1365-2230.2011.04102.x. [DOI] [PubMed] [Google Scholar]
- 46.Bordignon M, Belloni-Fortina A, Pigozzi B, Tarantello M, Alaibac M. Bullous pemphigoid during long-term TNF-alpha blocker therapy. Dermatology. 2009;219:357–8. doi: 10.1159/000243805. [DOI] [PubMed] [Google Scholar]
- 47.Toosi S, Bystryn JC. Does adalimumab cause bullous pemphigoid? Clin Exp Dermatol. 2010;35:795. doi: 10.1111/j.1365-2230.2010.03813.x. [DOI] [PubMed] [Google Scholar]
- 48.Ruocco V, Satriano RA, Guerrera V. “Two-step” pemphigus induction by ACE-inhibitors. Int J Dermatol. 1992;31:33–6. doi: 10.1111/j.1365-4362.1992.tb03517.x. [DOI] [PubMed] [Google Scholar]
- 49.Tufano MA, Baroni A, Buommino E, Ruocco E, Lombardi ML, Ruocco V. Detection of herpesvirus DNA in peripheral blood mononuclear cells and skin lesions of patients with pemphigus by polymerase chain reaction. Br J Dermatol. 1999;141:1033–9. doi: 10.1046/j.1365-2133.1999.03201.x. [DOI] [PubMed] [Google Scholar]
- 50.Maverakis E, van den Elzen P, Sercarz EE. Sercarz, Self-reactive T cells and degeneracy of T cell recognition: Evolving concepts-from sequence homology to shape mimicry and TCR flexibility. J Autoimmun. 2001;16:201–9. doi: 10.1006/jaut.2000.0493. [DOI] [PubMed] [Google Scholar]
- 51.Sercarz EE, Maverakis E. Recognition and function in a degenerate immune system. Mol Immunol. 2004;40(14-15):1003–8. doi: 10.1016/j.molimm.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 52.Drago F, Nozza P, Casazza S, Brusati C, Bandelloni R, Rebora A. Human herpesviruses in bullous pemphigoid lesions. Br J Dermatol. 2005;152:375–6. doi: 10.1111/j.1365-2133.2005.06331.x. [DOI] [PubMed] [Google Scholar]
- 53.Maverakis E, Menezes JS, Ametani A, Han M, Stevens DB, He Y, et al. Molecular mimics can induce a nonautoaggressive repertoire that preempts induction of autoimmunity. Proc Natl Acad Sci U S A. 2010;107:2550–5. doi: 10.1073/pnas.0914508107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robbins AC, Lazarova Z, Janson MM, Fairley JA. Pemphigus vulgaris presenting in a radiation portal. J Am Acad Dermatol. 2007;56(5 Suppl):S82–5. doi: 10.1016/j.jaad.2006.10.956. [DOI] [PubMed] [Google Scholar]
- 55.Tan SR, McDermott MR, Castillo CJ, Sauder DN. Pemphigus vulgaris induced by electrical injury. Cutis. 2006;77:161–5. [PubMed] [Google Scholar]
- 56.Isohashi F, Konishi K, Umegaki N, Tanei T, Koizumi M, Yoshioka Y. A case of bullous pemphigoid exacerbated by irradiation after breast conservative radiotherapy. Jpn J Clin Oncol. 2011;41:811–3. doi: 10.1093/jjco/hyr049. [DOI] [PubMed] [Google Scholar]
- 57.Maverakis E, Miyamura Y, Bowen MP, Correa G, Ono Y, Goodarzi H. Light, including ultraviolet. J Autoimmun. 2010;34:J247–57. doi: 10.1016/j.jaut.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muramatsu T, Yamashina Y, Shirai T, Ohnishi T. UVB irradiation reduces the expression of pemphigoid antigens in organ-cultured normal human skin. Arch Dermatol Res. 1994;286:142–4. doi: 10.1007/BF00374209. [DOI] [PubMed] [Google Scholar]
- 59.Srifi N, Benomar S, Zaghba N, Qasmi S, Senouci K, Elgueddari B, et al. Generalized bullous pemphigoid induced by radiotherapy. Ann Dermatol Venereol. 2011;138:311–4. doi: 10.1016/j.annder.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 60.Mul VE, Verschueren TA, van Geest AJ, Baumert BG. Bullous pemphigoid (BP) induced by radiotherapy. Radiother Oncol. 2007;82:105. doi: 10.1016/j.radonc.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 61.Brenner S, Tur E, Shapiro J, Ruocco V, D’Avino M, Ruocco E, Tsankov N, et al. Pemphigus vulgaris: Environmental factors. Occupational, behavioral, medical, and qualitative food frequency questionnaire. Int J Dermatol. 2001;40:562–9. doi: 10.1046/j.1365-4362.2001.01266.x. [DOI] [PubMed] [Google Scholar]
- 62.Ray DE, Richards PG. The potential for toxic effects of chronic, low-dose exposure to organophosphates. Toxicol Lett. 2001;120:343–51. doi: 10.1016/s0378-4274(01)00266-1. [DOI] [PubMed] [Google Scholar]
- 63.Costa LG. Interactions of neurotoxicants with neurotransmitter systems. Toxicology. 1988;49:359–66. doi: 10.1016/0300-483x(88)90019-4. [DOI] [PubMed] [Google Scholar]
- 64.Fania L, Zampetti A, Guerriero G, Feliciani C. Alteration of cholinergic system in keratinocytes cells produces acantholysis: A possible use of cholinergic drugs in pemphigus vulgaris. Antiinflamm Antiallergy Agents Med Chem. 2012;11:238–42. doi: 10.2174/1871523011202030238. [DOI] [PubMed] [Google Scholar]
- 65.Grando SA, Dahl MV. Nicotine and pemphigus. Arch Dermatol. 2000;136:1269. doi: 10.1001/archderm.136.10.1269. [DOI] [PubMed] [Google Scholar]