

Abstract
Metabolomics is undergoing tremendous growth and is being employed to solve a diversity of biological problems from environmental issues to the identification of biomarkers for human diseases. Nuclear magnetic resonance (NMR) and mass spectrometry (MS) are the analytical tools that are routinely, but separately, used to obtain metabolomics data sets due to their versatility, accessibility, and unique strengths. NMR requires minimal sample handling without the need for chromatography, is easily quantitative, and provides multiple means of metabolite identification, but is limited to detecting the most abundant metabolites (≥ 1 μM). Conversely, mass spectrometry has the ability to measure metabolites at very low concentrations (femtomolar to attomolar) and has a higher resolution (∼103-104) and dynamic range (∼103-104), but quantitation is a challenge and sample complexity may limit metabolite detection because of ion suppression. Consequently, liquid chromatography (LC) or gas chromatography (GC) is commonly employed in conjunction with MS, but this may lead to other sources of error. As a result, NMR and mass spectrometry are highly complementary, and combining the two techniques is likely to improve the overall quality of a study and enhance the coverage of the metabolome. While the majority of metabolomic studies use a single analytical source, there is a growing appreciation of the inherent value of combining NMR and MS for metabolomics. An overview of the current state of utilizing both NMR and MS for metabolomics will be presented.
Graphical abstract

1. Introduction
Metabolic profiling has existed for hundreds of years and was a common practice in ancient Chinese medicine [1]. At the turn of the century the term “metabolomics”1 was coined and ignited renewed interest in the field [2-4], which has led to rapid developments and advancements in metabolomics procedures and technologies [2-4]. In effect, metabolic profiling has transitioned from the use of smell and/or taste to the application of advanced spectrometric methods such as nuclear magnetic resonance (NMR) or mass spectrometry (MS) [5-8]. Consequently, metabolomics has become a cornerstone of systems biology [9, 10] since it enables unique insights that cannot be obtained from other “omics” methods [10-13]. Metabolomics directly relates a measurable chemical response to a biological event, thereby linking the genotype and phenotype of an organism [14]. Thus, the method is increasingly being used in nutrition science [15], environmental science [16], biomedical research [17] and precision medicine [18]. While interest has expanded exponentially in recent years (Fig. 1), the field of metabolomics is still in an early stage of growth and development, and faces many technical challenges [5, 7, 8, 19, 20]. Before it can become a routine tool for the life sciences, many of these current limitations need to be overcome. Similar to other “omics” methods, a single metabolomics study may contain an abundance of data making it difficult to correctly extract the relevant biological information [20-22]. Furthermore, the specific analytical platform chosen for metabolomics will uniquely limit the analysis because of spectral and chemical ambiguity, which, in turn, will yield an imperfect biological picture and incomplete coverage of the metabolome [23]. Sensitivity, resolution, dynamic range, ambiguous assignments, limits of detection, and various other issues are universal problems encountered by all analytical methods; there simply is not a single analytical source that will detect the entire metabolome.
Figure 1.

The chart plots the number of MS metabolomics (green), NMR metabolomics (blue), and combined NMR and MS metabolomics (red) studies published per year from 2001 to 2016. These data were obtained from a keyword search of all documents on PubMed using the key words “MS and metabolomics”, “NMR and metabolomics”, or “MS and NMR and metabolomics”.
Over three decades ago, the fundamental importance and benefit of employing several analytical methods to improve the quality of data analysis and advance compound elucidation was highly touted, which, at the time, produced an alphabet soup of hyphenated methods [24]. Although successful hyphenated techniques rarely expanded beyond combining a form of separation such as liquid chromatograph (LC), gas chromatograph (GC), or capillary electrophoresis (CE) with an analysis tool such as NMR or MS, the need for multiple analytical platforms for complex analysis was readily apparent [25]. This is especially true in the field of metabolomics [23]. Nevertheless, to date, the majority of metabolomics data sets have been acquired using only MS or NMR despite their fundamental complementarity [23]. Fortunately, there is a growing appreciation that combining MS and NMR data greatly improves the coverage of the metabolome and enhances the accuracy of metabolite identification [26-31]. For example, it is well-established that the structure elucidation of an unknown natural product is greatly facilitated by combining the unique information from NMR (e.g., chemical shifts, coupling constants, NOEs, spin systems, etc.) with MS (e.g., exact mass [molecular formula], molecular fragments) [32, 33].
A metabolomics study is defined by both the capabilities and limitations of the analytical method employed, and may be significantly hindered if only NMR or MS is used for the analysis. The important advantages of using NMR for metabolomics include a relatively high-throughput, nondestructive data acquisition, minimal sample handling, simple methods for metabolite quantitation, and redundant spectral information to improve the accuracy of metabolite identification [34-36]. Conversely, NMR is limited to detecting only the most abundant metabolites (≥ 1 μM), while MS has a much higher sensitivity and readily measures concentrations in the femtomolar to attomolar range. MS also boasts higher resolution (∼103-104) and dynamic range (∼103-104). Conversely, MS only detects metabolites that readily ionize, as a result of which upwards of 40% of chemical libraries are not observable by MS [37, 38]. Similarly, ion suppression is a well-known problem in MS, which further reduces the detection of ions of interest due to matrix effects [39]. Simply, the presence of other compounds in the sample or containments from external sources (e.g., plastics, buffers, solid phase, etc.) is a ‘matrix’ that reduces the ability of a specific compound to be ionized through a variety of proposed mechanisms [39]. For example, an ion suppressing agent from the matrix may simply out-compete the compound for available ions. Thus, ion suppression due to matrix effects is a significant concern for metabolomics given the complexity and heterogeneity of metabolomics samples. In effect, the presence of one metabolite may lead to other metabolites being undetected [40]. Thus, MS-based metabolomics typically involves chromatography [41, 42] to reduce peak overlap arising from the relatively narrow nominal mass and mass defect distribution of the metabolome [43]. But the use of chromatography may induce biologically irrelevant variations in the metabolome resulting from non-uniform metabolite derivatization, variable metabolite column recovery, metabolite decomposition during derivatization or separation, metabolite ion-suppression due to co-eluting matrix compounds, or misalignment of replicate retention times [44-48]. Simply, NMR and MS have distinct strengths and weaknesses and both uniquely benefit metabolomics.
In recent years, technical advancements in high field magnets, pulse sequences, and cryoprobe technology have led to some significant improvements in the sensitivity and resolution of NMR experiments [23]. This, in turn, has improved the quality of metabolomics data and has contributed to the observed increase in NMR-based metabolomics studies. Despite these advancements, one-dimensional (1D) 1H NMR spectra are still hindered by a significant amount of peak overlap, even for high field magnets, because of the limited chemical shift dispersion of metabolites [8, 49, 50]. Thus, these recent improvements in NMR still do not match the sensitivity and resolution advantages of MS [23, 51]. Analogous advancements in MS have also occurred, which have led to similar improvements in the quality of metabolomics data sets. High resolution spectrometry, accurate mass, and isotope labeling methods means that virtually every metabolite that is not an isomer produces a unique m/z and should be readily identified [52]. In fact, a majority of metabolomics studies routinely rely on MS (Fig. 1). Additionally, improvements in ionization methods and separation techniques have helped reduce matrix effects due to co-eluting compounds common in complex mixtures, but ion suppression remains a primary issue for MS metabolomics [11, 53]. Electrospray ionization mass spectrometry (ESI-MS) sensitivity is directly dependent on a compound's pKa and hydrophobicity, which can be negatively impacted by the heterogeneous composition of a metabolomics sample [54]. For example, if two compounds with different pKa values are present in the same electrospray droplet, only the compound with the lower pKa value may be protonated and detected. Thus, given the various limitations of both NMR and MS, no single analytical platform has the ability to analyze the entire metabolome alone [11, 55]. Instead, while some overlap exists, NMR and MS observe a highly complementary set of metabolites [56, 57]. Thus, applying both NMR and MS during a metabolomics study will allow for a more comprehensive coverage of the metabolome [57]. The combination of NMR and MS data will also improve the identification of unknown analytes and will increase the accuracy of identifying known metabolites [28, 31, 51, 57-60]. This occurs because metabolite identification will be based on distinct and confirmatory evidence.
A grand challenge in the field of metabolomics is the rapid and accurate identification of metabolites from the variety of complex biological samples routinely analyzed (e.g., tissues, serum and cell extracts) [61, 62]. As a consequence, nearly all published metabolomic studies contain at least one misidentified or unidentified metabolite [62]. This is an unfortunate and unavoidable outcome of our limited knowledge of the metabolome (the exact composition is currently unknown), the severe limitations in the software and databases available for metabolite identification, and the routine reliance on a single analytical method. Despite the routine application of NMR and MS to elucidate the structures of natural products for drug discovery [32, 33], most metabolic studies still rely on only NMR or MS spectral data for metabolite identification based on database searches [63]. Unfortunately, metabolic databases only contain, at most, a few thousand reference NMR or MS spectra of known metabolites. There is also minimal, if any, coordination between the various metabolomics databases. This leads to a significant amount of redundancy between databases, and also results in unique data being present in individual databases, requiring an investigator to search across multiple databases. Also, many databases are cumbersome and utilize simple search algorithms, which only allow for a single spectral category per query [63]. Finally, most metabolomics databases are limited to either NMR or MS reference data. Again, this is a result of the fact that the metabolomics field has evolved to support only a single analytical source. Thus, it is not possible to easily search metabolomics databases for simultaneous matches against NMR and MS spectral data. As a result, metabolomics will greatly benefit from the creation of a unified NMR and MS database and the merging of data. This is especially true given the growing recognition of the value of combining NMR and MS for metabolite assignments [28, 29, 51, 57, 58].
Multivariate statistical methods are normally applied to metabolomics data sets in order to simplify and expedite the data analysis [12, 64]. While chemometric techniques regularly streamline the process of data analysis, these advanced multivariate statistical techniques are routinely used incorrectly, lack proper validation, and have, unfortunately, lead to a proliferation of erroneous data in the scientific literature [65, 66]. This problem becomes compounded when an investigator is combining multiple analytical sources. In most cases, the samples, data, and analysis are done separately. The NMR and MS data sets are not integrated into a single chemometrics model because, until recently, the field lacked software capable of handling data from multiple analytical sources [57, 67, 68]. Thus, any observed changes in the NMR and MS spectra are not statistically correlated and, importantly, the metabolites and pathways separately identified by NMR and MS may not be biologically related. Only a minimal number of studies have been reported that actually utilized both NMR and MS in a single chemometrics model [69-72].
As outlined above, NMR and MS are routinely, but separately, used for metabolomics studies despite their complementary strengths. Nevertheless, there is a growing recognition of the benefits of combining the two techniques for metabolomics as evident by the increasing number of published manuscripts that utilized both NMR and MS (Fig. 1). NMR and MS have been combined in a number of ways that includes: 1) physically interfacing NMR and MS hardware, 2) chemical modification of samples by derivatization of metabolites with compounds that display unique characteristics for MS and NMR detection, 3) stable isotope tracing by isotopically labeling metabolites, 4) using combined cheminformatics techniques on MS and NMR data sets for an accurate and rapid analysis, or 5) unique data handling and data mining techniques which correlate trends in both data sets by using multivariate statistical-based methods [29, 30, 58]. Herein, we provide illustrative examples for each of these methods and present an overview of the various benefits derived from combining NMR and MS for the analysis of metabolomics samples.
2. Brief Overview of Metabolomics
2.1. Current State of NMR and MS Metabolomics
Both NMR- and MS-based metabolomics have been extensively reviewed in the recent scientific literature [3, 11, 13, 23, 73-80]. Similarly, a number of reviews describing the proper handling, preparation, and extraction of metabolic samples from urine, serum, cell cultures, tissue cultures, and a variety of other biological sources are readily available [36, 81-85]. So, only a very brief overview of the general procedure for metabolic profiling will be presented here, primarily to highlight important differences when NMR and MS are combined for a metabolomics study (Fig. 2) [86]. In principle, metabolomics is a relatively straightforward method: the metabolome is harvested or extracted from two or more groups (e.g., healthy vs. diseased), and an analytical technique (e.g., NMR or MS) is used to acquire a spectral profile of each metabolic sample. Then, multivariate statistical techniques (e.g., principal component analysis [PCA], orthogonal projections to latent structures [OPLS], etc.) are used to determine whether the metabolomes differ and, if they do, to identify the spectral features (i.e., the metabolites) defining group separation. A typical outcome of a PCA or OPLS model is a scores plot, where each NMR or MS spectrum has been reduced to a single point in PC-space. The relative clustering of the spectra in the scores plot identifies group membership. Similarly, a backscaled loadings plot is often generated from the OPLS model that is a pseudospectrum of the original NMR or MS data where the relative intensity and direction of the spectral peaks indicates the contribution and correlation of the peak to the group separation. Alternatively, a heatmap can be generated that compares all of the relative metabolite concentration changes per replicate. Following hierarchal clustering, if all or most of the replicates from the same group cluster together, then the heatmap identifies key metabolite concentration changes that define each group.
Figure 2.
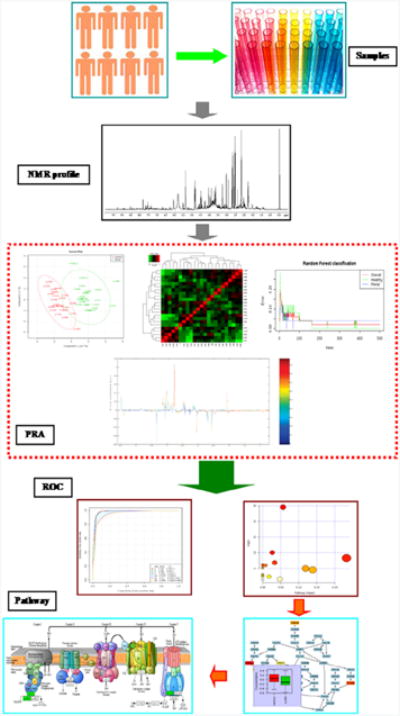
Illustration of the metabolomics work flow that combines advanced NMR spectroscopy techniques with multivariate statistics. Samples are collected in a uniform way to minimize variability and are analyzed by NMR profile to collect data on all metabolites potentially present in the sample. Pattern recognition approaches (PRA) include principal component analysis, partial least squares discriminant analysis, orthogonal projections to latent structures, heat map, support vector machines, and random forests method, and other modes aiming to highlight underlying trends and visualization tools such as contribution. Trend and box plots are used to further evaluate these. Receiver operating characteristic (ROC) curves are generally considered the method of choice for evaluating the performance of potential biomarkers. The markers are eventually placed in a metabolic pathway to provide insight on the biochemical phenomena. (Samples) Multiple replicate samples are obtained from cells, tissues, or biofluids (e.g., urine, plasma, etc.) for each group (e.g., healthy vs. disease). (NMR profile) A 1D 1H NMR spectrum is collected for each metabolomics sample, which becomes the data set. A mass spectrum can be used instead or in addition to the NMR spectrum. (PRA) Illustrations of typical multivariate statistical analysis of the metabolomics NMR data set. Clockwise from upper-left, a scores plot from a PCA model indicating two distinct clusters or groups are present in the data set. A heatmap shows the clustering of metabolite changes (x-axis) relative to each group replicate (y-axis). The relative color of each bin corresponds to the metabolite concentration difference between replicates. The heatmap identifies which set of metabolites are uniquely changing between each group. The result of a random forest classification is summarized by plotting the out-of-bag error rate or misclassifications versus the number of trees. The results indicate that the healthy and disease groups can be separated with an error rate of < 5% with a nominal number of trees. A back-scaled loadings plot, which is a pseudo 1D 1H NMR spectrum, is generated from an OPLS model. The relative intensity and direction of each peak conveys the importance and correlation of the NMR peak to the observed group separation in the corresponding scores plot. (ROC) Illustrations of the validation and the further analysis of the key metabolites identified from the multivariate statistical analysis that define the group separation. On the left is a ROC curve, which plots sensitivity (true positive rate) versus 1-specificity (false positive rate). The area under the curve is a measure of the accuracy of the model to correctly predict group membership. On the right is the pathway topology analysis produced by MetPA (http://metpa.metabolomics.ca) from the list of metabolites identified by the multivariate statistical analysis. MetPA assists in identifying the set of important metabolic pathways associated with the phenotype. (Pathways) Illustration of metabolic networks or signaling pathways identified from the observed metabolomic changes between the groups. Reproduced with permission from reference [86].
Since the numbers of replicates for a metabolomics data set are typically far fewer then the number of variables, overfitting the data, especially for supervised techniques like PLS or OPLS, is a serious concern [65, 66, 87]. In fact, a PLS/OPLS model can produce the appearance of a clear group separation even for noise or completely random data [88]. As a result, model validation is a critical final step before any biological interpretation of the model is reliable. CV-ANOVA [89], response permutation tests [87], and receiver operating characteristic (ROC) curves [90] are routinely used to provide standard p values and assess model validity. With a validated model, the observed metabolite changes can then be used to generate a metabolic network and identify the important metabolic pathways associated with the phenotype [91].
Unlike the proteome and genome, the metabolome is relatively unstable and is easily perturbed by the handling and processing of the sample. For example, metabolites have different enzyme turnover rates and different temperature stabilities [92-94]. Thus, a key concern is avoiding any changes in the metabolome that may be induced from sample preparation. Therefore, fast and uniform sample preparation protocols, rapid quenching of enzymatic activity, keeping samples cold throughout, and randomizing samples through the entire sample handling and data collection procedure are all important details that require optimization for a successful metabolomics study. Since biological samples are the materials that are analyzed, there is inherently a large natural variance to the data. Thus, to obtain statistical significance a maximal number of replicates, within practical constraints, is highly desirable [36]. Similarly, the data need to be properly normalized, scaled and aligned to account for both biological and instrument variance, and to remove bias due to the large range of metabolite concentrations. Again, various normalization, scaling and alignment algorithms are available and their utility for metabolomics has been previously reviewed [95-97]. Finally, the raw NMR or mass spectral data need to be properly processed before a reliable chemometrics model can be generated. For NMR, processing includes Fourier transformation, phasing, baseline correction, apodization, zero filling and chemical shift referencing. Similarly, MS requires centroiding, de-noising, de-isotoping, deconvolution and peak alignment. It cannot be overstated that the resulting chemometric models are incredibly sensitive to all aspects of the sample handling and data processing. Thus, changing processing details, such as a different apodization function or deconvolution algorithm will likely lead to a different chemometrics model and, potentially, a different biological interpretation. Consequently, great care must be taken in optimizing data processing protocols in order to avoid unintended biases in the data analysis. The complexity of the situation expands exponentially if two or more analytical sources are combined to generate a single chemometrics model simply because of the greater number of possible processing protocol combinations. Thus, while metabolomics is conceptually quite simple and straightforward to conduct, in practice it is difficult to do correctly because of the complex number of steps and choices available, with multiple sources of error encountered at each step of the process.
2.2 Analyte Compatibility for Analysis by NMR and MS Metabolomics
NMR and MS are the most popular analytical methods used for metabolomics because of the wide array of chemical species both techniques can probe and are likely to encounter in a complex and heterogeneous metabolite mixture. MS is generally coupled with a separation technique, with the majority of metabolomics studies utilizing LC-ESI-MS [11, 64, 98]. The coupling of MS with LC or GC is critical for reducing ion suppression, spectral complexity and spectral overlap. To further reduce ion suppression, samples are typically acidified and salt concentrations are reduced, usually with a desalting column [99]. Although NMR is occasionally paired with a separation method, the hardware for an online LC-NMR system can be somewhat crude or cumbersome, and the process of manually collecting fractions is extremely time-consuming. Moreover, a hybrid LC-NMR system does not provide a substantial benefit since individual metabolites are readily detectable by NMR without chromatographic separation [100].
Besides different chromatographic needs, MS and NMR require different sample conditions. NMR samples are commonly prepared in a buffered deuterated solvent to provide a lock signal and maintain a constant pH of 7.4 in order to minimize chemical shift deviations and replicate physiological conditions [9, 99]. In general, detecting a metabolite by NMR is not dependent on the sample condition. However, NMR is a relatively insensitive technique and requires maximizing metabolite concentrations to ensure detection. On average, NMR metabolomics requires a minimal sample volume of approximately 30 to 600 μL, with metabolite concentrations of approximately μM to mM. Because of the higher sensitivity of MS, the concentration requirements can be significantly reduced to as low as nM to pM, with sample volumes on the order of a few microliters. While MS is intrinsically more sensitive than NMR, detecting a specific metabolite does require the metabolite to be efficiently ionized. Different groups of metabolites will preferentially ionize under drastically different experimental conditions, which depend on mode of polarization, pH and the ionization efficiency of the specific metabolite. Thus, the detectability of a metabolite may vary dramatically depending on sample conditions. For example, 0.1% formic acid or acetic acid is commonly added to MS samples to enhance protonation and increase sensitivity. Conversely, a deuterated solvent used in NMR could complicate the analysis of an MS spectrum because exchangeable hydrogens would still be observed, but ambiguous mass shifts may occur. The exchangeable hydrogens are likely not observed in the NMR spectrum, potentially eliminating the ability to detect a given metabolite. Moreover, minimal sample handling is desired to reduce experimental errors. Thus, these necessary differences in sample conditions present a practical challenge, namely how can a single metabolomics sample be analyzed by two distinct analytical platforms?
To address the need for distinct NMR and MS sample preparations, Beltran et al. examined 12 extraction protocols and evaluated various solvents and temperatures to identify conditions compatible for use by both LC-ESI-MS and NMR [101]. A single metabolite extract from a liver tissue was used for the NMR and MS metabolomics studies. The combination of solvent conditions used for metabolite extraction ranged in polarity and consisted of a 2 mL combination of either: (i) 1:1 methanol/H2O, (ii) 1:1 acetonitrile/H2O, (iii) 7:2:1 methanol/chloroform/H2O, or (iv) 7:2:1 acetonitrile/chloroform/H2O. The extractions were also conducted at three different temperatures: −20 °C, 25 °C, or 60 °C. After each extraction, the metabolomics samples were dried and then dissolved in deuterated acetonitrile/H2O (2:8) before acquiring a 1D 1H NMR spectrum. A resulting PCA model comparing the range of solvent conditions revealed a large variance between the different extraction protocols. Interestingly, extraction temperature had a minor impact on group separation. Methanol/H2O was the most efficient extraction method and the least influenced by temperature. In contrast, acetonitrile/chloroform/H2O was the least efficient extraction method.
A very intriguing aspect of this study was the LC-MS analysis of metabolites in deuterated solvent. Pure metabolomics standards or liver metabolite extracts were prepared in either H2O or D2O (Fig. 3) [101]. In this manner, the impact of solvent exchange on NMR and MS spectral data was evaluated. D2O did not affect measurements of metabolite concentrations. In addition, D2O did not induce a mass shift or perturb the isotopic distribution. Metabolites with readily exchangeable hydrogens were also investigated and, surprisingly, no difference in the spectra due to D2O was observed. The lack of deuterium exchange in metabolites was attributed to the mobile phase; since the deuterated metabolomics sample was injected into an un-deuterated mobile phase, deuterated metabolites were readily back-exchanged to a protonated species.
Figure 3.

A) Scatter plot representing the area of each feature from the XCMS matrix of LC/MS data of liver samples reconstituted in H2O and D2O. A correlation coefficient (R2) of 0.997 indicates a high linear regression, which demonstrates that differences between the number and abundance of features detected in liver extracts reconstituted in H2O and D2O are insignificant. (B) Mass spectra of phenylalanine, tryptophan, and LysoPC (16:0) reconstituted in D2O (top) and H2O (bottom). Labile hydrogens are marked in red. Mass spectra show that the isotopic distributions of the compounds are not altered by D2O, indicating either slow H/D exchange of acidic protons in solution or fast back-exchange of labile deuterons in aqueous LC/MS buffers due to a total solvent accessibility of small molecule structures. Reproduced with permission from reference [101].
A similar study conducted by Marshall et al. identified a relatively straightforward and simple protocol to simultaneously investigate by both NMR and MS a metabolomics sample extracted from a cell lysate [57]. Human dopaminergic neuroblastoma cells were lysed by incubating with cold methanol (-80° C) for 15 minutes. The methanol was also used as the first metabolite extraction step. After removing the methanol supernatant, the cell debris were then washed with an 80%/20% methanol/ddH2O water mixture, followed by a second wash with 100% ddH2O. The supernatants from the three extractions were combined and then split into two portions: 1.8 mL for NMR and 200 μL for MS. The MS sample was then diluted tenfold with a 49.75:49.75:0.5 H2O/methanol/formic acid mixture containing 20 μM reserpine as an internal mass reference. The NMR sample was dried using a combination of RotoSpeed vacuum and lyophilization, and then resuspended in a 50 mM deuterated phosphate buffer (pH 7.2) with TMSP-d4 as a chemical shift reference. The separate NMR and MS metabolomics samples were then used to obtain a 1D 1H NMR spectrum and a direct-injection ESI-MS mass spectrum.
3. Approaches for combining NMR and MS for metabolomics
3.1. Interfacing NMR and MS Hardware
Natural product chemistry is a staple of drug discovery and the pharmaceutical industry, and has an obvious synergy with metabolomics since both fields are focused on the characterization of chemical entities extracted from a complex biological mixture [102, 103]. NMR and MS spectral data have been routinely used in tandem to characterize natural products and their secondary metabolites isolated from crude complex samples [102]. Since metabolites and natural products are essentially the same, these well-established protocols are an invaluable resource for characterizing the vast number of unknown metabolites in the metabolome. Natural product research is commonly guided by and focused on identification of biologically active fractions from complex mixtures, which are then analyzed by a sequential series of purification steps and MS profiling until a biologically active compound is isolated for structural determination by NMR and MS methods [104, 105]. In general, only a few nanograms or micrograms of a natural product are available for structural characterization. Therefore, sensitivity and efficiency are key concerns. In the case of NMR, the development of microcoils, microflow and cyroprobes have significantly improved the dynamic range and sensitivity of NMR and have thus greatly benefited the structural characterization of sample-limited natural products and metabolites [106-108]. Mass spectrometry has made similar advancements that have also benefited natural products chemistry and metabolomics by developing high-resolution mass spectrometers and tandem MS-MS methods to improve the accuracy and ease of structure elucidation [109-113].
The off-line combination of MS and NMR has clearly advanced natural products and metabolomics research, but an innovative on-line combination of the two instruments has the potential to significantly increase throughput, efficiency and sensitivity [59, 60]. HPLC fractions are routinely collected and separately analyzed by NMR and MS, but an on-line analysis allows for the simultaneous detection of metabolite data that are then easily cross-correlated between the two platforms while also reducing, or potentially eliminating, sample handling errors. This is especially true in regard to the manual manipulation of severely sample-limited natural products or metabolites. Additionally, the duplicate isolation of redundant or unwanted compounds is a large issue in the natural products field. It is simply undesirable to expend valuable resources to re-discover known natural products. Dereplication is the commonly employed process of examining active fractions to recognize and eliminate previously identified natural products to avoid unnecessary compound isolation [114]. Dereplication routinely occurs by rapidly comparing experimental NMR and/or MS spectral data against spectral libraries of known natural products [60]. Employing an LC-NMR-MS instrument for dereplication would significantly reduce the time requirements, lower the amount of sample required and increase identification accuracy [60].
Previously, physically siting an instrument in close proximity to an NMR magnet was difficult, as it caused severe spectral distortions, required long sample transfer lines, and demanded significant laboratory space [59]. Fortunately, interfacing LC, NMR, and MS instrumentation has been greatly simplified by the advent of shielded magnets [115-117]. The stray fields have been significantly reduced for shielded magnets, and, in some cases, the 5 gauss line lies within the magnet's dewar. This permits a close arrangement of instruments without incurring any magnetic interference or disturbing field homogeneity. The availability of bench-top mass spectrometers has also aided in the coupling of multiple platforms. An MS instrument with a small footprint and superior resolution makes it possible to site an LC-NMR-MS spectrometer in a smaller laboratory space then previously possible. Moreover, the proximity of the instruments simplifies sample transfer between them. For example, an NMR and mass spectrometer can be interfaced to LC by using a post-column splitter or the Bruker NMR-Mass Spectrometry Interface (BNMI). The post-column splitter simultaneously directs LC flow to both the NMR and MS spectrometers in a 9 to 1 ratio, respectively. BNMI is a valve-switching interface that is a computer-controlled splitter and double dilutor, which allows for proton–deuterium exchange to occur for NMR, and the optimal selection of solvent polarity for MS. Additionally, BNMI has a loop storage mode, which allows for a portion of the LC eluent to be temporarily stored in a sample loop while waiting for the NMR instrument to be available. A storage mode allows for NMR acquisition times that are significantly longer than the LC experiment to maximize NMR signal-to-noise.
Recently, Lin et al. described an LC-NMR-MS approach, which significantly reduced sample size, increased concentration sensitivity by 10-fold, and enhanced mass sensitivity by 1000-fold [59, 98]. The method significantly outperformed traditional offline LC-NMR-MS by the addition of a nanoSplitter for nanospray LC-MS and a microcoil flow probe for NMR analysis. In addition, the analysis of the LC flow normally occurs with a 9:1 split between NMR and MS, respectively. But, instead, the improved performance required an LC flow that was split 98% to NMR and 2% to MS [59]. The LC-NMR-MS system exhibited a 93% sample recovery and yielded a limit of detection as low as 50-ng (RSD 1.17%) for NMR. Similarly, four natural products (ambiguine A, I, E, and hapalindole H) from a cyanobacterial extract were readily detected by LC-MS at a concentration of only 30 μg/mL. Thus, interfacing LC with parallel NMR and MS analysis may greatly benefit metabolomics by improving throughput, by increasing sensitivity and coverage, and by reducing the amount of sample required for a study.
3.2. Chemical Modification
The diverse and complementary set of information obtained from NMR and mass spectral data is the major reason for combining the two methods for metabolomics. However, combining two distinct data sets also increases the complexity of the analysis. The NMR and MS results obtained from a single heterogeneous sample are not easily correlated [30]. For example, it is not a trivial task to definitively assign an NMR chemical shift and a MS m/z value to a specific metabolite. Simply, there is no information in the NMR or MS spectrum that indicates that the chemical shift and m/z value are from the same metabolite. The lack of a correlation may also appear to produce contradictory results. For example, the presence of a metabolite may only be supported by one method or the two methods may appear to predict vastly different concentrations. As a result, the complexity of analyzing NMR and MS spectra may hinder an interpretation or yield erroneous results. Consequently, a novel twist on the well-established method of chemical derivation has been employed to overcome this lack of a correlation between NMR and MS spectral data [30]. A chemical agent is introduced into a complex mixture that reacts with a specific chemical moiety or functional group within the metabolite. The resulting chemical modification is then visible by both NMR and MS.
Chemical modification has frequently been used for LC-MS and GC-MS [118-121]. Traditionally, compound derivatization has been used to separate stereoisomers or to improve ionization efficiency in LC-MS. Chemical modification for GC-MS is generally used to promote the volatility of compounds [122]. Fariba et al. exploited the benefits of chemical modification by using 15N-cholamine, a so-called “smart tag”, to specifically label carboxyl-containing metabolites [30, 123]. A permanent charge and an 15N isotope label are incorporated into the metabolites through a condensation reaction [30]. Each chemically labeled metabolite would then have a unique set of 1H and 15N chemical shifts that can be easily detected with a 2D 1H-15N HSQC NMR spectrum. Only metabolites labeled with 15N-cholamine are observed in the NMR spectrum because of the low natural abundance of 15N (0.37%). Similarly, the incorporation of a permanent charge into the metabolite significantly improved its ionization efficiency. Sensitivity enhancements exceeding three orders of magnitude were observed for labeled metabolites, which could be easily detected in the positive mode compared to unlabeled metabolites in the negative mode. Thus, the increased MS sensitivity and the unique 1H-15N chemical shifts establish a correlation between the NMR and MS data for a labeled metabolite. The smart-tag approach was successfully demonstrated with a standard mixture of 48 metabolites that each contain a carboxyl-group and are prevalent in human biofluids (Fig. 4) [30]. The experiment was successfully repeated with human serum and urine samples. The chemical shift assignments obtained from the standard metabolite mixture was used to quickly and accurately assign the 48 metabolites in the biofluids. Thus, 15N-cholamine was shown to be useful for correlating data across multiple analytical platforms through chemical derivatization, and as a valuable approach to aid in the metabolite profiling of complex biological mixtures.
Figure 4.
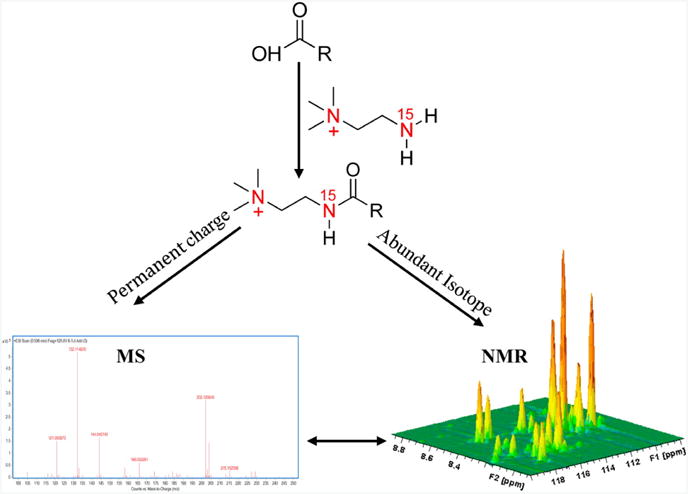
Schematic figure illustrating the “smart isotope tag” approach used to detect the same metabolites using NMR and MS with high sensitivity. Tagging carboxyl-containing metabolites with 15N-cholamine enables their enhanced detection by both NMR and MS. Reproduced with permission from reference [30].
3.3. Stable Isotope Tracers
The ability to simultaneously measure hundreds of chemical species from a complex biological sample is the primary reason NMR and MS are routinely used in metabolomics, but the overwhelming amount of data can also confound the analysis. Consequently, the majority of metabolomics studies are end-point or single time-point measurements despite the obvious advantage of flux analysis. Fortunately, MS and NMR can de-convolute these complex data sets based on their unique ability to identify and distinguish between different isotopes. Thus, NMR and MS metabolomics analysis can be greatly simplified by using stable isotope tracers. Stable Isotope-Resolved Metabolomics (SIRM) has been used to monitor metabolite flux, to reveal novel metabolic networks, and has recently been shown to correlate data across MS and NMR platforms [124-130]. In this manner, SIRM may overcome common limitations encountered with steady-state metabolite profiling. SIRM also enables the combined use of NMR and MS for metabolomics, but, more importantly, the SIRM approach greatly benefits from combining both NMR and MS [126-129, 131].
SIRM uses stable isotope-enriched nutrients containing 13C-carbons or 15N-nitrogens, which are provided to an organism as the primary source of carbon or nitrogen. The 13C-carbons or 15N-nitrogens are distributed throughout the organism's metabolome based on active metabolic processes. The resulting 13C- or 15N-labeled metabolites are readily detected by NMR and MS. In this manner, SIRM reveals the unique flow of 13C-carbons or 15N-nitrogens through metabolic pathways in order to highlight systematic changes due to environmental stress, genetic mutations, a disease state, a drug treatment, or numerous other factors. Importantly, SIRM allows for monitoring the flow of position-specific carbons or nitrogens between metabolites (i.e., isotopomer or isotopologue probing). In essence, it is possible to decipher the chemical source of each carbon or nitrogen in a given metabolite (Fig. 5) [132]. Moreover, the analysis can be fine-tuned by repeating the experiments with different labeled metabolites (e.g., glucose, pyruvate, etc.) or different labeling within a metabolite (e.g., [1-13C] glucose, [2-13C] glucose, etc.). Identification of position-specific carbon or nitrogen labeling is relatively straightforward by NMR since, in general, each carbon or nitrogen has a unique chemical shift. For example, consider the distinct carbon chemical shifts observed for L-alanine: 178.5 ppm (C′), 53.4 ppm (Cα) and 19.0 ppm (Cβ). Conversely, it is not possible to identify position-specific carbon or nitrogen labeling by MS except in the case of a compound containing a single carbon or nitrogen, or by employing MSn fragmentation analysis. Instead, the number of isotopically labeled carbons or nitrogens is easily determined by MS from an observed mass shift. In addition, the abundance of each isotopomer is readily obtainable from an MS spectrum by comparing the relative intensity of each molecular-ion peak. Conversely, obtaining the number and abundance of each isotopomer and isotopologue is not easily obtained from an NMR spectrum.
Figure 5.

Metabolic model of liver acetate oxidative metabolism used to estimate hepatic TCA cycle flux (VTCA) and anaplerosis (VANA). Carbon positional enrichment denoted in red occurs during the initial incorporation of label from 1-13C-acetate to glutamate on the first pass through the TCA cycle. Positional enrichment denoted in blue occurs during the 2nd pass through the TCA cycle, with label originating from internal scrambling at succinate or from bicarbonate (HCO3−)/13CO2 via anaplerosis. Reproduced with permission from reference [145].
Lane et al. used direct infusion FT-ICR-MS combined with 1D and 2D NMR methods to identify isotopomers of glycerophospholipids (GPL) derived from [U-13C]-glucose in breast cancer MCF7-LCC2 cell extracts [124]. An algorithm was also presented that accounts for the contribution of natural abundance 13C following the incorporation of 13C-carbon derived from glucose [133]:
| (1) |
where, IM +i;N A is the expected intensity of the ith isotopologue peak, N A13c is the 13C natural abundance (1.1%), cMax is the total number of carbons in the molecule, k is the total number of 13C carbons, n is the number of 13C carbons incorporated from a labeling source, k-n is the number of natural abundant 13C carbons.
Analysis of the 1D 1H NMR spectra identified phosphatidylcholines with approximately two double bonds as the major GPL present in the MCF7-LCC2 cell extracts (Fig. 6A-C). Based on the relative intensity of the assigned GPL NMR resonances, it was determined that the choline head groups were not 13C labeled, but the fatty acyl chains and the glycerol moieties were predominately derived from [U-13C]-glucose. Specifically, the glycerol moiety was determined to be 44 ± 1.2% 13C, and the C2, C3 and C4 positions of the fatty acids were determined to have an average 13C incorporation of 46 ± 4%. These results indicated that nearly 50% of GPL was newly synthesized in 24 hours. The FT-ICR-MS data (Fig. 6D-F) complemented the NMR results by identifying the individual GPL species [i.e., PC (34:1)] and the abundance of the corresponding 13C isotopologues as a function of time. Specifically, the m0 isotopologue decreased and the m0+3, m0+2n + m0+3+2n isotopologues sequentially increased as 13C-carbon from glucose was incorporated into PC (34:1). The odd number isotopologues were derived from 13C glycerol and unlabeled fatty acids, while conversely; even number isotopologues resulted from unlabeled glycerol and 13C fatty acids. Interestingly, the abundance of the n = 10 to 20 fatty acid chains increased with time, but only a minimal mass shift was observed. A 12C acetate pool persisted for twenty-four hours from fatty acid turnover and internal triglyceride stores. In fact, only 30 to 50% of the 34 carbons were incorporated into PC (34:1) from 13C acetyl CoA. Consistent with the NMR results, the mass spectral data indicates that 46% of glycerol and 44% of the fatty acyl moieties are 13C-labeled. Moreover, based on these measurements, isotopologue distributions of GPLs over a twenty-four period were accurately measured and simulated. Thus, the authors clearly demonstrate the value of combining NMR and MS to monitor the synthesis of various GPLs from pools of metabolite. The combination of MS and NMR yielded positional isotope labeling information, 13C isotopologue distributions, and enabled the accurate and efficient identification of GPL species. SIRM using both NMR and MS greatly improves the analysis of metabolomic flux (i.e., metabolite synthesis and turnover).
Figure 6.

High resolution NMR spectra of a methanolic extract of LCC2 cells. Glycerophospholipids were extracted from LCC2 cells grown in the presence of 10 mM [U-13C]-glucose for 24 h. (A) 1D 1H NMR spectrum and (B) TOCSY spectrum. The TOCSY spectrum was recorded at 18.8 T 293 K with 50 ms mixing time at a B1 field strength of 9 kHz. The data were processed with one linear prediction and zerofilling in t1 and apodized using an unshifted Gaussian function in both dimensions. (C) 1D 1H NMR spectra, top: 1D 13C-edited 1H (HSQC) spectrum, bottom: high resolution 1H NMR spectrum. High resolution FT-ICR mass spectrum of a methanolic extract of LCC2 cells. (D) FT-ICR-MS profile spectrum of an LCC2 methanol extract after 24 h labeling with [U-13C]-glucose. A close up of the m/z region from 760 to 782 is shown. The accurate masses (better than 1 ppm) at high resolution (>100,000 at measured mass) enable assignment of the GPLs and their isotopologues. Masses were externally calibrated, and secondarily calibrated with respect to internal standard reserpine; intensities have been arbitrarily scaled to 100 units for m0 at m/z = 760.5860. (E) Mass distribution of PC 34:1 normalized to the total intensity as a function of time. The distribution at 0 h is indistinguishable from the expected natural abundance intensity. Line graphs are used here for clarity only; no values are implied between data points. (F) Time courses of selected mass peaks. (■) m0, (□) m0+3, (•) Σ(m0+2n); (○) Σ(m0+3+2n). The m0+3 intensities were fitted to a(1-exp(−kt)) with a = 0.11 ± 0.008 and k = 0.19 ± 0.04 h−1 . Reproduced with permission from reference [124].
3.4. Combined Cheminformatics Methods
The effective handling and analysis of large amounts of information (i.e., “Big data”) presents unique challenges [134-137]. The field of metabolomics faces similar concerns, which make it difficult to handle the large amounts of information with traditional methods such as database management, basic statistical methods, or simple manual analysis [138]. Combined cheminformatic approaches present a valuable alternative to accelerate the accurate processing of “omics” data sets. Since a considerable amount of data may be generated by combining NMR and MS, combined cheminformatics approaches are increasingly being employed during metabolomics studies. Recently, the SUMMIT and NMR/MS translator methods were developed for the rapid and accurate identification of metabolites [28, 29]. SUMMIT MS/NMR and the NMR/MS translator combine NMR and MS to elucidate the structures of unknown metabolites from complex mixtures. NMR/MS translator combines COLMAR [139] database search queries and experimental NMR and MS spectral data to accelerate accurate metabolite identification [28]. NMR/MS translator uses 1D 1H NMR, or 2D 1H-13C HSQC and 2D 1H-13C HSQC TOCSY chemical shifts to perform a COLMAR database search and return a list of possible metabolite candidates. The query candidate list is then used to produce a simulated mass spectrum for each possible metabolite, which includes possible adducts, fragments, and isotope distributions. These simulated mass spectra are then compared against an experimental metabolomics mass spectrum to make metabolite assignments. In effect, potential metabolites identified by NMR are confirmed by MS. As a proof of concept, the NMR/MS translator was used to successfully analyze a model mixture of 26 metabolites with 2D 1H-13C HSQC NMR spectra and DI-ESI-MS positive and negative mode spectra. The NMR/MS translator was further validated using a set of human urine samples from healthy volunteers. A total of 98 urine metabolites were identified by the NMR/MS translator, which included 8 metabolites that were not previously observed in a comprehensive study of human urine. Importantly, only 48 of these metabolites were correctly identified using MS data alone, including MS/MS fragmentation patterns. The NMR/MS translator approach automates the metabolite assignment and avoids labor-intensive manual analysis, which enhances coverage, improves consistency, and increases throughput.
SUMMIT MS/NMR is an alternative high throughput approach that combines MS with NMR data to identify unknown metabolites in a complex biological sample (Fig. 7) [29]. The SUMMIT MS/NMR approach relies on the acquisition of a high-resolution mass spectrum, in which each m/z peak is converted into a molecular formula. The list of molecular formulas is then used to generate a set of all feasible structures (e.g., a structural manifold) with the ChemSpider database [140], which are used to predict an NMR spectrum using MestReNova 9.0.1 (Mestrelab Research, Santiago de Compostela, Spain). The COLMAR algorithm [139] is then used to compare the experimental NMR data against the database of predicted NMR spectra. Importantly, the experimental 2D 1H–13C HSQC NMR spectrum is deconvoluted into subspectra corresponding to the individual components of the mixture using connectivity information derived from 2D 1H–1H TOCSY, 2D 1H–13C HSQC-TOCSY, and 2D 1H–13C HMBC spectra. The potential metabolites are rank-ordered based on the relative agreement between the experimental NMR subspectrum and the simulated NMR spectra.
Figure 7.

Schematic representation of the SUMMIT MS/NMR strategy for the identification of metabolites in complex metabolomic mixtures by the combined use of mass spectrometry and 1D 1H NMR spectroscopy. High-resolution MS yields the unique molecular formulas of the metabolites present in the mixture (left). For each molecular formula, all possible structures are generated, representing the total structural manifold depicted as the sum of the three local manifolds (green, red, blue; middle), each belonging to a different mass. Next, NMR chemical shifts are predicted for all manifold structures. Comparison of the predicted with the experimental NMR chemical shifts (right) allows identification of the structures that are present in the mixture, requiring neither an NMR nor an MS metabolomics database [28, 29, 58]. Reproduced with permission from reference [29].
SUMMIT MS/NMR was validated using a DI-ESI-MS spectrum of a model mixture containing 10 metabolites [29]. The 50 largest m/z peaks were selected, which yielded 22 molecular formulas, 362 potential structures, and 4772 predicted 2D 1H–13C HSQC NMR spectra. SUMMIT MS/NMR ranked 6 of the 10 metabolites as the top hit and three other metabolites were identified as the second-best hit. The remaining metabolite did not ionize. For the three metabolites identified as second best, the top hits were structurally very similar (i.e., allo-isoleucine instead of leucine). The approach was repeated using an MS spectrum of a polar extract of an E. coli lysate. The 500 largest m/z peaks were selected corresponding to 56 molecular formulas and 13872 structures and 1H–13C HSQC spectra. A total of 21 metabolites were accurately and rapidly identify by SUMMIT MS/NMR, and then confirmed using a set of 2D 1H-13C NMR experiments. Thus, both SUMMIT MS/NMR and NMR/MS translator clearly illustrate the inherent value of combining NMR and MS to enhance metabolomics.
3.5. Multivariate Statistical Methods
A well-known problem with metabolomics data is the presence of confounding factors that may complicate the identification of group membership. For example, the analysis of urine or serum to identify biomarkers may be masked by metabolites associated with age, diet, ethnicity, gender, or race, among other factors. Multivariate statistical methods are able to cope with these multiparametric data sets and extract group membership [141]. For a detailed and comprehensive review of multivariate statistics and its application for metabolomics see Worley et al. [12]. Chemometric techniques can be divided into supervised or unsupervised methods. In metabolomics, unsupervised methods are commonly used to identify global trends or group membership. Alternatively, supervised methods are highly valuable for identifying the spectral features (or metabolites) that primarily contribute to the differentiation between groups. PCA, PLS, and OPLS are the chemometric methods commonly used in metabolomics based on a single analytical source. The limited availability of chemometric methods applicable to multiple analytical sources is one reason NMR and MS have not been commonly combined for metabolomics.
Nevertheless, multivariate statistical techniques have been previously applied for the combined analysis of MS and NMR data sets. Chen et al. generated individual PCA models for NMR and MS data sets and then combined the scores from each analysis into a three dimensional (3D) scores plot. The combined scores yielded a greater between-class separation than the original NMR or MS scores alone. Unfortunately, such an analysis ignores the highly informative correlations that exist between the two data sets. Gu et al. replaced the binary class designation of an MS data set with the first principal component (PC1) from a PCA model generated from NMR data to produce a subsequent OPLS-DA model for the MS data [70]. Again, a greater class separation was observed when the MS OPLS-DA model was generated with the NMR PC1 compared to the binary classification [57]. Nevertheless, such an analysis carries no statistical guarantee of success for any data set.
Recently, it was shown that a chemometrics model generated by integrating NMR and MS metabolomics data provided better group separation and a greater level of model interpretability than with NMR or MS data sets alone [57]. Marshall et al. combined 1D 1H NMR and DI-ESI-MS. Multiblock methods are similar to traditional PLS and PCA, but provide a means for analyzing data from multiple analytical sources [142-144]. The spectral observations from each analytical method are placed into separate “blocks,” which allows for the generation and simultaneous usage of within-block and between-block data correlations. Since the blocks share common trends, a model based on the between-block correlations will provide a better agreement with the biological groups. In effect, better discrimination between groups is expected by combining NMR and MS data than would be achieved from only the individual data sets. DI-ESI-MS and 1D 1H NMR spectra were collected on cell lysates obtained from human dopaminergic neuroblastoma cells (SK-N-SH) treated with different neurotoxins: rotenone, 6-hydroxydopamine (6-OHDA), 1-methyl-4-phenylpyridinium (MPP+), or paraquat. The PCA model produced from the 1D 1H NMR data set yielded only two groups corresponding to the untreated controls and cells treated with the different neurotoxins. In effect, NMR detected no difference in the metabolome of neuronal cells after treatment with the different neurotoxins. The PCA model generated from the MS data set produced a modest separation between four groups.
The untreated controls, MPP+, and paraquat treatment each formed a separate group. Both the rotenone and 6-OHDA cell treatment were clustered together and formed the fourth group (Fig. 8). The MB-PCA and MB-PLS models generated from both the DI-ESI-MS and 1D 1H NMR data sets yielded five distinct groups corresponding to each neurotoxin treatment and the untreated controls. This clearly demonstrated that each neurotoxin induced dopaminergic neuronal cell death through a distinct molecular mechanism. A detailed analysis of the metabolic impact of paraquat revealed that paraquat “hijacks” the pentose phosphate pathway (PPP) to increase NADPH-reducing equivalents and stimulate paraquat redox cycling, oxidative stress, and cell death [57, 71]. Thus, a successful outcome for a metabolomics study was critically dependent on combining NMR and MS data.
Figure 8.

Scores generated from (A) PCA of 1H NMR, (B) PCA of DI-ESI-MS, and (C) MB-PCA of 1H NMR and DI-ESI-MS. Separations between classes are greatly increased upon combination of the two data sets via MB-PCA. Symbols designate the following classes: Control (
 ), Rotenone (
), Rotenone (
 ), 6-OHDA (
), 6-OHDA (
 ), MPP+ (
), MPP+ (
 ), and Paraquat (
), and Paraquat (
 ). Corresponding dendrograms are shown in (D-F). The statistical significance of each node in the dendrogram is indicated by a p value. Reproduced with permission from reference [57].
). Corresponding dendrograms are shown in (D-F). The statistical significance of each node in the dendrogram is indicated by a p value. Reproduced with permission from reference [57].
4. Conclusion
Metabolomics is an invaluable tool of systems biology and has made significant contributions to several diverse fields, including drug discovery, disease diagnosis, nutrition, environmental studies, and personalized medicine. To date, the majority of metabolomics data sets have been acquired using either MS or NMR separately. However, it is well known that combining MS and NMR data greatly improves the coverage of the metabolome and enhances the accuracy of metabolite identification. Consequently, combining NMR and MS techniques for metabolomics is a growing trend that will greatly benefit the quality and accuracy of metabolomics data. Herein we have reviewed several methodologies for integrating NMR and MS for the analysis of metabolomics samples. As demonstrated throughout this review, combining NMR and MS greatly enhances and improves the outcomes of metabolomics studies.
Highlights.
Metabolomics routinely relies on only a single analytical source
Combining NMR and MS improves the quality of a metabolomics study
Both NMR and MS are required for accurate metabolite assignments
Acknowledgments
We would like to thank Dr. Eric D. Dodds, Dr. Rodrigo Franco, Dr. Aracely Garcia-Garcia, Dr. Yuting Huang, Dr. Shulei Lei, and Dr. Bradley Worley for their contributions to the metabolic studies presented in this manuscript. This manuscript was supported in part by funds from the National Institute of Health (R01 AI087668, R21 AI087561, R01 CA163649, P20 RR-17675, P30 GM103335), the University of Nebraska, the Nebraska Tobacco Settlement Biomedical Research Development Fund, and the Nebraska Research Council. The research was performed in facilities renovated with support from the National Institutes of Health (RR015468-01).
Glossary of Abbreviations
- 1D
One-Dimensional
- 2D
Two-Dimensional
- 3D
Three-Dimensional
- 6-OHDA
6-hydroxydopamine
- ANOVA
ANalysis Of Variance
- BNMI
Bruker
- NMR
Mass Spectrometry Interface
- CE
Capillary Electrophoresis
- COLMAR
Complex Mixture Analysis by NMR
- CV-ANOVA
ANOVA of the Cross-Validated residuals
- ddH2O
double-distilled water
- DI-ESI-MS
Direct Infusion ElectroSpray Ionisation Mass Spectrometry
- ESI
ElectroSpray Ionisation
- GC
Gas Chromatograph
- GPL
GlyceroPhosphoLipids
- HMBC
Heteronuclear Multiple-Bond Correlation
- HSQC
Heteronuclear Single Quantum Coherence spectroscopy
- FT-ICR-MS
Fourier Transform Ion Cyclotron Resonance Mass Spectrometry
- LC
Liquid Chromatograph
- LysoPC (16:0)
Lysophospholipid (16:0)
- MB-PCA
Multi-Block-Principal Component Analysis
- MB-PLS
Multi-Block-Projections to Latent Structures
- MetPA
METabolomics Pathway Analysis
- MPP+
1-methyl-4-phenylpyridinium
- MS
Mass Spectrometry
- MSn
multiple-stage mass spectrometry where n is the number of product ion stages (n=2,3, etc.)
- m/z
mass to charge ratio NADPH, Nicotinamide Adenine Dinucleotide Phosphate
- NMR
Nuclear Magnetic Resonance
- OPLS
Orthogonal Projections to Latent Structures
- OPLS-DA
Orthogonal Projections to Latent Structures- Discriminate Analysis
- PC (34
1), PhosphatidylCholine 34:1
- PC
Principal Component
- PC1
first Principal Component
- PCA
Principal Component Analysis
- pKa
negative logarithm of an acid dissociation constant
- PLS
Projections to Latent Structures
- PPP
Pentose Phosphate Pathway
- PRA
Pattern Recognition Approaches
- ROC
Receiver Operating Characteristic
- RSD
Relative Standard Deviation
- SIRM
Stable Isotope-Resolved Metabolomics
- TCA cycle
TriCarboxylic Acid cycle
- TMSP-d4
3-(TriMethylSilyl)Propionic-2,2,3,3-d4
- TOCSY
Total Correlation SpectroscopY
- XCMS
various forms (X) of Chromatography Mass Spectrometry
Footnotes
The term metabonomics is often used interchangeably with metabolomics and the primary distinction between the two terms is historical instead of scientific. The word metabolome was first used by Oliver et al. in 1998 to define, similar to the terms proteome or transcriptome, the collection of metabolites present in a cell, tissue or organism. Consequently, metabolomics as defined by Fiehn in 2001 is the ‘comprehensive and quantitative analysis of all metabolites’. The term metabonomics was defined in 1999 by Nicholson et al. as ‘the quantitative measurement of the dynamic multiparametric metabolic response of living systems to pathophysiological stimuli or genetic modification’.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wachtel-Galor S, Benzie IFF. Herbal Medicine: An Introduction to Its History, Usage, Regulation, Current Trends, and Research Needs. 2011 [PubMed] [Google Scholar]
- 2.Oliver SG, Winson MK, Kell DB, Baganz F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998;16:373–378. doi: 10.1016/s0167-7799(98)01214-1. [DOI] [PubMed] [Google Scholar]
- 3.Nicholson JK, Lindon JC, Holmes E. ‘Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica. 1999;29:1181–1189. doi: 10.1080/004982599238047. [DOI] [PubMed] [Google Scholar]
- 4.Fiehn O. Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Comp Funct Genomics. 2001;2:155–168. doi: 10.1002/cfg.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunn WB, Bailey NJ, Johnson HE. Measuring the metabolome: current analytical technologies. Analyst. 2005;130:606–625. doi: 10.1039/b418288j. [DOI] [PubMed] [Google Scholar]
- 6.Bijland LR, Bomers MK, Smulders YM. Smelling the diagnosis A review on the use of scent in diagnosing disease. The Journal of Medicine. 2013;71 [PubMed] [Google Scholar]
- 7.Wolfender JL, Marti G, Thomas A, Bertrand S. Current approaches and challenges for the metabolite profiling of complex natural extracts. J Chromatogr A. 2015;1382:136–164. doi: 10.1016/j.chroma.2014.10.091. [DOI] [PubMed] [Google Scholar]
- 8.Bingol K, Brüschweiler R. Multidimensional approaches to NMR-based metabolomics. Anal Chem. 2014;86:47–57. doi: 10.1021/ac403520j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Likic VA, McConville MJ, Lithgow T, Bacic A. Systems biology: the next frontier for bioinformatics. Adv Bioinformatics. 2010:268925. doi: 10.1155/2010/268925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wishart DS. Current progress in computational metabolomics. Brief Bioinform. 2007;8:279–293. doi: 10.1093/bib/bbm030. [DOI] [PubMed] [Google Scholar]
- 11.Dettmer K, Aronov PA, Hammock BD. Mass spectrometry-based metabolomics. Mass Spectrom Rev. 2007;26:51–78. doi: 10.1002/mas.20108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Worley B, Powers R. Multivariate Analysis in Metabolomics. Curr Metabolomics. 2013;1:92–107. doi: 10.2174/2213235X11301010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gebregiworgis T, Powers R. Application of NMR metabolomics to search for human disease biomarkers. Comb Chem High Throughput Screen. 2012;15:595–610. doi: 10.2174/138620712802650522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiehn O. Metabolomics - the link between genotypes and phenotypes. Plant Mol Biol. 2002;48:155–171. [PubMed] [Google Scholar]
- 15.Astarita G, Langridge J. An Emerging Role for Metabolomics in Nutrition Science. J Nutrigenet Nutrigenomics. 2013;6:181–200. doi: 10.1159/000354403. [DOI] [PubMed] [Google Scholar]
- 16.Jones DP. Sequencing the exposome: A call to action. Toxicol Rep. 2016;3:29–45. doi: 10.1016/j.toxrep.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim SJ, Kim SH, Kim JH, Yoo HJ, Hwang S. Understanding Metabolomics in Biomedical Research. Endocrinol Metab (Seoul) 2016;31:7–16. doi: 10.3803/EnM.2016.31.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beger RD, Dunn W, Schmidt MA, Gross SS, Kirwan JA, Cascante M, Brennan L, Wishart DS, Oresic M, Hankemeier T, Broadhurst DI, Lane AN, Suhre K, Kastenmuller G, Sumner SJ, Thiele I, Fiehn O, Kaddurah-Daouk R. Metabolomics enables precision medicine: [A White Paper, Community Perspective] Metabolomics. 2016;12:1–15. doi: 10.1007/s11306-016-1094-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gowda GAN, Raftery D. Can NMR solve some significant challenges in metabolomics? J Magn Reson. 2015;260:144–160. doi: 10.1016/j.jmr.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gowda GAN, Raftery D. Biomarker Discovery and Translation in Metabolomics. Curr Metabolomics. 2013;1:227–240. doi: 10.2174/2213235X113019990005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindon JC, Nicholson JK. The emergent role of metabolic phenotyping in dynamic patient stratification. Expert Opin Drug Metab Toxicol. 2014;10:915–919. doi: 10.1517/17425255.2014.922954. [DOI] [PubMed] [Google Scholar]
- 22.Rhee EP, Gerszten RE. Metabolomics and cardiovascular biomarker discovery. Clin Chem. 2012;58:139–147. doi: 10.1373/clinchem.2011.169573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Markley JL, Bruschweiler R, Edison AS, Eghbalnia HR, Powers R, Raftery D, Wishart DS. The future of NMR-based metabolomics. Curr Opin Biotechnol. 2017;43:34–40. doi: 10.1016/j.copbio.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirschfeld T. The Hy-phen-ated Methods. Anal Chem. 1980;52:297A–312A. [Google Scholar]
- 25.Zhang A, Sun H, Wang P, Han Y, Wang X. Modern analytical techniques in metabolomics analysis. Analyst (Cambridge, U K) 2012;137:293–300. doi: 10.1039/c1an15605e. [DOI] [PubMed] [Google Scholar]
- 26.Barding GA, Beni S, Fukao T, Bailey-Serres J, Larive CK. Comparison of GC-MS and NMR for Metabolite Profiling of Rice Subjected to Submergence Stress. J Proteome Res. 2013;12:898–909. doi: 10.1021/pr300953k. [DOI] [PubMed] [Google Scholar]
- 27.Dai H, Xiao C, Liu H, Hao F, Tang H. Combined NMR and LC-DAD-MS Analysis Reveals Comprehensive Metabonomic Variations for Three Phenotypic Cultivars of Salvia miltiorrhiza Bunge. J Proteome Res. 2010;9:1565–1578. doi: 10.1021/pr901045c. [DOI] [PubMed] [Google Scholar]
- 28.Bingol K, Brüschweiler R. NMR/MS Translator for the Enhanced Simultaneous Analysis of Metabolomics Mixtures by NMR Spectroscopy and Mass Spectrometry: Application to Human Urine. J Proteome Res. 2015;14:2642–2648. doi: 10.1021/acs.jproteome.5b00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bingol K, Bruschweiler-Li L, Yu C, Somogyi A, Zhang F, Brüschweiler R. Metabolomics Beyond Spectroscopic Databases: A Combined MS/NMR Strategy for the Rapid Identification of New Metabolites in Complex Mixtures. Anal Chem. 2015;87:3864–3870. doi: 10.1021/ac504633z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tayyari F, Gowda GA, Gu H, Raftery D. 15N-cholamine--a smart isotope tag for combining NMR- and MS-based metabolite profiling. Anal Chem. 2013;85:8715–8721. doi: 10.1021/ac401712a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baker JM, Ward JL, Beale MH. Combined NMR and flow injection ESI-MS for Brassicaceae metabolomics. Methods Mol Biol. 2012;860:177–191. doi: 10.1007/978-1-61779-594-7_12. [DOI] [PubMed] [Google Scholar]
- 32.Prichystal J, Schug KA, Lemr K, Novak J, Havlicek V. Structural Analysis of Natural Products. Anal Chem. 2016;88:10338–10346. doi: 10.1021/acs.analchem.6b02386. [DOI] [PubMed] [Google Scholar]
- 33.Yang Z. Online hyphenated liquid chromatography-nuclear magnetic resonance spectroscopy-mass spectrometry for drug metabolite and nature product analysis. J Pharm Biomed Anal. 2006;40:516–527. doi: 10.1016/j.jpba.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 34.Pan Z, Raftery D. Comparing and combining NMR spectroscopy and mass spectrometry in metabolomics. Anal Bioanal Chem. 2007;387:525–527. doi: 10.1007/s00216-006-0687-8. [DOI] [PubMed] [Google Scholar]
- 35.t'Kindt R, Scheltema RA, Jankevics A, Brunker K, Rijal S, Dujardin JC, Breitling R, Watson DG, Coombs GH, Decuypere S. Metabolomics to unveil and understand phenotypic diversity between pathogen populations. PLoS Negl Trop Dis. 2010;4:e904. doi: 10.1371/journal.pntd.0000904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halouska S, Zhang B, Gaupp R, Lei S, Snell E, Fenton RJ, Barletta RG, Somerville GA, Powers R. Revisiting Protocols for the NMR Analysis of Bacterial Metabolomes. Journal of Integrated OMICS. 2013;2:120–137. doi: 10.5584/jiomics.v3i2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Copeland JC, Zehr LJ, Cerny RL, Powers R. The Applicability of Molecular Descriptors for Designing an Electrospray Ionization Mass Spectrometry Compatible Library for Drug Discovery. Comb Chem High Throughput Screening. 2012;15:806–815. doi: 10.2174/138620712803901180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moy FJ, Haraki K, Mobilio D, Walker G, Tabei K, Tong H, Siegel MM, Powers R. MS/NMR: A Structure-Based Approach for Discovering Protein Ligands and for Drug Design by Coupling Size Exclusion Chromatography, Mass Spectrometry, and Nuclear Magnetic Resonance Spectroscopy. Anal Chem. 2001;73:571–581. doi: 10.1021/ac0006270. [DOI] [PubMed] [Google Scholar]
- 39.Antignac JP, de Wasch K, Monteau F, De Brabander H, Andre F, Le Bizec B. The ion suppression phenomenon in liquid chromatography-mass spectrometry and its consequences in the field of residue analysis. Anal Chim Acta. 2005;529:129–136. [Google Scholar]
- 40.Metz TO, Page JS, Baker ES, Tang K, Ding J, Shen Y, Smith RD. High-resolution separations and improved ion production and transmission in metabolomics, TrAC. Trends Anal Chem. 2008;27:205–214. doi: 10.1016/j.trac.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crockford DJ, Holmes E, Lindon JC, Plumb RS, Zirah S, Bruce SJ, Rainville P, Stumpf CL, Nicholson JK. Statistical Heterospectroscopy, an Approach to the Integrated Analysis of NMR and UPLC-MS Data Sets: Application in Metabonomic Toxicology Studies. Anal Chem. 2006;78:363–371. doi: 10.1021/ac051444m. [DOI] [PubMed] [Google Scholar]
- 42.Kuehnbaum NL, Britz-Mc Kibbin P. New Advances in Separation Science for Metabolomics: Resolving Chemical Diversity in a Post-Genomic Era. Chem Rev. 2013;113:2437–2468. doi: 10.1021/cr300484s. [DOI] [PubMed] [Google Scholar]
- 43.Kell DB. Metabolomics and systems biology: making sense of the soup. Curr Opin Microbiol. 2004;7:296–307. doi: 10.1016/j.mib.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 44.Canelas AB, ten Pierick A, Ras C, Seifar RM, van Dam JC, van Gulik WM, Heijnen JJ. Quantitative evaluation of intracellular metabolite extraction techniques for yeast metabolomics. Anal Chem. 2009;81:7379–7389. doi: 10.1021/ac900999t. [DOI] [PubMed] [Google Scholar]
- 45.Kanani H, Chrysanthopoulos PK, Klapa MI. Standardizing GC-MS metabolomics. J Chromatogr B: Anal Technol Biomed Life Sci. 2008;871:191–201. doi: 10.1016/j.jchromb.2008.04.049. [DOI] [PubMed] [Google Scholar]
- 46.Xu F, Zou L, Ong CN. Multiorigination of Chromatographic Peaks in Derivatized GC/MS Metabolomics: A Confounder That Influences Metabolic Pathway Interpretation. J Proteome Res. 2009;8:5657–5665. doi: 10.1021/pr900738b. [DOI] [PubMed] [Google Scholar]
- 47.Taylor PJ. Matrix effects: the Achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clin Biochem. 2005;38:328–334. doi: 10.1016/j.clinbiochem.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 48.Kopka J. Current challenges and developments in GC-MS based metabolite profiling technology. J Biotechnol. 2006;124:312–322. doi: 10.1016/j.jbiotec.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 49.Marion D, Driscoll PC, Kay LE, Wingfield PT, Bax A, Gronenborn AM, Clore GM. Overcoming the overlap problem in the assignment of 1H NMR spectra of larger proteins by use of three-dimensional heteronuclear 1H-15N Hartmann-Hahn-multiple quantum coherence and nuclear Overhauser-multiple quantum coherence spectroscopy: application to interleukin 1 beta. Biochemistry. 1989;28:6150–6156. doi: 10.1021/bi00441a004. [DOI] [PubMed] [Google Scholar]
- 50.Mahrous EA, Farag MA. Two dimensional NMR spectroscopic approaches for exploring plant metabolome: A review. J Adv Res. 2015;6:3–15. doi: 10.1016/j.jare.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bingol K, Brüschweilerr-Li L, Li D, Zhang B, Xie M, Brüschweiler R. Emerging new strategies for successful metabolite identification in metabolomics. Bioanalysis. 2016;8:557–573. doi: 10.4155/bio-2015-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lei Z, Huhman DV, Sumner LW. Mass spectrometry strategies in metabolomics. J Biol Chem. 2011;286:25435–25442. doi: 10.1074/jbc.R111.238691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iwasaki Y, Sawada T, Hatayama K, Ohyagi A, Tsukuda Y, Namekawa K, Ito R, Saito K, Nakazawa H. Separation technique for the determination of highly polar metabolites in biological samples. Metabolites. 2012;2:496–515. doi: 10.3390/metabo2030496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cech NB, Enke CG. Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom Rev. 2001;20:362–387. doi: 10.1002/mas.10008. [DOI] [PubMed] [Google Scholar]
- 55.Tugizimana F, Steenkamp PA, Piater LA, Dubery IA. Multi-platform metabolomic analyses of ergosterol-induced dynamic changes in Nicotiana tabacum cells. PLoS One. 2014;9:e87846. doi: 10.1371/journal.pone.0087846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ling YS, Liang HJ, Chung MH, Lin MH, Lin CY. NMR- and MS-based metabolomics: various organ responses following naphthalene intervention. Mol Biosyst. 2014;10:1918–1931. doi: 10.1039/c4mb00090k. [DOI] [PubMed] [Google Scholar]
- 57.Marshall DD, Lei S, Worley B, Huang Y, Garcia-Garcia A, Franco R, Dodds ED, Powers R. Combining DI-ESI-MS and NMR Datasets for Metabolic Profiling. Metabolomics. 2015;11:391–402. doi: 10.1007/s11306-014-0704-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bingol K, Brüschweiler R. Two elephants in the room. Current Opinion in Clinical Nutrition and Metabolic Care. 2015;18:471–477. doi: 10.1097/MCO.0000000000000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin Y, Schiavo S, Orjala J, Vouros P, Kautz R. Microscale LC-MS-NMR platform applied to the identification of active cyanobacterial metabolites. Anal Chem. 2008;80:8045–8054. doi: 10.1021/ac801049k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Corcoran O, Spraul M. LC–NMR–MS in drug discovery. Drug Discov Today. 2003;8:624–631. doi: 10.1016/s1359-6446(03)02749-1. [DOI] [PubMed] [Google Scholar]
- 61.Johnson CH, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms. Nat Rev Mol Cell Biol. 2016;17:451–459. doi: 10.1038/nrm.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bird SS, Sheldon DP, Gathungu RM, Vouros P, Kautz R, Matson WR, Kristal BS. Structural characterization of plasma metabolites detected via LC-electrochemical coulometric array using LC-UV fractionation, MS, and NMR. Anal Chem. 2012;84:9889–9898. doi: 10.1021/ac302278u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alonso A, Marsal S, Julia A. Analytical methods in untargeted metabolomics: state of the art in 2015. Front Bioeng Biotechnol. 2015;3:23. doi: 10.3389/fbioe.2015.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dunn WB, Ellis DI. Metabolomics: Current analytical platforms and methodologies. TrAC Trends in Anal Chem. 2005;24:285–294. [Google Scholar]
- 65.Worley B, Powers R. PCA as a predictor of OPLS-DA model reliability. Current Metabolomics. 2016;4:97–103. doi: 10.2174/2213235X04666160613122429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brereton RG. A short history of chemometrics: a personal view. J Chemom. 2014;28:749–760. [Google Scholar]
- 67.Worley B, Powers R. MVAPACK: A Complete Data Handling Package for NMR Metabolomics. ACS Chem Biol. 2014;9:1138–1144. doi: 10.1021/cb4008937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Worley B, Powers R. A sequential algorithm for multiblock orthogonal projections to latent structures. Chemometr Intell Lab Syst. 2015;149:33–39. doi: 10.1016/j.chemolab.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen H, Pan Z, Talaty N, Raftery D, Cooks RG. Combining desorption electrospray ionization mass spectrometry and nuclear magnetic resonance for differential metabolomics without sample preparation. Rapid Commun Mass Spectrom. 2006;20:1577–1584. doi: 10.1002/rcm.2474. [DOI] [PubMed] [Google Scholar]
- 70.Gu H, Pan Z, Xi B, Asiago V, Musselman B, Raftery D. Principal component directed partial least squares analysis for combining nuclear magnetic resonance and mass spectrometry data in metabolomics: Application to the detection of breast cancer. Anal Chim Acta. 2011;686:57–63. doi: 10.1016/j.aca.2010.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lei S, Zavala-Flores L, Garcia-Garcia A, Nandakumar R, Huang Y, Madayiputhiya N, Stanton RC, Dodds ED, Powers R, Franco R. Alterations in Energy/Redox Metabolism Induced by Mitochondrial and Environmental Toxins: A Specific Role for Glucose-6-Phosphate-Dehydrogenase and the Pentose Phosphate Pathway in Paraquat Toxicity. ACS Chem Biol. 2014;9:2032–2048. doi: 10.1021/cb400894a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anandhan A, Lei S, Levytskyy R, Pappa A, Panayiotidis MI, Cerny RL, Kalimonchuk O, Powers R, Franco R. Glucose Metabolism and AMPK Signaling Regulate Dopaminergic Cell Death Induced by gene (α-synuclein)-Envronment(paraquat) Interactions. Molecular neurobiology. doi: 10.1007/s12035-016-9906-2-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Powers R. NMR metabolomics and drug discovery. Magn Reson Chem. 2009;47:S2–S11. doi: 10.1002/mrc.2461. [DOI] [PubMed] [Google Scholar]
- 74.Bollard ME, Stanley EG, Lindon JC, Nicholson JK, Holmes E. NMR-based metabonomic approaches for evaluating physiological influences on biofluid composition. NMR Biomed. 2005;18:143–162. doi: 10.1002/nbm.935. [DOI] [PubMed] [Google Scholar]
- 75.Dunn WB, Broadhurst DI, Atherton HJ, Goodacre R, Griffin JL. Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem Soc Rev. 2011;40:387–426. doi: 10.1039/b906712b. [DOI] [PubMed] [Google Scholar]
- 76.Nicholson JK, Lindon JC, Holmes E. [Metabonomics]: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica. 1999;29:1181–1189. doi: 10.1080/004982599238047. [DOI] [PubMed] [Google Scholar]
- 77.Wishart DS. Quantitative metabolomics using NMR, TrAC. Trends Anal Chem. 2008;27:228–237. [Google Scholar]
- 78.Chughtai K, Heeren RMA. Mass Spectrometric Imaging for Biomedical Tissue. Analysis, Chem Rev. 2010;110:3237–3277. doi: 10.1021/cr100012c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Katajamaa M, Oresic M. Data processing for mass spectrometry-based metabolomics. J Chromatogr A. 2007;1158:318–328. doi: 10.1016/j.chroma.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 80.Villas-Boas SG, Mas S, Aakesson M, Smedsgaard J, Nielsen J. Mass spectrometry in metabolome analysis. Mass Spectrom Rev. 2005;24:613–646. doi: 10.1002/mas.20032. [DOI] [PubMed] [Google Scholar]
- 81.Dona AC, Jimenez B, Schafer H, Humpfer E, Spraul M, Lewis MR, Pearce JTM, Holmes E, Lindon JC, Nicholson JK. Precision High-Throughput Proton NMR Spectroscopy of Human Urine, Serum, and Plasma for Large-Scale Metabolic Phenotyping. Anal Chem. 2014;86:9887–9894. doi: 10.1021/ac5025039. [DOI] [PubMed] [Google Scholar]
- 82.Emwas AH, Luchinat C, Turano P, Tenori L, Roy R, Salek RM, Ryan D, Merzaban JS, Kaddurah-Daouk R, Zeri AC, Nagana Gowda GA, Raftery D, Wang Y, Brennan L, Wishart DS. Standardizing the experimental conditions for using urine in NMR-based metabolomic studies with a particular focus on diagnostic studies: a review. Metabolomics. 2015;11:872–894. doi: 10.1007/s11306-014-0746-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Troyer D. Biorepository standards and protocols for collecting, processing, and storing human tissues. Methods Mol Biol (Totowa, NJ, U S) 2008;441:193–220. doi: 10.1007/978-1-60327-047-2_13. [DOI] [PubMed] [Google Scholar]
- 84.Villas-Boas SG, Hoejer-Pedersen J, Aakesson M, Smedsgaard J, Nielsen J. Global metabolite analysis of yeast: Evaluation of sample preparation methods. Yeast. 2005;22:1155–1169. doi: 10.1002/yea.1308. [DOI] [PubMed] [Google Scholar]
- 85.Dunn WB, Broadhurst D, Begley P, Zelena E, Francis-Mc Intyre S, Anderson N, Brown M, Knowles JD, Halsall A, Haselden JN, Nicholls AW, Wilson ID, Kell DB, Goodacre R. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat Protoc. 2011;6:1060–1083. doi: 10.1038/nprot.2011.335. [DOI] [PubMed] [Google Scholar]
- 86.Zhang Ah, Sun H, Qiu S, Wang Xj. NMR-based metabolomics coupled with pattern recognition methods in biomarker discovery and disease diagnosis. Magn Reson Chem. 2013;51:549–556. doi: 10.1002/mrc.3985. [DOI] [PubMed] [Google Scholar]
- 87.Westerhuis JA, Hoefsloot HCJ, Smit S, Vis DJ, Smilde AK, van Velzen EJJ, van Duijnhoven JPM, van Dorsten FA. Assessment of PLSDA cross validation. Metabolomics. 2008;4:81–89. [Google Scholar]
- 88.Kjeldahl K, Bro R. Some common misunderstandings in chemometrics. J Chemom. 2010;24:558–564. [Google Scholar]
- 89.Eriksson L, Trygg J, Wold S. CV-ANOVA for significance testing of PLS and OPLS models. J Chemom. 2008;22:594–600. [Google Scholar]
- 90.Lasko TA, Bhagwat JG, Zou KH, Ohno-Machado L. The use of receiver operating characteristic curves in biomedical informatics. J Biomed Inform. 2005;38:404–415. doi: 10.1016/j.jbi.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 91.Xia J, Wishart DS. MetPA: a web-based metabolomics tool for pathway analysis and visualization. Bioinformatics. 2010;26:2342–2344. doi: 10.1093/bioinformatics/btq418. [DOI] [PubMed] [Google Scholar]
- 92.Heijnen JJ. Impact of thermodynamic principles in systems biology. Adv Biochem Eng/Biotechnol. 2010;121:139–162. doi: 10.1007/10_2009_63. [DOI] [PubMed] [Google Scholar]
- 93.Arrivault S, Guenther M, Ivakov A, Feil R, Vosloh D, van Dongen JT, Sulpice R, Stitt M. Use of reverse-phase liquid chromatography, linked to tandem mass spectrometry, to profile the Calvin cycle and other metabolic intermediates in Arabidopsis rosettes at different carbon dioxide concentrations. Plant J. 2009;59:826–839. doi: 10.1111/j.1365-313X.2009.03902.x. [DOI] [PubMed] [Google Scholar]
- 94.Breier M, Wahl S, Prehn C, Fugmann M, Ferrari U, Weise M, Banning F, Seissler J, Grallert H, Adamski J, Lechner A. Targeted metabolomics identifies reliable and stable metabolites in human serum and plasma samples. PLoS One. 2014;9:e89728/89721–e89728/89711. doi: 10.1371/journal.pone.0089728. 89711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van den Berg RA, Hoefsloot HCJ, Westerhuis JA, Smilde AK, van der Werf MJ. Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics. 2006;7:142. doi: 10.1186/1471-2164-7-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Craig A, Cloarec O, Holmes E, Nicholson JK, Lindon JC. Scaling and Normalization Effects in NMR Spectroscopic Metabonomic Data Sets. Anal Chem. 2006;78:2262–2267. doi: 10.1021/ac0519312. [DOI] [PubMed] [Google Scholar]
- 97.Vu TN, Laukens K. Getting your peaks in line: a review of alignment methods for NMR spectral data. Metabolites. 2013;3:259–276. doi: 10.3390/metabo3020259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Andrews CL, Yu CP, Yang E, Vouros P. Improved liquid chromatography-mass spectrometry performance in quantitative analysis using a nanosplitter interface. J Chromatogr A. 2004;1053:151–159. [PubMed] [Google Scholar]
- 99.Emwas AH, Roy R, McKay RT, Ryan D, Brennan L, Tenori L, Luchinat C, Gao X, Zeri AC, Gowda GA, Raftery D, Steinbeck C, Salek RM, Wishart DS. Recommendations and Standardization of Biomarker Quantification Using NMR-Based Metabolomics with Particular Focus on Urinary Analysis. J Proteome Res. 2016;15:360–373. doi: 10.1021/acs.jproteome.5b00885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Appiah-Amponsah E, Owusu-Sarfo K, Gowda GA, Ye T, Raftery D. Combining Hydrophilic Interaction Chromatography (HILIC) and Isotope Tagging for Off-Line LC-NMR Applications in Metabolite Analysis. Metabolites. 2013;3:575–591. doi: 10.3390/metabo3030575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Beltran A, Suarez M, Rodríguez MA, Vinaixa M, Samino S, Arola L, Correig X, Yanes O. Assessment of Compatibility between Extraction Methods for NMR- and LC/MS-Based Metabolomics. Anal Chem. 2012;84:5838–5844. doi: 10.1021/ac3005567. [DOI] [PubMed] [Google Scholar]
- 102.Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discovery. 2015;14:111–129. doi: 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- 103.David B, Wolfender JL, Dias DA. The pharmaceutical industry and natural products: historical status and new trends. Phytochemistry Reviews. 2014;14:299–315. [Google Scholar]
- 104.Kellogg JJ, Todd DA, Egan JM, Raja HA, Oberlies NH, Kvalheim OM, Cech NB. Biochemometrics for Natural Products Research: Comparison of Data Analysis Approaches and Application to Identification of Bioactive Compounds. J Nat Prod. 2016;79:376–386. doi: 10.1021/acs.jnatprod.5b01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bucar F, Wube A, Schmid M. Natural product isolation--how to get from biological material to pure compounds. Nat Prod Rep. 2013;30:525–545. doi: 10.1039/c3np20106f. [DOI] [PubMed] [Google Scholar]
- 106.Krunic A, Orjala J. Application of high-field NMR spectroscopy for characterization and quantitation of submilligram quantities of isolated natural products. Magn Reson Chem. 2015;53:1043–1050. doi: 10.1002/mrc.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Molinski TF. Nanomole-scale natural products discovery. Curr Opin Drug Discovery Dev. 2009;12:197–206. [PubMed] [Google Scholar]
- 108.Olson DL, Norcross JA, O'Neil-Johnson M, Molitor PF, Detlefsen DJ, Wilson AG, Peck TL. Microflow NMR: concepts and capabilities. Anal Chem. 2004;76:2966–2974. doi: 10.1021/ac035426l. [DOI] [PubMed] [Google Scholar]
- 109.Nicolaou KC, Snyder SA. Chasing molecules that were never there: Misassigned natural products and the role of chemical synthesis in modern structure elucidation. Angew Chem, Int Ed. 2005;44:1012–1044. doi: 10.1002/anie.200460864. [DOI] [PubMed] [Google Scholar]
- 110.Biemann K. High-resolution mass spectrometry of natural products. Pure Appl Chem. 1964;9:95–118. [Google Scholar]
- 111.Cheng KW, Wong CC, Wang M, He QY, Chen F. Identification and characterization of molecular targets of natural products by mass spectrometry. Mass Spectrom Rev. 2010;29:126–155. doi: 10.1002/mas.20235. [DOI] [PubMed] [Google Scholar]
- 112.Cooks RG, Ifa DR, Sharma G, Tadjimukhamedov FK, Ouyang Z. Perspectives and retrospectives in mass spectrometry: one view. Eur J Mass Spectrom. 2010;16:283–300. doi: 10.1255/ejms.1073. [DOI] [PubMed] [Google Scholar]
- 113.Biemann K. Structure Determination of Natural Products by Mass Spectrometry. Annu Rev Anal Chem. 2015;8:1–19. doi: 10.1146/annurev-anchem-071114-040110. [DOI] [PubMed] [Google Scholar]
- 114.Gaudencio SP, Pereira F. Dereplication: racing to speed up the natural products discovery process. Nat Prod Rep. 2015;32:779–810. doi: 10.1039/c4np00134f. [DOI] [PubMed] [Google Scholar]
- 115.Duarte IF, Godejohann M, Braumann U, Spraul M, Gil AM. Application of NMR spectroscopy and LC-NMR/MS to the identification of carbohydrates in beer. J Agric Food Chem. 2003;51:4847–4852. doi: 10.1021/jf030097j. [DOI] [PubMed] [Google Scholar]
- 116.Shockcor JP, Unger SE, Savina P, Nicholson JK, Lindon JC. Application of directly coupled LC-NMR-MS to the structural elucidation of metabolites of the HIV-1 reverse-transcriptase inhibitor BW935U83. J Chromatogr B Biomed Sci Appl. 2000;748:269–279. doi: 10.1016/s0378-4347(00)00360-1. [DOI] [PubMed] [Google Scholar]
- 117.Spraul M, Freund AS, Nast RE, Withers RS, Maas WE, Corcoran O. Advancing NMR sensitivity for LC-NMR-MS using a cryoflow probe: application to the analysis of acetaminophen metabolites in urine. Anal Chem. 2003;75:1536–1541. doi: 10.1021/ac026203i. [DOI] [PubMed] [Google Scholar]
- 118.Iwasaki Y, Nakano Y, Mochizuki K, Nomoto M, Takahashi Y, Ito R, Saito K, Nakazawa H. A new strategy for ionization enhancement by derivatization for mass spectrometry. J Chromatogr B: Anal Technol Biomed Life Sci. 2011;879:1159–1165. doi: 10.1016/j.jchromb.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 119.Santa T. Derivatization reagents in liquid chromatography/electrospray ionization tandem mass spectrometry. Biomed Chromatogr. 2011;25:1–10. doi: 10.1002/bmc.1548. [DOI] [PubMed] [Google Scholar]
- 120.Halket JM, Waterman D, Przyborowska AM, Patel RKP, Fraser PD, Bramley PM. Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J Exp Bot. 2005;56:219–243. doi: 10.1093/jxb/eri069. [DOI] [PubMed] [Google Scholar]
- 121.Halket JM, Zaikin VG. Derivatization in mass spectrometry-1. Silylation. Eur J Mass Spectrom. 2003;9:1–21. doi: 10.1255/ejms.527. [DOI] [PubMed] [Google Scholar]
- 122.Brattoli M, Cisternino E, Dambruoso PR, de Gennaro G, Giungato P, Mazzone A, Palmisani J, Tutino M. Gas chromatography analysis with olfactometric detection (GC-O) as a useful methodology for chemical characterization of odorous compounds. Sensors (Basel) 2013;13:16759–16800. doi: 10.3390/s131216759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gowda GA, Tayyari F, Ye T, Suryani Y, Wei S, Shanaiah N, Raftery D. Quantitative analysis of blood plasma metabolites using isotope enhanced NMR methods. Anal Chem. 2010;82:8983–8990. doi: 10.1021/ac101938w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lane AN, Fan TW, Xie Z, Moseley HN, Higashi RM. Isotopomer analysis of lipid biosynthesis by high resolution mass spectrometry and NMR. Anal Chim Acta. 2009;651:201–208. doi: 10.1016/j.aca.2009.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lane AN, Fan TWM, Bousamra M, Higashi RM, Yan J, Miller DM. Stable Isotope-Resolved Metabolomics (SIRM) in Cancer Res. with Clinical Application to NonSmall Cell Lung Cancer. OMICS: A Journal of Integrative Biology. 2011;15:173–182. doi: 10.1089/omi.2010.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fan TWM, Lane AN. Applications of NMR spectroscopy to systems biochemistry. Prog Nucl Magn Reson Spectrosc. 2016;92-93:18–53. doi: 10.1016/j.pnmrs.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fan TWM, Lane AN, Higashi RM, Farag MA, Gao H, Bousamra M, Miller DM. Altered regulation of metabolic pathways in human lung cancer discerned by (13)C stable isotope-resolved metabolomics (SIRM) Mol Cancer. 2009;8:41. doi: 10.1186/1476-4598-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fan TWM, Yuan P, Lane AN, Higashi RM, Wang Y, Hamidi AB, Zhou R, Guitart X, Chen G, Manji HK, Kaddurah-Daouk R. Stable isotope-resolved metabolomic analysis of lithium effects on glial-neuronal metabolism and interactions. Metabolomics. 2010;6:165–179. doi: 10.1007/s11306-010-0208-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Higashi RM, Fan TWM, Lorkiewicz PK, Moseley HNB, Lane AN. Stable Isotope-Labeled Tracers for Metabolic Pathway Elucidation by GC-MS and FT-MS. 2014;1198:147–167. doi: 10.1007/978-1-4939-1258-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lane AN, Higashi RM, Fan TWM. Preclinical models for interrogating drug action in human cancers using Stable Isotope Resolved Metabolomics (SIRM) Metabolomics. 2016;12:1–15. doi: 10.1007/s11306-016-1065-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fan TWM, Lane AN, Higashi RM, Yan J. Stable isotope resolved metabolomics of lung cancer in a SCID mouse model. Metabolomics. 2011;7:257–269. doi: 10.1007/s11306-010-0249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fan TWM, Lorkiewicz PK, Sellers K, Moseley HNB, Higashi RM, Lane AN. Stable isotope-resolved metabolomics and applications for drug development. Pharmacol Ther. 2012;133:366–391. doi: 10.1016/j.pharmthera.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Moseley HN. Correcting for the effects of natural abundance in stable isotope resolved metabolomics experiments involving ultra-high resolution mass spectrometry. BMC Bioinf. 2010;11:139. doi: 10.1186/1471-2105-11-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Callebaut W. Scientific perspectivism: A philosopher of science's response to the challenge of big data biology. Stud Hist Philos Biol Biomed Sci. 2012;43:69–80. doi: 10.1016/j.shpsc.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 135.Dolinski K, Troyanskaya OG. Implications of Big Data for cell biology. Mol Biol Cell. 2015;26:2575–2578. doi: 10.1091/mbc.E13-12-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gligorijevic V, Malod-Dognin N, Przulj N. Integrative methods for analyzing big data in precision medicine. Proteomics. 2016;16:741–758. doi: 10.1002/pmic.201500396. [DOI] [PubMed] [Google Scholar]
- 137.Tenenbaum JD, Sansone SA, Haendel M. A sea of standards for omics data: sink or swim? J Am Med Inform Assoc. 2014;21:200–203. doi: 10.1136/amiajnl-2013-002066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Anagnostopoulos I, Zeadally S, Exposito E. Handling big data: research challenges and future directions. The Journal of Supercomputing. 2016;72:1494–1516. [Google Scholar]
- 139.Robinette SL, Zhang F, Bruschweiler-Li L, Bruschweiler R. Web Server Based Complex Mixture Analysis by NMR. Anal Chem. 2008;80:3606–3611. doi: 10.1021/ac702530t. [DOI] [PubMed] [Google Scholar]
- 140.Pence HE, Williams A. ChemSpider: An Online Chemical Information Resource. J Chem Educ. 2010;87:1123–1124. [Google Scholar]
- 141.Livera AMD, Sysi-Aho M, Jacob L, Gagnon-Bartsch JA, Castillo S, Simpson JA, Speed TP. Statistical Methods for Handling Unwanted Variation in Metabolomics Data. Anal Chem. 2015;87:3606–3615. doi: 10.1021/ac502439y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wold S, S W, Hellbern S, Lundstedt T, Siostrom M. PLS modeling with latent variables in two or more dimensions. Proceedings PLS Model Building, Theory and Applications Symposium Frankfurt am Main; September 23-25, 1987. [Google Scholar]
- 143.Smilde AK, Westerhuis JA, de Jong S. A framework for sequential multiblock component methods. J Chemom. 2003;17:323–337. [Google Scholar]
- 144.Westerhuis JA, Kourti T, Macgregor JF. Analysis of multiblock and hierarchical PCA and PLS models. J Chemom. 1998;12:301–321. [Google Scholar]
- 145.Befroy DE, Perry RJ, Jain N, Dufour S, Cline GW, Trimmer JK, Brosnan J, Rothman DL, Petersen KF, Shulman GI. Direct assessment of hepatic mitochondrial oxidative and anaplerotic fluxes in humans using dynamic 13C magnetic resonance spectroscopy. Nat Med. 2014;20:98–102. doi: 10.1038/nm.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]