

Abstract
From plant research to biomedicine, proteome analysis plays a critical role in many areas of biological inquiry. Steady improvement in mass spectrometer (MS) technology has transformed the speed and depth of proteome analysis. Proteomes of simple organisms can now be sequenced to near completion in just over an hour. Comparable coverage of mammalian proteomes, however, still requires hours or even days of analysis. Here we ask why current technology fails to achieve comprehensive and rapid analysis of the more complex mammalian proteomes. We propose that further advancements in MS technology alone are unlikely to solve this problem and suggest that concomitant improvements in peptide separation technology will be critical.
Advancing myriad applications – from basic biological studies to precision medicine – will require technologies for rapid and comprehensive proteome profiling. Dominating the field, liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) delivers increasingly promising results (Riley et al, 2016). The key tenets of this strategy, however, have been in place for nearly twenty years (Pandey & Mann, 2000). Proteins are digested into smaller, more manageable peptides, which are separated using chromatography and then presented to the MS. The eluting peptide ions are converted from the condensed phase to the gas phase using electrospray ionization. Following injection into the MS, their overall mass-to-charge ratio is recorded. In order of abundance, each unique peptide species is isolated and subjected to fragmentation, either by collisions, ion/ion reactions, or photons. The resulting product ions are then measured to create a tandem MS (MS/MS) scan. These MS/MS scans are then searched against a database of in silico generated spectra, scored, and used to generate a list of confidently identified peptides and consequently proteins.
Twenty years ago, state-of-the-art MS systems could acquire up to one MS/MS scan per second. In this scenario, the rate-limiting step was the collection of MS/MS spectra (Link et al. 1999). That is, a 60-minute LC-MS/MS experiment could not possibility produce more than 3,600 peptide identifications, as only this many MS/MS spectra could be collected during that period (i.e., 60 MS/MS per minute × 60 minutes). To achieve greater proteomic analysis depth, the field moved toward multi-dimensional separations like MudPit (Link et al, 1999; Washburn et al, 2001). Fractionation of a proteome digest into 80 samples, each of which could be subjected to a 60-minute LC-MS/MS experiment, allowed one to collect considerably more MS/MS spectra (nearly 300,000 in this example). Proteome depth increased as a consequence – but so too did the duration of the proteomics experiment.
Mass spectrometer performance has since steadily improved in nearly every category, including mass accuracy, mass resolving power, scan speed, and sensitivity. With these improvements and other advances, a modern mass spectrometer now sequences up to 19 peptides per second, and near complete coverage of the whole yeast proteome can be achieved in just over one hour of analysis (Hebert et al, 2014). Deep sequencing of more complex mammalian proteomes, however, still requires two-dimensional chromatography, mandating considerably extended analysis time (Richards et al, 2015). Because these experiments take several hours or even days to conduct, systems-wide studies of mammalian proteomes are not routine. We reason that to permit the practical analysis of hundreds or even thousands of mammalian proteomes we must substantially reduce the amount of analysis time required. This rather obvious goal will not raise controversy. However, there is no general consensus on which technological innovations should be pursued to achieve it. Here we present a largely overlooked but, in our opinion, primary obstacle to achieving rapid, whole mammalian proteome analysis— peptide separations.
Single-shot proteomics
As MS systems have evolved to enable faster scanning, those seeking to further increase throughput have pursued single-shot proteomics, where the complex mixture of peptides is separated in only one dimension and analyzed via MS. Figure 1A highlights some prominent single-shot studies. First, we note the chromatographic format and performance is primarily uniform across these works; however, the MS scan rate, defined as the number of MS/MS scans taken per second, is steadily increasing over time, as new technology is released and adopted. What is not as obvious from the figure is an exciting concomitant trend of increasing proteomic sampling depth over this same period. Specifically, the 2011 work of Thakur et al detected just under 3,000 yeast proteins from an eight-hour analysis. The very next year that same lab reduced the separation duration to four hours and detected nearly 4,000 yeast proteins (Nagaraj et al, 2012). This remarkable improvement was primarily afforded by the adoption of a new MS platform offering an increase in MS/MS acquisition rate, high resolution, and accurate mass. Two years later, by using an even faster scanning Orbitrap hybrid (20 Hz), our laboratory further increased throughput for yeast proteomics by identifying 4,000 proteins in just over one hour of analysis (Hebert et al, 2014). A similar upward trend is observed among the studies aimed at human proteomes, although even the latest works report identification of only about half of the expressed proteins (Pirmoradian et al, 2013; Scheltema et al, 2014).
Figure 1. The dynamic relationship between MS/MS scan rate and peptide separations in single-shot proteomics.
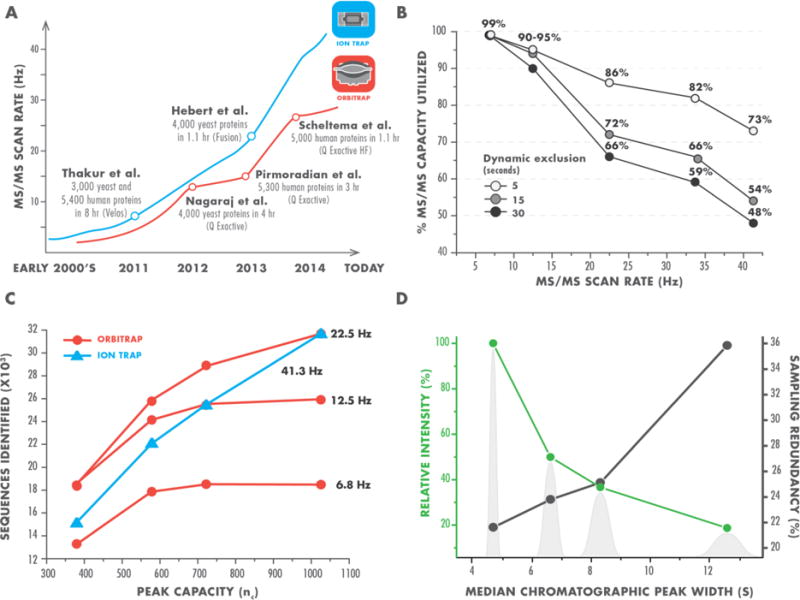
Note that plot axes of panel B & C do not show zero values.
A. Historic improvements in MS/MS scan rate of commonly used mass analyzers (ion trap in blue and Orbitrap in red). Landmark single-shot proteomics studies associated with the emergence of new MS instrumentation are highlighted.
B. Utilization of MS/MS capacity at different scan rates. We calculated a maximum number of MS/MS scans a mass spectrometer could theoretically obtain at specific scan rate and compared it to the actual number of MS/MS scans the instrument acquired during analysis of a complex peptide mixture over the same time window. Dynamic exclusions is a parameter of MS instruments that restricts repeated selection of a precursor for a tandem scan over a defined time period. Data obtained with dynamic exclusion settings of 5 (white), 15 (light gray), and 30 (black) seconds are shown. At the fastest scan rates, even with extremely short dynamic exclusion, the mass spectrometer does not acquire the maximum theoretical number of MS/MS scans.
C. Synergistic effect of MS/MS scan rate and peak capacity on peptide detection. We separated a mixture of tryptic yeast peptides over a 90 minute gradient using various chromatographic columns and analyzed it at different MS/MS scan rates. At slower scan rates considerable improvements in the quality of peptide separation only marginally increase the number of identified peptides. However, at scan rates >20 Hz the number of detected peptides exhibits pronounced dependency on the quality of separation, more than doubling at 41.3 Hz with the increased peak capacity (in blue).
D. Two example benefits of higher quality peptide separations. More effective chromatographic columns produce peaks that are narrower and consequently have higher relative intensity (green). Narrower elution profiles also reduce redundant sampling of precursors (black), as measured by sampling redundancy - the ratio of unique peptide identifications to peptide spectrum matches. Background gray peak shapes are for illustration purposes and based on experimental measurements of median peak widths (in seconds), obtained using four different capillary columns.
Complementing the increase in scan rate of all instruments are improvements in analysis sensitivity, sample preparation techniques, and data processing. The relative contributions of these factors must not be overlooked, but they are more difficult to assess and likely secondary to the astounding progression in MS performance.
Based on this trajectory, we fully anticipated that further gains in single-shot proteomics would be readily achieved by the next-generation MS systems featuring faster data acquisition, unprecedented sensitivity, and ion transfer efficiency. Upon acquirement and use of the newest Orbitrap hybrid instrument (Fusion Lumos®), capable of collecting high quality MS/MS spectra at a rate of 40 Hz, we were dismayed to discover that our prediction did not prove true. Single-shot analysis of both human and yeast proteomes were minimally improved—far less than one would expect upon doubling MS/MS acquisition rate.
Have we sampled everything?
To understand why this latest technology did not continue the trend towards performance improvement, we took a closer look at the literature and our data. Specifically, we wondered how many eluting peptides were presented to the MS system, what percentage was sampled, and whether all of the system’s sequencing capacity was utilized. In 2011 Mann and co-workers pondered these questions and reported that a standard LC-MS analysis produced ~100,000 peptide-like features and estimated that an MS/MS rate of 25 Hz was needed for their complete sampling (Michalski et al, 2011). To evaluate this theoretical prediction, we measured the percentage of MS/MS scanning capacity utilized at various scan rates (Figure 1B). We reasoned that if insufficient precursors were available for sampling, then MS/MS scans would not be triggered and the cumulative number of acquired MS/MS scans would be lower than the maximum value achievable at a given scan rate. Overall we observed a strong correlation with reduced utilization of MS/MS capacity as scan rate increased, even at a very low dynamic exclusion setting of five seconds, which permits redundant sampling of many peptide-like precursors. These data both confirm the Mann prediction and explain our unexpected results. We therefore conclude that to exploit the capabilities of new MS technology and, ultimately, to continue improving single-shot proteomic analysis, we must increase the number of detectable peptide features. We believe that the most promising way to achieve this aim is through enhancing the quality of peptide separations.
Separation quality matters
To survey the quality of single-shot proteomic separations in the published literature we obtained raw data and calculated chromatographic peak capacity (nc; Neue, 2005) for many published proteomic experiments, including those shown in Figure 1A (refer to Supplementary Table S1 for the complete list). These experiments had nc values ranging from 350 to over 1,000 (Supplementary Figure S1), with the highest values drawn from the milestone works displayed in Figure 1A. By varying column length and particle diameter, we fabricated capillary columns that performed across this range (Supplementary Table S2). Note our highest performing chromatography setup delivered separations with nc of ~1,025, commensurate with the highest nc observed in the milestone single-shot articles (~1,015).
Using these columns, we analyzed a whole yeast proteome digest to characterize the dynamic interplay between MS/MS scan rate and separation quality. Figure 1C shows that as the scan rate increases so does sensitivity of the analysis to separation quality. For example, at lower MS/MS acquisition rates, high-quality separations provide little benefit. When collecting MS/MS spectra at <10 Hz, a cutting-edge rate just a few years ago, the significance of chromatographic quality is greatly diminished with doubling of peak capacity resulting in detection of virtually no additional peptides. The most recent quadrupole-ion trap-Orbitrap hybrid is, however, capable of >40 Hz MS/MS acquisition, and operation in this mode exhibits the strongest performance correlation with nc. The number of identified sequences increases by greater than two fold exclusively due to chromatographic improvements.
Several benefits of the higher quality peptide separations help put these observations in perspective (Figure 1D). First, the narrower peptide elution profiles serve to greatly reduce redundant sampling. Second, as the chromatographic peak area remains relatively constant, a reduced peak width is accompanied with increased ion intensity, boosting sensitivity of the analysis. Further, because ionization suppression diminishes ion flux, reducing the number of co-eluting peptides increases dynamic range and lowers the occurrence of problematic “chimeric” spectra (Michalski et al, 2011). Lastly, poor separations adversely impact the ability of the mass spectrometer to identify precursor monoisotopic peaks, obstructing their selection for a tandem scan (McAlister et al, 2014).
In search of better separations
In 2004 the field of liquid chromatography underwent a revolution: commercial ultra-high pressure pumps (UPLC, up to 10,000 PSI) became available and afforded use of fully porous sub-two micron separation particles (Jorgenson, 2010; Gritti & Guiochon, 2012). Now over a decade old, this technology still remains at the core of proteomic analyses and was used by all aforementioned landmark single-shot proteome studies. Meanwhile, as we have discussed, considerable gains in MS acquisition rate have caught up with and outpaced the relatively constant front half of the LC-MS/MS experiment, so that only recently has the problematic mismatch between the MS and chromatographic performance fully emerged.
The general importance of separations is recognized by other researchers. Many improved and alternative approaches have been introduced in the last years, including capillary electrophoresis (Fonslow & Yates, 2009), superficially porous and sub-micron particles (Blue & Jorgenson, 2015), use of slower flow rates (Köcher et al, 2014), HPLC systems with higher-pressure capabilities (i.e., > 20,000 psi) (Shen et al, 2005), and monolithic columns (Iwasaki et al, 2012; Horie et al, 2014; Zhang et al, 2015), among others. Due to a variety of drawbacks, such as robustness, load capacity, ease of implementation, and availability, these technologies have not been broadly adopted by the proteomics community or provided superior performance for single-shot analysis. We and others view the use of longer columns (Liu et al, 2007; Fountain et al, 2009; Pirmoradian et al, 2013), packed with smaller particles, as the most direct path towards improved peptide separations. This, however, demands LC systems and fittings that can withstand very high pressures (Grinias et al, 2016), currently unavailable to non-specialists.
Understanding the interplay between separations and MS sampling rate impacts proteomics at every level. When collecting data on older, slower systems, one need not be overly concerned with separation quality. However, those acquiring faster systems will best leverage their investment by careful implementation of state-of-the-art separations. In our opinion, quality of chromatographic separations is currently the key but underappreciated bottleneck limiting the speed and depth of single-shot proteomic analysis. Our hope is that researchers across multiple disciplines—i.e., separation scientists and proteomic practitioners—will become aware of this roadblock and employ their respective expertise to enable rapid and comprehensive analysis of human proteome at last.
Supplementary Material
Acknowledgments
We thank Graeme McAlister and Mike Westphall for helpful discussions. We gratefully acknowledge support from NIGMS grants GM118110 and GM108538 (awarded to J.J.C.).
References
- Blue LE, Jorgenson JW. 1.1μm Superficially porous particles for liquid chromatography. Part II: Column packing and chromatographic performance. Journal of Chromatography A. 2015;1380:71–80. doi: 10.1016/j.chroma.2014.12.055. [DOI] [PubMed] [Google Scholar]
- Fountain KJ, et al. Effects of extra-column band spreading, liquid chromatography system operating pressure, and column temperature on the performance of sub-2-μm porous particles. Journal of Chromatography A. 2009;1216(32):5979–5988. doi: 10.1016/j.chroma.2009.06.044. [DOI] [PubMed] [Google Scholar]
- Grinias KM, et al. Development of a 45 kpsi Ultrahigh Pressure Liquid Chromatography Instrument for Gradient Separations of Peptides Using Long Microcapillary Columns and Sub-2μm Particles. Journal of Chromatography A. 2016 doi: 10.1016/j.chroma.2016.09.053. CHROMA-357925, article in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritti F, Guiochon G. The current revolution in column technology: How it began, where is it going? Journal of Chromatography A. 2012;1228:2–19. doi: 10.1016/j.chroma.2011.07.014. [DOI] [PubMed] [Google Scholar]
- Hebert AS, et al. The one hour yeast proteome. Molecular & Cellular Proteomics: MCP. 2014;13(1):339–47. doi: 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie K, et al. Hydrophilic Interaction Chromatography Using a Metre-Scale Monolithic Silica Capillary Column for Proteomics LC-MS. Analytical Chemistry. 2014;86:3817–3824. doi: 10.1021/ac4038625. [DOI] [PubMed] [Google Scholar]
- Iwasaki M, et al. Human proteome analysis by using reversed phase monolithic silica capillary columns with enhanced sensitivity. Journal of Chromatography A. 2012;1228:292–297. doi: 10.1016/j.chroma.2011.10.059. [DOI] [PubMed] [Google Scholar]
- Jorgenson JW. Capillary liquid chromatography at ultrahigh pressures. Annual Rev Anal Chem. 2010;3:129–150. doi: 10.1146/annurev.anchem.1.031207.113014. [DOI] [PubMed] [Google Scholar]
- Köcher T, et al. Development and performance evaluation of an ultra-low flow nano liquid chromatography-tandem mass spectrometry set-up. Proteomics. 2014;14:1999–2007. doi: 10.1002/pmic.201300418. [DOI] [PubMed] [Google Scholar]
- Link AJ, et al. Direct analysis of protein complexes using mass spectrometry. Nature Biotechnology. 1999;17(7):676–682. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- Liu H, et al. Effects of column length, particle size, gradient length and flow rate on peak capacity of nano-scale liquid chromatography for peptide separations. Journal of Chromatography A. 2007;1147(1):30–36. doi: 10.1016/j.chroma.2007.02.016. [DOI] [PubMed] [Google Scholar]
- McAlister G, et al. Exploring the uncharted depths of the complex human proteome using the Orbitrap Fusion mass spectrometer. 62nd Annual Meeting of the American Society of Mass Spectrometry and Allied Topics; Baltimore, MD. 2014. [Google Scholar]
- Michalski A, Cox J, Mann M. More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. Journal of Proteome Research. 2011;10(4):1785–1793. doi: 10.1021/pr101060v. [DOI] [PubMed] [Google Scholar]
- Nagaraj N, et al. System-wide Perturbation Analysis with Nearly Complete Coverage of the Yeast Proteome by Single-shot Ultra HPLC Runs on a Bench Top Orbitrap. Molecular & Cellular Proteomics. 2012;11:13722–13722. doi: 10.1074/mcp.M111.013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neue UD. Theory of peak capacity in gradient elution. Journal of Chromatography A. 2005;1079(1–2):153–161. doi: 10.1016/j.chroma.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Pandey A, Mann M. Proteomics to study genes and genomes. Nature. 2000;405(6788):837–846. doi: 10.1038/35015709. [DOI] [PubMed] [Google Scholar]
- Pirmoradian M, et al. Rapid and deep human proteome analysis by single-dimension shotgun proteomics. Molecular & Cellular Proteomics: MCP. 2013;12(11):3330–8. doi: 10.1074/mcp.O113.028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards AL, Merrill AE, Coon JJ. Proteome sequencing goes deep. Current Opinion in Chemical Biology. 2015;24:11–17. doi: 10.1016/j.cbpa.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley NM, Hebert AS, Coon JJ. Proteomics Moves into the Fast Lane. Cell Systems. 2016;2(3):142–143. doi: 10.1016/j.cels.2016.03.002. [DOI] [PubMed] [Google Scholar]
- Scheltema RA, et al. The Q Exactive HF, a Benchtop Mass Spectrometer with a Pre-filter, High Performance Quadrupole and an Ultra-High Field Orbitrap Analyzer. Molecular & Cellular Proteomics: MCP. 2014:3698–3708. doi: 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur SS, et al. Deep and highly sensitive proteome coverage by LC-MS/MS without prefractionation. Molecular & cellular proteomics: MCP. 2011;10(8):M110.003699. doi: 10.1074/mcp.M110.003699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MP, Wolters D, Yates JR. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nature Biotechnology. 2001;19(3):242–7. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- Zhang Z, et al. Integrated strong cation-exchange hybrid monolith coupled with capillary zone electrophoresis and simultaneous dynamic pH junction for large-volume proteomic analysis by mass spectrometry. Talanta. 2015;138:117–122. doi: 10.1016/j.talanta.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.