

Abstract
Chimeric antigen receptor (CAR)-modified T cells with anti-CD19 specificity are a highly effective novel immune therapy for relapsed/refractory acute lymphoblastic leukemia (ALL). Cytokine release syndrome (CRS) is the most significant and life-threatening toxicity. To improve understanding of CRS, we measured cytokines and clinical biomarkers in 51 CTL019-treated patients. Peak levels of 24 cytokines, including IFNγ, IL6, sgp130, and sIL6R in the first month after infusion were highly associated with severe CRS. Using regression modeling, we could accurately predict which patients would develop severe CRS with a signature composed of three cytokines. Results validated in an independent cohort. Changes in serum biochemical markers, including C-reactive protein and ferritin, were associated with CRS but failed to predict development of severe CRS. These comprehensive profiling data provide novel insights into CRS biology, and importantly represent the first data that can accurately predict which patients have a high probability of becoming critically ill.
Keywords: Cytokine, tocilizumab, chimeric antigen receptor, acute lymphoblastic leukemia, biomarker
Introduction
Chimeric antigen receptor (CAR)-modified T cells with specificity against CD19 have demonstrated considerable promise against highly refractory hematologic malignancies. Dramatic clinical responses with complete remission rates as high as 90% have been reported in children and adults with relapsed/refractory acute lymphoblastic leukemia (ALL).(1–4) At our center, patients have been treated with CTL019, engineered T cells composed of an anti-CD19 single chain variable fragment (scFv), CD3ζ activation domain, and 4-1BB costimulatory domain. Marked in vivo CAR T-cell proliferation (100 to 100,000×) leads to efficacy, but can lead to toxicity, including cytokine release syndrome (CRS).(2) CRS is the most common potentially severe toxicity associated with CAR T cells.(1–5) CRS is not unique to CAR T cells and occurs with other therapies that engage T cells to kill cancer cells, including bi-specific T-cell engaging (BiTE™) antibodies such as blinatumomab.(6, 7)
Despite the frequency of CRS post-infusion of CAR T cells, relatively little is known about the underlying biology of the syndrome. Improved understanding of CRS may lead to better recognition, improved treatment, and perhaps the ability to prevent or abrogate the most serious complications of CRS. The ability to predict which patients will become critically ill with severe CRS is vital to the development of CAR T cell therapy, yet there are no published accurate predictors for severe CRS. Our group previously demonstrated that CRS can be successfully ameliorated with the IL6R inhibitor, tocilizumab, and its use has become commonplace after T-cell engaging therapies by our group and others.(1–4) Despite its efficacy, the mechanism of tocilizumab in alleviating CRS remains poorly defined. Currently, tocilizumab is used to treat CRS after symptoms become severe. It is unknown whether tocilizumab can prevent CRS or, if used too early, could decrease the efficacy of the CAR T cells.
To better characterize and potentially predict CRS, we evaluated data from 39 children and 12 adults with refractory/relapsed ALL treated with CTL019. We obtained clinical and comprehensive biomarker data, measuring 43 different cytokines, chemokines, and soluble receptors (hereafter collectively called ‘cytokines’) as well as a number of other laboratory markers. Serial measurements from these patients allowed us to make a number of novel observations that improve our understanding of the biology of CRS and will directly affect clinical practice.
Key results to be discussed herein include: (1) a prediction model for severe CRS; (2) an overall description of the timing and pattern of cytokine rise and fall after treatment with CAR T cells; (3) a comprehensive comparison of cytokine profiles between patients who develop severe CRS versus not, which reveals significant details of the underlying biology of severe CRS; (4) analysis showing that patients who develop severe CRS develop clinical, laboratory, and cytokine profiles that mirror hematophagocytic lymphohistiocytosis (HLH)/macrophage activation syndrome (MAS); and, (5) a characterization of the effects of tocilizumab on CRS, establishing the toxicity of CRS is mediated by trans-IL6 signaling that is rapidly abrogated after tocilizumab treatment in the majority of patients.
Results
Clinical Description of Patients
A total of 51 patients with ALL, 39 patients in the pediatric cohort, age 5–23, and 12 in the adult cohort, age 25–72, were treated at CHOP and PENN, respectively (Supplemental Table 5). The two cohorts were defined based on the clinical trials and treating institutions (see Supplemental Methods). 47 patients (37 pediatric; 10 adults) had B ALL in first to fourth relapse, 1 child had relapsed T-ALL with aberrant CD19 expression, and 3 patients (1 pediatric; 2 adults) had primary refractory B-ALL. 31 patients (27 pediatric; 4 adults) (61%) had relapsed after prior allogeneic hematopoietic stem cell transplant (SCT). 4 patients (all pediatric) had previously been treated with blinatumomab, a CD19 BITE antibody. No patient was treated with any other CD19-directed therapy prior to CTL019. Data on response to CTL019 in the first 30 patients (25 children and 5 adults) were recently published, demonstrating a 90% complete remission (CR) rate and 6 month event free survival (EFS) of 67%.(2)
Clinical Description of Cytokine Release Syndrome (CRS)
48 of 51 patients (94%) developed CRS; the three that did not were children. Patients with CRS typically presented with flu-like illness. The majority of patients developed mild (grade 1–2) (18/51; 35%) to moderate (grade 3) CRS (16/51; 31%), and 14 patients (27%) developed severe (grade 4–5) CRS (12 grade 4 and 2 grade 5) (Table 1). For patients who developed fever, start of CRS was defined as the day with the first fever >= 38.0°C (100.5°F) relative to infusion of CTL019. Stop of CRS was defined as 24 hours without fever or vasoactive medications, indicating recovery from shock. Four patients developed CRS without fever: start and stop of CRS were defined based on the first day with flu-like symptoms and the first 24-hour period without symptoms, respectively. Additional details are included in Table 1 and Supplemental Results.
Table 1.
Clinical factors related to cytokine release syndrome, by grade (N = 48).
| Clinical Factor | Total Cohort (N = 48) |
Total Cohort | Children (Age <25) | Adults (Age ≥25) | |||
|---|---|---|---|---|---|---|---|
| Grade 1–3 (N = 34) |
Grade 4–5 (N = 14) |
Grade 1–3 (N = 25) |
Grade 4–5 (N = 11) |
Grade 1–3 (N = 9) |
Grade 4–5 (N = 3) |
||
| Days to CRS: Start | 1 (0 – 10) | 2.5 (0 – 10) | 1 (0 – 6) | 4 (0 – 10) * | 1 (0 – 2) * | 1 (0 – 7) | 1 (0 – 6) |
| Stop | 10 (5 – 24) | 9 (5 – 24) | 11.5 (5 – 16) | 9 (5 – 24) * | 11 (9 – 15) * | 10 (6 – 20) | 15 (5 – 16) |
| Days to Fever (N=41): Start | 1 (0 – 10) | 2 (0 – 10) | 1 (0 – 6) | 4 (0 – 10) | 1 (0 – 2) | 1 (0 – 7) | 2 (0 – 6) |
| Stop | 8 (5 – 23) | 8 (5 – 23) | 9 (5 – 15) | 7 (5 – 23) | 8 (5 – 12) | 8 (6 – 13) | 13 (5 – 15) |
| Total Days Febrile | 6 (1 – 17) | 6 (1 – 17) | 8 (3 – 13) | 5 (1 –17) | 7 (3 – 12) | 6.5 (6 – 10) | 9 (3 – 13) |
| Intubation Yes | 9 (19%) | 0 (0%) | 9 (64%) | 0 (0%) | 6 (55%) | 0 (0%) | 3 (100%) |
| No | 39 (81%) | 34 (100%) ** | 5 (36%) ** | 25 (100%) ** | 5 (45%) ** | 9 (100%) * | 0 (0%) * |
| Days to Intubation: Start | 6 (3 – 13) | 6 (3 – 13) | 6 (4 – 10) | 10 (3 – 13) | |||
| Stop | 15 (5 – 66) | 15 (5 – 66) | 15 (10 – 66) | 15 (5 –16) | |||
| Vasoactives Yes | 20 (42%) | 6 (18%) | 14 (100%) | 3 (12%) | 11 (100%) | 3 (33%) | 3 (100%) |
| No | 28 (58%) | 28 (82%) ** | 0 (0%) ** | 22 (88%) ** | 0 (0%) ** | 6 (67%) | 0 (0%) |
| Days to Vasoactives: Start | 5 (1 – 13) | 4.5 (1 – 7) | 5 (2 – 13) | 7 (2 – 7) | 5 (2 – 10) | 3 (1 – 6) | 10 (4 – 13) |
| Stop | 10 (2 – 16) | 8 (2 – 10) | 10.5 (5 – 16) | 9 (8 – 10) | 10 (8 – 14) | 5 (2 – 8) | 15 (5 – 16) |
| Encephalopathy Yes | 13 (27%) | 5 (15%) | 8 (57%) | 5 (20%) | 8 (73%) | 0 (0%) | 0 (0%) |
| No | 34 (71%) | 29 (85%) * | 5 (36%) * | 20 (80%) * | 2 (18%) * | 9 (100%) | 3 (100%) |
| Unknown | 1 (2%) | 0 (0%) | 1 (7%) | 0 (0%) | 1 (9%) | ||
| Infection Yes | 6 (13%) | 2 (6%) | 4 (29%) | 2 (8%) | 2 (18%) | 0 (0%) | 2 (67%) |
| No | 42 (88%) | 32 (94%) | 10 (71%) | 23 (92%) | 9 (82%) | 9 (100%) | 1 (33%) |
Continuous variables are summarized as median (range), categorical variables as number (percent). Event timing is provided as time since infusion in days p < 0.05 (bold);
p < 0.01;
p < 0.001.
Days: exact Wilcoxon; categorical: Fisher exact (Unknown category not included in calculation).
Nine patients required mechanical ventilation and twenty patients required vasoactive medications for either distributive (19/20) or cardiogenic shock (1/20). Fourteen patients required high dose vasoactives as defined in the Supplemental Table 1B. Only 6 patients developed a documented co-morbid infection and only 2 of these infections, both in adults, were clinically consistent with sepsis (Supplemental Results and Supplemental Table 6). Clinical factors related to CRS are summarized in Table 1. Three adults died in the first 30 days after CTL019 treatment. Some children with severe CRS developed organomegaly, including hepato- and splenomegaly and a number of patients developed encephalopathy. Additional details are provided in the Supplemental Results and Supplemental Table 4.
Laboratory Description of CRS
We serially evaluated laboratory markers of inflammation and organ failure in patients treated with CTL019 (see Supplemental Table 2 for time points). Baseline ferritins (N=48) were elevated in the majority of patients (median: 1580 mg/dl, range 232–14,673) as a consequence of systemic inflammation and/or iron overload. Only six children out of the 37 measured and no adults had baseline ferritins <500mg/dl. Peak ferritins (defined as highest value in first month after CTL109 infusion) were very high in all patients regardless of grade but the median was significantly higher in patients with grade 4–5 CRS (p <0.001): grade 0–3 CRS (median 8,290mg/dl, range 280 – 411,936) and grade 4–5 CRS (median 130,000mg/dl, range 11,200 – 299,000). Similar trends were seen in adults and children (Table 2). All patients with grade 4–5 CRS had a peak ferritin >10,000mg/dl, a value that is considered sensitive and specific for macrophage activation/HLH syndrome in children.(8, 9) Thirty patients, including 20 of 39 (51%) children and 10 of 12 (83%) adults with grade 0–3 CRS had a peak ferritin >10,000mg/dl.
Table 2.
Clinical biomarkers related to cytokine release syndrome, by grade (N = 51). Unless otherwise noted, the median (range) for the peak observed value over one month is displayed
| Biomarker | Total Cohort (N = 51) |
Total Cohort | Children | Adults | |||
|---|---|---|---|---|---|---|---|
| Grade 0–3 (N = 37) |
Grade 4–5 (N = 14) |
Grade 0–3 (N = 28) |
Grade 4–5 (N = 11) |
Grade 1–3 (N = 9) |
Grade 4–5 (N = 3) |
||
| Ferritin (ng/ml) | 23,439 (280 – 411,936) | 8,290 (280 – 411,936)**§ | 130,000C (11,200 – 299,000)**§ | 4,455 (280 – 411,936) ** | 123,000 (11,200–299,000)** | 35,246 (2,191–220,657)§ | >100,000, 266,190, 234,811†§ |
| LDH ‡ (U/L) | 1,673 (158 – 23,778) | 852 (158 – 19,104) * | 5,594 (1,648 – 23,778) * | 881 (545 – 19,104) * | 5,898 (1,697–23,778)* | 679 (158 – 9,581) | 1,902 (1,648 – 7,386) |
| CRP ‡ (mg/dl) | 20.1C (0.7 – 56.5) | 16.2 (0.7 – 56.5)*§ | 22.9C (>16.0 – 37.1) *§ | 14.7 (0.7 – 56.5) * | 26.6 (20.2 – 37.1) * | 22.4 (12.2 – 32.0)§ | >16, 17.1, 22.9 §† |
| ALT (U/L) | 107 (26 – 1238) | 95 (26 – 748) | 198 (50 – 1,238) | 82 (29 – 748) | 190 (50 – 1,238) | 146 (26 – 396) | 284 (84 –504) |
| AST (U/L) | 149 (19 – 1,427) | 103 (19 – 1,424)** | 389 (98 – 1,427) ** | 98 (35 – 1,060) ** | 318 (119 – 1,427) ** | 103 (19 – 1,424) | 867 (98 – 938) |
| BUN (mg/dl) | 18 (9 – 202) | 17 (9 – 72) ** | 44 (12 – 202) ** | 17 (9 – 54) * | 42 (12 – 202) * | 17 (11 – 72) | 55 (31 – 61) |
| Cr (mg/dl) | 0.7 (0.2 – 5) | 0.7 (0.2 – 4) * | 1 (0.4 – 5) * | 0.5 (0.2 – 1.7) * | 0.9 (0.4 – 3.4) * | 0.9 (0.7 – 4) | 1.8 (1.04 – 4.95) |
| Fibrinogen min ‡ (mg/dl) <150 N (%) | 18/36 (50%) | 7/23 (30%) * | 11/13 (85%) * | 2/15 (13%) ** | 9/10 (90%) ** | 5/8 (63%) | 2/3 (67%) |
| Fibrinogen min ‡ (mg/dl) | 159 (23 – 624) | 241 (23 – 624) ** | 77 (23 – 221) ** | 288 (93 – 624) ** | 76 (23 – 221)** | 124 (23 – 278) | 128 (64 – 175) |
| PT (sec) | 17.1 (13.5 – 45.4) | 15.7 (13.5 – 27.5) ** | 23.1 (17.1 – 45.4) ** | 15.1 (13.5 – 20.6) ** | 24.4 (17.1 – 33.7) ** | 18.0 (14.5 – 27.5) | 19.3 (17.7 – 45.4) |
| PTT (sec) | 44.1 (25.7 – 92.0) | 39.8 (25.7 – 64.4) * | 54.2 (37.2 – 92.0) * | 34.4 (25.7 – 64.4) ** | 58.1 (40.6 – 92.0) ** | 44.1 (32.4 – 58.1) | 43.5 (37.2 – 52.2) |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; BUN = Blood urea nitrogen; Cr = Creatinine; CRP = C-Reactive Protein; LDH = Lactate dehydrogenase; PT = prothrombin time; PTT = partial thromboplastin time
p< 0.05 (bold);
p < 0.01,
p < 0.001. Peaks or min: exact Wilcoxon for continuous; Fisher exact for discrete
Wilcoxon test for right-censored data
Total N = 50, 49, 36, 39 and 38 for LDH, CRP, Fibrinogen, PT and PTT respectively
Actual values
Estimated from Kaplan-Meier curve due to censored data at the upper limit of detection
Baseline CRP was elevated in a majority of patients (median 1.20 mg/dl, range 0.12–29.4). Three children and 2 adults did not have CRP tested at baseline. 24 of 36 children (67%) and 1 of 10 adults had a baseline CRP >1mg/dl. Similar to ferritin, 1-month peak CRP was very high in the majority of patients with grade 4–5 CRS (median 22.9, range 16.0–37.1) and grade 0–3 CRS (median 16.2, range 0.7–56.5), with a statistically significant median difference in grade 4–5 versus grade 0–3 CRS (p=0.010). Similar trends were seen in adults and children (Table 2). Consistent with generalizable inflammation and hypotension, ALT, AST, BUN, LDH and Cr markedly increased in the majority of patients with CRS with a statistically significant increase in grade 4–5 versus 0–3 CRS (Table 2). While peak values of these clinical labs correlated with severity of CRS, none of these labs were helpful at predicting CRS in the first 3 days. Early CRP elevation was associated with grade 4–5 CRS (p=0.02), but, contrary to another published report,(3) we did not find early assessment of CRP in the first three days following CTL019 infusion was useful in predicting severity of CRS (AUC=0.73). For example, considering CRP as a screen for high-risk cases, a CRP >6.8mg/dl would have identified only 72% of the cases and had a positive predictive value (PPV) of 43%. Similarly, early ferritin elevation was associated with grade 4–5 CRS, but it was not useful in predicting CRS. Additional details on CRP and ferritin are included in Supplemental Figure 1 and Supplemental Results.
Fibrinogen <150mg/dl is used in the diagnostic criteria for HLH as it is a sensitive marker of the syndrome.(10) We found a strong association with low fibrinogen and grade 4 CRS in the pediatric cohort but not adults (Table 2). Children became mildly coagulopathic with more significant coagulopathy with severe CRS (Table 2). Adults also developed hypofibrinogenemia and mild coagulopathy; however, this was seen across CRS grades (Table 2). Additional details are provided in Supplemental Results. While bleeding was rare, understanding the coagulopathy has direct clinical implications as many of the patients required cryoprecipitate in addition to fresh frozen plasma to maintain hemostasis.
Cytokine profiles
In order to understand the biology of CRS after CAR T cell therapy, we performed serial cytokine assessment on the 51 patients. We compared median baseline values from 50 patients with ALL (one subject did not have a baseline value) with a 10-patient normal donor cohort (Supplemental Table 7). Of note, we found a number of cytokines, including sIL2Rα and MCP1 were consistently elevated in most patients with ALL compared to the normal donors and significant by Holm’s adjusted p-value. We determined if certain cytokines were associated with baseline disease burden in children (bone marrows were not collected at the time of infusion in many adults). Only EGF, IL12 and IL13 were associated with higher disease burden at baseline by Holm’s p-value (Supplemental Table 8).
We compared cytokine profiles in patients who had severe CRS with patients who did not. We found peak levels of 24 cytokines, including IFNγ, IL6, IL8, sIL2Rα, sgp130, sIL6R, MCP1, MIP1α, MIP1β, and GM-CSF sent in the first month after CTL019 were associated with grade 4–5 CRS compared to grade 0–3 CRS and significant by the Holm’s-adjusted p-value (Supplemental Table 9 and Figure 1[panels a and b]). As a sensitivity analysis, we repeated this analysis with a reduced set of cytokine measures, keeping only one measure per target assessment window specified in the protocol, to equalize the number of measurements between subjects (see Supplemental Methods and Supplemental Table 10). Results were similar, 23 cytokines were significant –all amongst the previously found 24. Only IL1RA, the weakest significant result of the original 24, did not remain significant. Supplemental Table 9 also presents the median peak cytokine values for the pediatric and adult cohorts, separately.
Figure 1. Cytokines associated with severe CRS.
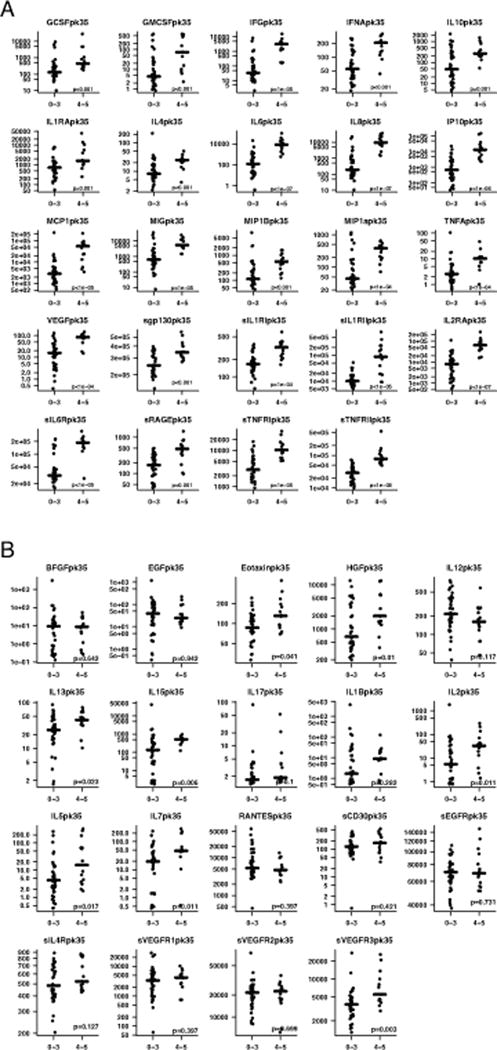
Serial cytokine assessment of 43 cytokines was performed in 51 patients treated with CTL019. Cytokine profiles were compared in patients who developed severe CRS with patients who did not. Figure 1 depicts peak values of cytokines over the first month. (A). 24 cytokines, including IFNγ, IL6, IL8, sIL2Rα, sgp130, sIL6R, MCP1, MIP1α, and GM-CSF were highly associated with CRS4-5 compared to CRS0-3, significant by Holm’s adjusted p-value. (B). The 19 cytokines that were not statistically different by CRS severity based on Holm’s adjusted p-value.
Certain cytokines peaked earlier than others in patients with severe CRS. Understanding the timing of the rise and fall improves understanding of not only the underlying biology but also has potential therapeutic relevance. IFNγ and sgp130, for example, rise very early. These two cytokines were the only ones differently elevated for severe versus non-severe CRS in the first 3 days after infusion and prior to patients becoming critically ill after adjustment for multiple comparisons (Holm’s) (Supplemental Figure 2, Supplemental Table 11). In contrast, while IL6 is the cytokine most strongly associated with severe CRS over the first month early IL6 levels (days 0–3) were not different by CRS after adjustment for multiple comparisons. Supplemental Table 11 summarizes the peak value of all cytokines in the first 3 days after infusion by CRS severity.
We found severity of CRS was weakly associated with the peak CAR T cell expansion by copies/microgram qPCR over one month (p=0.058); however the peak in the first 3 days post infusion was not associated with CRS severity. (Supplemental Figure 3 [panels a and b])
Predictive modeling
We developed and analyzed 16 predictive models (8 from combined cohort and 8 from pediatric only cohort) fit based on data from the main (discovery) cohort of 51 patients (Supplemental Table 12). Table 3 lists the best overall regression models and decision tree models for both the combined and pediatric only cohorts. With the forward-selected logistic regression model, we accurately predicted which patients developed grade 4–5 CRS using IFNγ, sgp130, and sIL1RA with sensitivity 86% (95% CI: 57, 98), specificity 89% (95% CI: 73, 97), and AUC=0.93 (95% CI: 0.86, 1.0) (Figure 2a and Table 3). Using a decision tree a combination of sgp130, MCP1, and Eotaxin had sensitivity 86% (95% CI: 57, 98) and specificity 97% (95% CI: 85, 100) (Figure 2b and Table 3). For the pediatric cohort, the modeling was even more accurate; the forward-selected logistic regression model including IFNγ, IL13, and MIP1α had sensitivity 100% (95% CI: 72, 100), specificity 96% (95% CI: 81, 100) (AUC=0.98; 95% CI: 0.93, 1.0) (Figure 2c and Table 3). Our group and others have previously published that disease burden prior to infusion can predict severe CRS.(2, 3) In the pediatric cohort only, a bone marrow aspirate was collected immediately prior to infusion. We found disease burden did not improve the predictive accuracy of the models over the cytokines alone using regression modeling; however, it was identified as an important predictive variable in the pediatric cohort using the decision tree modeling. A combination of IL10 and disease burden had sensitivity 91% (95% CI: 59, 100), and specificity 96% (95% CI: 81,100) (Figure 2d and Table 3). As many trials are not measuring disease burden, we note that a classifier build on predictors from our top candidate logistic model included a combination of IFNγ and MIP1α and had sensitivity 82% (95% CI: 48, 98) and specificity 93% (95% CI: 76, 99) (Figure 2e and Table 3). Supplemental Results include examples of application of the models and further discussion of the strengths and weaknesses of the different models. Supplemental Figure 4 (panels a–d) depicts the best single and 2 variable regression models for CRS prediction.
Table 3.
| Model+ | Equation | Discovery Accuracy | Validation accuracy |
|---|---|---|---|
| A: Top logistic regression model for the combined cohort | Predict low risk if expit{13.8712*log10(sgp130Pk3) + 2.4626*log10(IFNγPk3) −1.6559*log10(IL1RAPk3)-75.3502} ≤ 0.3623 | Sens=12/14 Spec=31/35 |
Sens=2/2 Spec=6/10 |
| B: Top tree model for combined cohort | Predict low risk if sgp130pk3 < 218179 or failing that if MCP1Pk3<4636.52 & EotaxinPk3>29.09 | Sens= 12/14 Spec=34/35 |
Sens=2/2 Spec =7/10 |
| C: Top logistic regression model for the pediatric cohort | Predict low risk if expit{ 8.483*log10(IFNγPk3) - 5.599*log10(IL13Pk3) - 16.343*log10(MIP1αPk3) +15.742} ≤ 0.3288 | Sens=11/11 Spec=26/27 |
Sens=1/2 Spec=8/10 |
| D: Top tree model for pediatric cohort | Predict low risk if IL10Pk3 < 11.7457, or failing that if burden <51% | Sens=10/11 Spec=26/27 |
Sens=2/2 Spec=9/10 |
| E: Best pediatric classifier using covariates from top pediatric regression model | Predict low risk if IFNγPk3<27.6732 or failing that if MIP1αPk3>=30.1591 and IFNγPk3 < 94.8873 | Sens=9/11 Spec=25/27 |
Sens=1/2 Spec=10/10 |
The expit function converts the logistic regression score to the modeled probability of being a case. expit(x) = exp(x)/{exp(x) + 1}; Pk=peak; sens = sensitivity; spec = specificity
The designation of top regression model considered the overall accuracy in the sensitivity and specificity (prioritizing sensitivity) in the discovery cohort, and preferring models with no fold change factors unless there was an appreciable increase in accuracy with those factors added since they required an extra (baseline) measurement for the cytokines, which did not occur. Supplemental Table 12 provides the full list of models considered. The best tree (classifier) model for the combined and pediatric cohorts stood out as simultaneously maximizing the sensitivity and specificity among the candidate tree models in these respective discovery cohorts. Additionally, in the pediatric cohort we considered the best classifier that did not use burden (model E), a clinical factor that may not be available in some trials.
Figure 2. Cytokine profiles can predict severe CRS.
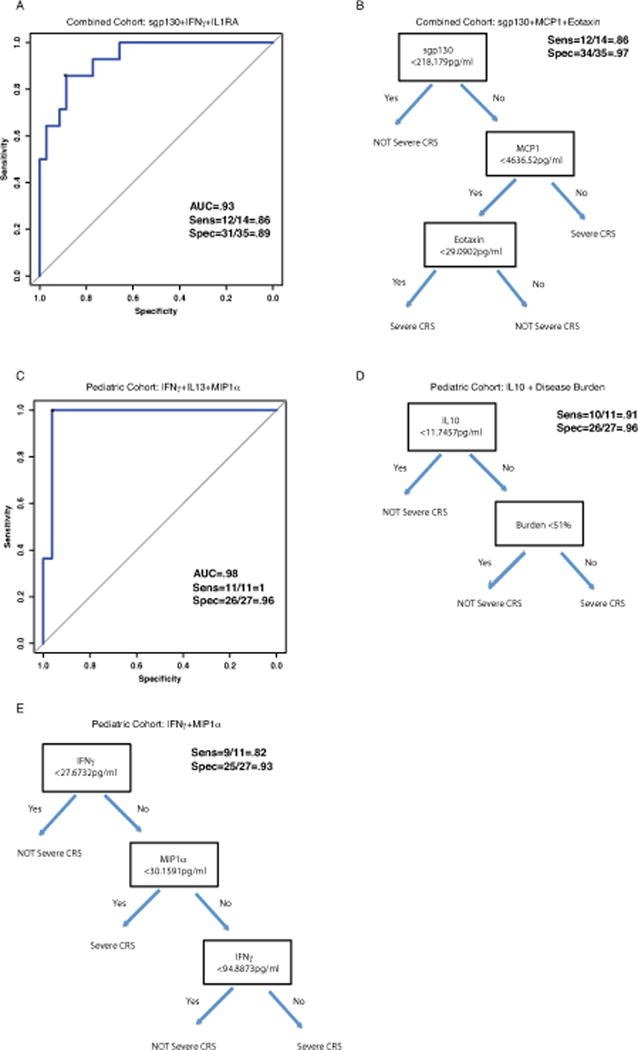
Cytokines were analyzed from the first 3 days after infusion, sent before patients developed severe CRS. Logistic and classification tree models were used to identify predictors of severe CRS. With a 3-variable regression model, found by forward selection, we accurately predicted which patients developed severe CRS using IFNγ, sgp130, and IL1RA. Figure 2A depicts ROC curve for the 3-variable regression models in the combined cohort. With a decision tree model, we accurately predicted which patients developed severe CRS using sgp130, MCP1, and Eotaxin (Figure 2B). For the pediatric cohort, using a 3-variable regression model we accurately predicted severe CRS with a combination of IFNγ, IL13, and MIP1α (Figure 2C). In the pediatric cohort only, a bone marrow aspirate was collected immediately prior to infusion. We found disease burden was associated with CRS severity but did not improve the predictive accuracy of the models over the cytokines alone. A combination of a single cytokine, IL10 and disease burden using decision tree modeling was very accurate for CRS prediction (Figure 2D). Without disease burden, using decision tree modeling a combination of IFNγ and MIP1α was accurate in the pediatric only cohort (Figure 2E).
We then tested the accuracy of the models using our validation cohort of 12 additional pediatric patients. Clinical details on the 12 patients in the validation cohort are listed in Supplemental Table 13. We found all of the models performed extremely well in the validation cohort (s). Thus, the validation cohort did not change the model rank order.
Hemophagocytic syndrome/Macrophage activation syndrome as a consequence of CTL019
19 cytokines studied in our patients have been studied in children with HLH.(11–14) We found a nearly identical pattern of those cytokines having 1-month peak values differentially elevated in HLH patients also elevated in patients with versus without severe CRS (Supplemental Table 9, Figure 3 [panels a and b]). There were statistically significant differences in IFNγ, IL10, sIL2Rα, IL6, IL8, IP10, MCP1, MIG and MIP1β in CRS grade 4–5 vs CRS grades 0–3. All of these cytokines are expected to be elevated in patients with HLH.(11–14) No significant differences by Holm’s adjusted p-value were seen in IL1β, IL2, IL5, IL7, IL12, IL13, and IL17, cytokines expected to be normal in HLH based on published work.(11–14) GM-CSF and TNFα were differentially elevated in our study in those with severe CRS. GM-CSF and TNFα have been demonstrated to be elevated with HLH in some studies while normal in others.(13, 15–17) IL4 is typically normal in patients with HLH.(11, 12) It was differentially elevated in our study in the patients with severe CRS, although the levels were low in both groups.
Figure 3. Severe CRS has a cytokine pattern that mirrors hemophagocytic syndrome.
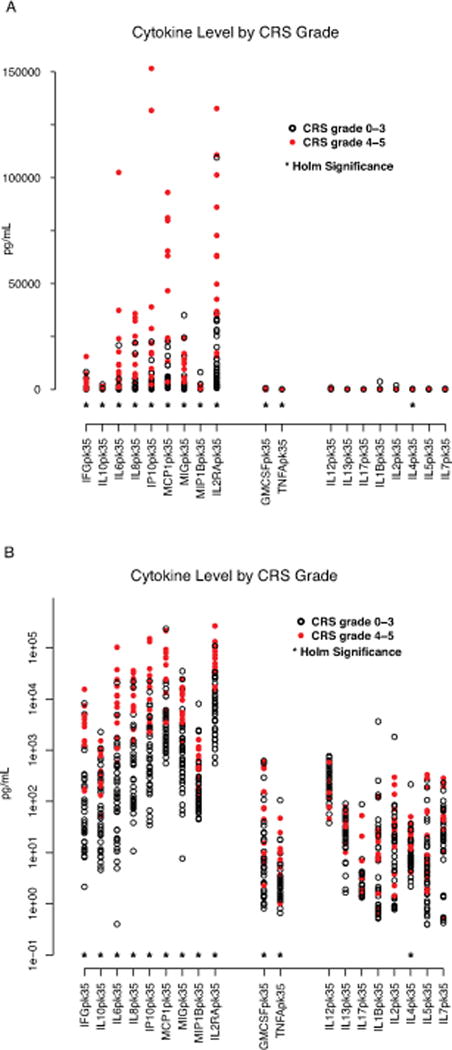
19 of the tested cytokines have previously been studied in children macrophage activation syndrome (MAS)/hemophagocytic syndrome (HLH). A near identical pattern of cytokines differentially elevated in HLH were also elevated in patients with CRS4-5 compared with CRS0-3. Figure 3 depicts cytokines are clustered into three groups. Those on left, including IFNγ, IL10, IL6, IL8, IP10, MCP1, MIP1β, and IL2Rα are expected to be elevated in HLH and were also found to be differently elevated in patients with severe CRS. Those in middle, including TNFα and GM-CSF have been found to be elevated in some patients with HLH and normal in others. Those on the right are cytokines expected to be normal in HLH. * = statistically significant by Holm’s adjustment. (A) Data presented in linear scale. (B) Data presented in log 10 scale.
IL6 signaling and IL6 directed therapy
21 patients were treated with tocilizumab for CRS. 7 of 15 subjects with grade 3 CRS and all 14 subjects with grade 4–5 CRS received tocilizumab. 10 of the 21 subjects (4 pediatric; 6 adults) received more than one dose. 12 patients were also treated with corticosteroids and 2 patients received etanercept. No patient received siltuximab (see Supplemental Table 14). Tocilizumab was given a median of 5 days after infusion with CTL019 (range 2 to 12 days). Supplemental Table 15 details the time to initiation of tocilizumab relative to infusion, first fever, use of vasoactives, and intubation, if applicable. Response to tocilizumab was rapid. Many patients became afebrile immediately after the first dose. Most patients were able to wean vasoactives over the 24–36 hours after receiving tocilizumab and they were stopped a median of 4.5 days after tocilizumab was given (Supplemental Table 15). While all of the children with CRS survived and responded to tocilizumab, 3 adults treated with CTL019 died.
Figure 4 depicts the levels of 4 cytokines (IFNγ, IL6, sIL6R, and sgp130) over time in the 14 grade 4–5 subjects treated with tocilizumab. After first dose of tocilizumab, there was generally a transient rise in IL6 levels, followed by a rapid decrease. sIL6R generally increased and continued to remain elevated for at least 2–3 weeks after tocilizumab, and sgp130 appeared to increase in some patients but not in others after tocilizumab (see Figure 4 for additional details).
Figure 4. Tocilizumab improves hypercytokinemia in patients with severe CRS.
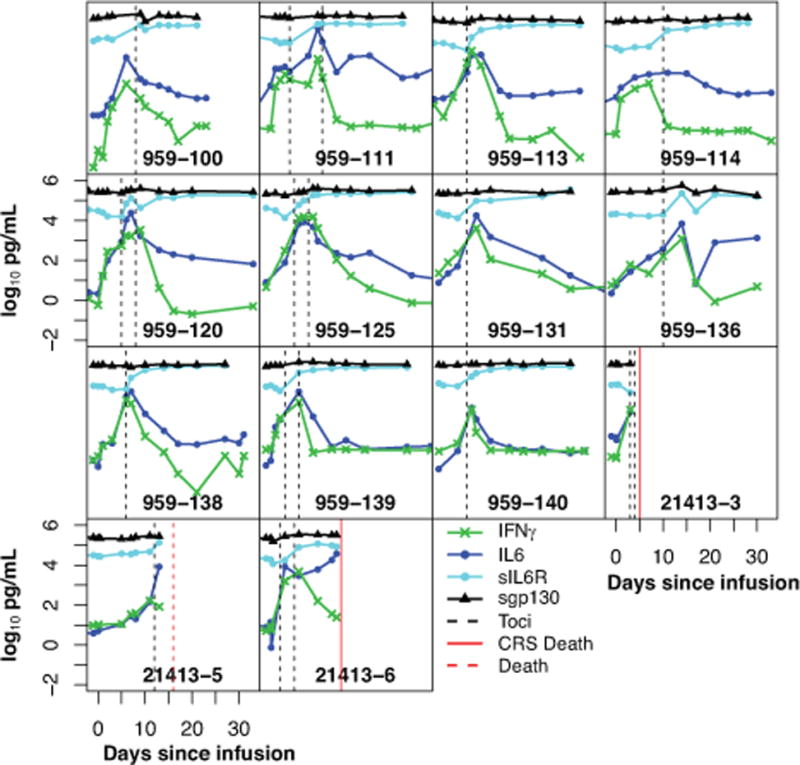
14 of 51 patients developed severe CRS and all were treated with tocilizumab. Cytokines were measured serially. Figure 4 depicts the levels of 4 cytokines starting from day of infusion over the first month. Hashed lines depict time of tocilizumab administration. After tocilizumab treatment, there was generally an apparent transient rise in IL6, followed by a rapid decrease. INFγ also decreased rapidly after tocilizumab administration in most patients. sIL6R increased in all and sgp130 levels increased in most patients after tocilizumab
Discussion
In this study, we have made a number of novel observations. First, we developed models that can predict which patients treated with CAR T cells are likely to become critically ill before they become ill, potentially allowing us to make early interventions that could reduce morbidity or mortality. Second, we established that concentrations of sIL6R and sgp130 are likely clinically and biologically relevant, as this is the first work that has systemically evaluated sIL6R and sgp130 after CAR T cells. Third, we identified 24 distinct cytokines that are differentially expressed in patients with versus without severe CRS, adding new insight into the biology underlying severe CRS. Finally, we confirmed our previously published but untested hypothesis that patients who develop severe CRS develop a clinical, laboratory, and biomarker profile consistent with secondary HLH.
The most common and potentially severe toxicity seen across trials using CAR-modified T cell therapy is CRS. Data from our group and others suggest a correlation between development of CRS and response to CAR T cells.(3, 4) Nevertheless, there does not appear to be a strong association between the degree of CRS and outcome.(2) Similar to data with other T-cell engaging therapies, including blinatumomab, our group and others have previously found that the severity of CRS may be associated with disease burden at the time of treatment.(2, 3) While this association exists, as we have demonstrated herein, disease burden alone is not sufficient to predict which patients will develop severe CRS. The PPV of high disease burden alone was poor as only 10 of 23 patients with an M3 marrow (>25% blasts) developed severe CRS. However, low disease burden does have a strong negative predictive value (NPV). In our pediatric cohort, only one of 15 patients who had a marrow which demonstrated <5% blasts at time of CTL019 infusion developed severe CRS. Baseline disease burden was not obtained in most adults and our pediatric trials moving forward are no longer assessing disease burden at the time of infusion. Our data demonstrate that the risk of CRS can be predicted accurately without the need for assessment of disease burden at the time of infusion.
Severe CRS is a potentially life-threatening toxicity. Indeed, 2 adults in our series died as a consequence of CRS. The ability to predict which patients may develop severe CRS prior to its development may be helpful in mitigating toxicity, as cytokine-directed therapy could be instituted before a patient becomes critically ill. Patients predicted to develop severe CRS could be more closely monitored to allow early initiation of aggressive supportive care. In contrast, the ability to predict which patients are unlikely to develop severe CRS can prevent unnecessary early hospitalization and/or exposure to unneeded cytokine-directed therapy. Accordingly, the models we have developed using a small number of cytokines to predict severity of CRS with both high sensitivity and specificity have direct clinical and therapeutic relevance. It is not known if early intervention or prevention of CRS will limit efficacy. Prospective trials initiating early intervention based on cytokine profile models will need to be carried out carefully.
We did not find any standard clinical laboratory tests were helpful in predicting CRS severity as many (ferritin, CRP, LDH, AST, ALT, BUN, and CR) peaked after patients became ill. Unlike prior reports by another group, we did not find early assessment of CRP could accurately predict severity of CRS.(3) Future work will determine if the early predictive cytokine profiles we have identified are also relevant to other T-cell engaging therapies such as BiTEs, as well as after cytotoxic T lymphocytes targeted at viruses.(7, 18)
In addition to developing accurate predictive models, we have made a number of key insights into the biologic understanding of CRS. Analyzing cytokines sent before patients developed severe CRS, we demonstrated sgp130 and IFNγ were strongly associated with later development of severe CRS. We confirmed our earlier observation that IFNγ, IL6, and sIL2Rα show a marked differential increase in patients with severe CRS as compared with patients without severe CRS. We found marked differences in a number of additional cytokines not previously studied after CAR T cell therapy. Generally, cytokines that were differentially elevated based on CRS grade included either cytokines released from activated T cells (sIL2Rα, IFNγ, IL6, sIL6R, GM-CSF) or activated monocytes/macrophages (IL1RA, IL10, IL6, IP10, MIG, INFα, MIP1α, MIP1β, sIL6R), as well as chemokines that are chemotactic for monocytes/macrophages (MCP1, MIP1β), and cytokines that are often elevated after tissue damage and inflammation (IL8, GCSF, GMCSF, VEGF, IL6, sRAGE).(13–15, 19, 20)
We found that patients who develop severe CRS develop a clinical phenotype that resembles MAS/HLH, as well as laboratory evidence of abnormal macrophage activation, including elevated ferritin, low fibrinogen, and a cytokine profile that mirrors that seen in genetic forms of HLH. Xu and colleagues demonstrated that an IFNγ level of 75pg/ml and an IL10 level >60pg/ml has 98.9% specificity and 93% sensitivity for HLH when measuring cytokine levels in critically ill children (with and without malignancies) with either sepsis or HLH.(11) IFNγ is not expected to be elevated in patients with sepsis. All patients with CRS 4–5 in our series had an IFNγ > 75pg/ml and IL10 >60pg/ml. IL4 is the only cytokine tested that was an outlier from our a priori hypothesis; however, the absolute values were very small in all patients. Thus, we hypothesize IL4 is likely not clinically or biologically relevant in our cohort. Future work will determine if there is any genotype-phenotype association between the development of MAS/HLH after CAR T cells and mutations in genes that predispose to the development of HLH, including PRF1.
IL6-directed therapy is the cornerstone of cytokine-based therapy after treatment with CAR T cells. It has been shown to be effective and, importantly, does not appear to decrease efficacy of the CAR T cells.(2–4) That said, as IL6 does not appreciably rise prior to the development of CRS, clinical assessment of IL6 in the first few days after infusion will not help determine which patients will develop severe CRS or require IL6 directed therapy. It is unknown whether early treatment with tocilizumab prior to development of CRS would be of benefit. Tocilizumab has a very long half-life (11–14 days).(21) Thus, if given early, drug would be present at the time of IL6 peak and could in theory prevent severe CRS.
This study demonstrates the importance of trans-IL6 signaling in CRS. IL6 signals through two mechanisms, either via the membrane-bound or soluble IL6 receptor (sIL6R).(20, 22) In classical IL6 signaling, IL6 binds to its membrane-bound receptor. Most cells do not express IL6R and are not responsive to classical IL6 signaling.(23) In trans-IL6 signaling, sIL6R binds IL6 and the complex associates with membrane-bound gp130.(24) Membrane-bound gp130 is associated with JAK1, JAK2, and TYK2.(20, 22) Accordingly, IL6-trans signaling activates the Jak/Stat pathway. IL6-trans signaling occurs in cells that do NOT express the IL6R. Normally, high levels of soluble gp130 (sgp130) and sIL6R in the blood serve as a buffer, blocking IL6-trans signaling. In healthy persons, IL6 levels are typically on the order of pg/ml, yet sIL6R and sgp130 levels are typically 1000× higher at ng/ml levels.(20, 22) Consequently, IL6-trans signaling only occurs when IL6 levels rise from pg/ml to ng/ml levels. IL6-trans signaling can be blocked either by lowering IL6 levels, blocking the interaction of IL6 with IL6R, raising sIL6R levels, raising sgp130 levels, or blocking the interaction of IL6-IL6R with sgp130.(20, 22) Tocilizumab is an anti-IL6R monoclonal antibody. In other IL6-mediated diseases, IL6 levels often go up and sIL6R levels either increase or decrease after treatment as the interaction between IL6R and IL6 is blocked.(21, 23, 24) After treatment with tocilizumab, there appeared to be a transient rise in IL6 followed by a rapid decrease in our CRS cohort. sIL6R levels also appeared to increase significantly after tocilizumab, because the complex of sIL6R and tocilizumab cannot be cleared by the kidney due to its size. These data suggest tocilizumab is blocking IL6-trans signaling through multiple mechanisms: blocking the interaction of IL6R with IL6, raising sIL6R to increase the IL6 buffer, and eventually lowering IL6 levels. Of note, collection of samples was not uniform between patients before and after treatment with tocilizumab, as tocilizumab was given on different days relative to time of infusion and some patients received more than one dose. Uno and colleagues recently published data that suggest that patients with rheumatoid arthritis who have elevated sgp130 levels are more likely to respond to tocilizumab as higher sgp130 levels will neutralize more IL-6/sIL-6R complexes, leaving less complexes that need to be neutralized by tocilizumab. Thus, we believe the high levels of sgp130 that are seen in our patients prior to treatment with tocilizumab are clinically and biologically relevant; however, future work investigating the importance and function of trans-IL6 signaling is needed.(25)
Other agents that target IL6 signaling are either commercially available or in clinical development, including direct IL6 inhibitors such as siltuximab and the IL6 trans-signaling blocker sgp130Fc.(20, 21, 23, 24) These agents have the potential to be effective for CRS, but future studies are needed. Our extensive cytokine profiling does not support the use of TNFα blockade after CAR T cells. While some of the soluble TNF-receptors were markedly elevated in patients with severe CRS, peak levels of TNFα were quite low. Interestingly, it has recently been shown that induction of shedding of TNF-receptors leads to complete unresponsiveness of TNFα target cells.(26) Published studies demonstrating efficacy of TNFα blockade used inhibitors in diseases with elevated serum or tissue TNFα levels. While some TNFα blocking agents such as etanercept also target TNF-receptors, it is unknown whether targeting TNF-receptors in patients with low levels of TNFα is efficacious. For patients who become critically ill after CAR T cells and do not respond to IL6 blockade, Jak/Stat, IFNγ, or sIL2Rα inhibitors could potentially be effective in ameliorating CRS symptoms. Unfortunately, these would likely affect the function of the CAR T cells.
Confounding variables that can affect cytokine production should always be considered when interpreting cytokine patterns to understand disease biology or develop predictive models. Mild differences in baseline cytokine values can occur in healthy normal subjects based on age, gender, and ethnic background.(27) Disease-related factors, including the type of malignancy or disease burden can also affect cytokine production. It is also important to evaluate both relative and absolute changes in cytokine production. Differences between populations are sometimes reported as fold-changes without consideration of the absolute values, but this can be misleading, as statistically significant differences may not be biologically or clinically meaningful. We considered values in the context of the degree of variation seen in healthy populations and the levels reported in patients with inflammatory diseases and/or infection. We considered both the absolute and relative differences between groups.
Despite several important observations, our study has several limitations. While we describe CRS after CAR T cells in the largest cohort of patients to date, the total number of patients with grade 4–5 CRS was relatively small. Nevertheless, our findings had adjustments for multiple comparisons and our prediction models remained accurate in an independent validation cohort. Our data reflect patients treated at two centers with CAR T cell products generated using the same manufacturing process, and it is unknown if our models will be generalizable. The only laboratory biomarkers that were robust for CRS prediction were cytokines and testing for cytokines is not available with rapid turnaround in many clinical laboratories. Our data allow the design of a focused panel of analytes that can be used to predict and track CRS. Common Terminology Criteria for Adverse Events (CTCAE) grading scales do not adequately or accurately define CRS after T-cell engaging therapies. Thus, different sites and different publications use different grading scales, which can make comparisons between studies challenging. Lee and colleagues and Davila and colleagues also published CRS grading scales for patients treated with CAR T cells (see Supplemental Tables 16 and 17).(3, 28) We have included a comparison of our CRS grading scale to other published grading scales in the Supplemental Discussion. The grading systems are similar enough that the predictive models we have developed are relevant in the other grading systems. Regardless of “numerical grade” our models identify patients who develop life-threatening complications of CRS (mechanical ventilation and/or decompensated shock).
The need for predictive models is to distinguish patients who become critically ill from CRS with those who do not. We made the demarcation based on patients who developed life-threatening complications of CRS, including decompensated shock or respiratory failure (grade 4–5). Patients with grade 0–2 CRS developed only mild illness. Grade 3 CRS in contrast represents a heterogeneous clinical spectrum, as some patients only required IV fluids or minimal supplemental oxygen, whereas others required low-dose vasoactive medications or developed more significant hypoxemia. Moving forward with certain trials, it may be important to loosen our definition of “severe CRS” and include patients who became very ill but did not develop life-threatening CRS. We performed additional analyses sub-dividing patients with grade 3 CRS based on the need for any vasoactive medications or significant oxygen requirement (>=40% FI02) into two groups (3a and 3b) and re-split our cohort into severe and not severe defined as CRS 0–3a vs CRS 3b-5. We performed logistic regression and decision tree modeling to develop new models with this alternate categorization and also tested the accuracy of our “original” models using the alternate categorization. These additional models and the additional analyses are included in the Supplemental Results and as Supplemental Tables 18 and 19 and Supplemental Figures 5a–e.
We studied the biology of CRS and investigated if certain cytokines measured early could predict CRS severity. Additional variables not studied herein may predict severe CRS and these will be investigated in future work. These include T cell phenotype of the product, T cell function of the product, CD19 polymorphisms that may differentially activate CTL019, tumor expression of CD19 or PD-L1, and immune gene polymorphisms. We have previously published that products generated from the majority of patients show high cytolytic activity and produce very similar in vitro levels of most cytokines.(29) Since there is little variability in the ex vivo composition and cytokine production of the CTL019 product, but considerable heterogeneity in CRS in patients, we hypothesize we would likely not find differences in the CTL019 product that will correlate with severity of CRS.
In conclusion, these data represent the largest and most comprehensive analysis to date of the clinical and biologic manifestations of CRS after CAR T cell therapy. We have identified and characterized cytokines that are associated with severe CRS and cytokines that can predict which patients will likely develop severe CRS before it happens. Early prediction will allow trials to determine if early intervention will mitigate toxicity without impacting efficacy. Based on the exciting efficacy seen with CAR T cells in early phase trials, their use is rapidly expanding from a select number of tertiary care institutions to a larger number of centers. Accordingly, understanding and reducing toxicity is paramount and these data provide significant novel information that may help achieve that goal.
Methods
We collected clinical and laboratory data on 39 patients with ALL treated consecutively with CTL019 at the Children’s Hospital of Philadelphia (CHOP) from April 2012 through September 2014 on a phase 1/2a clinical trial (NCT01626495). We collected clinical and laboratory data on 12 adults treated with CTL019 at the Hospital of the University of Pennsylvania (PENN) between March 2013 through August 2014 on two trials (NCT02030847 and NCT01029366). Additional details on the trial design and CTL019 product are provided in Supplemental Methods. Written informed consent was obtained from all subjects or their legal guardian according to the Declaration of Helsinki and all protocols were approved by their respective institutional review boards (IRBs) at CHOP and PENN. We also collected a limited set of clinical and laboratory data on an additional 12 consecutive patients treated with CTL019 at CHOP from October 2014 to May 2015 on NCT01626495. This data set provided a validation cohort for our predictive models. CRS was graded as previously described and as defined in Supplemental Table 1A and 1B.(2, 5, 29) This grading scale was developed a priori and before any data analysis. Cytokine markers were measured on serum samples from 10 healthy volunteers (see Supplement for details).
All data were decoded and maintained in secure databases. 43 unique cytokines and a panel of clinical laboratory tests, including chemistries, ferritin and C-reactive protein (CRP) were serially monitored. CTL019 cells were serially measured in peripheral blood by quantitative PCR.(2) Analysis was restricted to the first month after infusion of CTL019. A detailed description of the laboratory tests, cytokines, and collection time-points is included in Supplemental Methods and Supplemental Table 2. Baseline bone marrow aspirate and biopsies were collected in the pediatric cohort (see Supplemental Methods). MRD was performed in the CLIA and CAP approved Children’s Oncology Group Western Flow Cytometry Reference Laboratory at the University of Washington (Seattle, WA) as previously described.(30, 31)
Statistical Methods
Clinical, laboratory, and cytokine markers associated with CRS are summarized overall and by occurrence of severe CRS (grade 4–5) for the pediatric, adult, and combined cohorts. For markers measured serially, values were summarized both as the peak over the first 3 days and over the month post-infusion in order to capture early and overall peak values of these biomarkers during the period when patients experienced CRS. In addition, relative changes from baseline (fold-changes) were evaluated during the first 3 days after infusion. The month was defined as the first 35 days, allowing for a 1-week window beyond the expected 28-day evaluation. Between-group comparisons were performed using Fisher’s exact test for discrete and exact Wilcoxon rank-sum test for continuous factors. Fibrinogen and CRP, due to a few values recorded as exceeding a limit of detection that was within range of other observed values, were analyzed with the generalized Wilcoxon test for right-censored data. Values less than the lower limit of detection were recorded as half the lower limit. Further details are provided in Supplemental Methods. All statistical tests were two-sided and generally done at the 0.05 level. Consideration for multiple comparisons was given when examining hypotheses for the 43 cytokine biomarkers, as described below. Statistical analyses were performed using R (version 3.2.1; R Development Core Team, Vienna, Austria) and SAS (version 9.4; SAS Institute Inc, Cary, NC) software.
Cytokine analysis
Several hypotheses regarding cytokine levels were investigated, and statistical associations were declared significant only if they remained significant at the 0.05 level, after the Holm’s adjustment for the 43 multiple comparisons.(32) All hypotheses that were tested were developed a priori. Comparisons of cytokine levels analyzed in this manner included: 1-month peak and 3-day peak between those with versus without severe CRS, patient baseline between those with high versus low disease burden and baseline patient versus healthy adult subjects; 1-month and 3-day peak analyses were performed for the combined, adult and pediatric cohorts separately. Multiple published studies have demonstrated that while cytokines can vary between children and adults with disease or after antigen stimulation, baseline values in normal healthy children and adults are similar.(33–35) Thus, we did not include a separate healthy pediatric cohort. In order to rule out that any variation around the sampling frequency of cytokine levels between patients could be biasing these comparisons, 1-month analyses were repeated including measurements from a reduced, common sampling schedule that was shared by nearly all subjects (details provided in the Supplemental Methods and Results). We hypothesized that patients treated with T-cell engaging therapies, including CTL019, who experience severe CRS, develop abnormal macrophage activation with secondary hemophagocytic lymphohistiocytosis (HLH). We made this hypothesis based on clinical symptomatology and marked hyperferritinemia. Numerous studies have shown ferritin >10,000ng/ml is highly sensitive and specific for HLH in children.(33, 34) To establish whether the patients with severe CRS were manifesting HLH (diagnostic criteria in s), we compared the cytokine profiles from patients who developed severe CRS with published reports of cytokine profiles in patients with primary HLH associated with a genetic predisposition. Of the 43 tested cytokines, 19 have been previously studied in children with HLH.(14–17) We reconsidered this subset of cytokines for the association with severe CRS to compare the HLH pattern, with Holm’s adjustment for 19 multiple comparisons.
In order to understand which factors may be most intrinsically involved with CRS syndrome and the immune system’s initial response, we sought to develop a prediction model for severe CRS that considered clinical and laboratory factors measured within the first 3 days post-infusion. Models were kept small due to the limited number of severe CRS cases (14 overall and 11 in the pediatric cohort). Candidate variables included those factors for which the 3-day peak was missing in no more than 2/14 cases and 10% overall: ALT, AST, BUN, CR, Ferritin, qPCR, LDH, the 43 cytokines markers, as well CRS-defining symptoms in the first 2 days post-infusion (yes/no) and age at infusion. 3-day peak fold change was also considered for the 43 cytokines and baseline disease burden was an additional candidate variable for the pediatric cohort. Two patients (both in pediatric cohort) developed severe CRS on Day 3; however, all data included in the models was collected at least 12 hours prior to the development of severe CRS (see Supplemental Appendix and Supplemental Table 4 for additional details). Logistic regression and classification tree models were fit in the combined and pediatric cohorts, hereafter referred to as the discovery cohort. The adult cohort was too small to model separately. For the logistic regression models, forward selection using the Akaike information criterion (AIC) was used to select the final models. The deviance statistic was used to select the tree models. Further details are provided in the Supplemental Methods. Models were validated in an independent cohort of 12 pediatric patients, referred to as the validation cohort.
Supplementary Material
Statement of Significance.
Cytokine release syndrome (CRS) is the most common severe toxicity seen after CAR T cell treatment. We developed models that can accurately predict which patients are likely to develop severe CRS before they become critically ill, which improves understanding of CRS-biology and may guide future cytokine-directed therapy.
Acknowledgments
Financial Support: Financial support was provided by a grant from Novartis (to Dr. June), by grants from the National Institutes of Health (R01CA165206, to Dr. June; R01CA193776, to Dr. Teachey; R01CA102646 and R01CA116660 to Dr. Grupp; and, K23GM110496 to Dr. Weiss), the Pennsylvania Department of Health (to Dr. Grupp), the Leukemia and Lymphoma Society (to Drs. Grupp and Teachey), the Jeffrey Jay Weinberg Memorial Foundation (to Dr. Grupp), and the Children’s Hospital of Philadelphia Hematologic Malignancy Research Fund (to Dr. Teachey), a Stand Up To Cancer–St. Baldrick’s Pediatric Dream Team translational research grant (SU2C-AACR-DT1113 to Drs. Grupp, Teachey, Barrett, and Maude), St. Baldrick’s Foundation Scholar Awards (to Dr. Maude and to Dr. Barrett), and a Research Scholar Grant from the American Cancer Society (RSG-14-022-01-CDD to Dr. Teachey). Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research.
Footnotes
Conflict of Interest Statement. Carl June, Stephan Grupp, David Teachey, Simon Lacey, Pamela Shaw, J. Joseph Melenhorst, Noelle Frey, Shannon Maude, Fang Chen, Vanessa Gonzalez, Edward Pequignot, Bruce Levine and David Porter received research funding from Novartis. Shannon Maude and Susan Rheingold perform consultancy for Novartis. Carl June, David Porter, and Bruce Levine have patents and royalties, Zhaohui Zheng has royalties, and Stephan Grupp has a patent in the field of cell and gene therapy that are managed in accordance with University of Pennsylvania policy and oversight.
References
- 1.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. The New England journal of medicine. 2013;368:1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. The New England journal of medicine. 2014;371:1507–17. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–28. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. The New England journal of medicine. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119:6226–33. doi: 10.1182/blood-2012-01-400515. [DOI] [PubMed] [Google Scholar]
- 7.Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121:5154–7. doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen CE, Yu X, Kozinetz CA, McClain KL. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatric blood & cancer. 2008;50:1227–35. doi: 10.1002/pbc.21423. [DOI] [PubMed] [Google Scholar]
- 9.Switala JR, Hendricks M, Davidson A. Serum ferritin is a cost-effective laboratory marker for hemophagocytic lymphohistiocytosis in the developing world. J Pediatr Hematol Oncol. 2012;34:e89–92. doi: 10.1097/MPH.0b013e31824227b9. [DOI] [PubMed] [Google Scholar]
- 10.Janka G, zur Stadt U. Familial and acquired hemophagocytic lymphohistiocytosis. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program. 2005:82–8. doi: 10.1182/asheducation-2005.1.82. [DOI] [PubMed] [Google Scholar]
- 11.Xu XJ, Tang YM, Song H, Yang SL, Xu WQ, Zhao N, et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr. 2012;160:984–90 e1. doi: 10.1016/j.jpeds.2011.11.046. [DOI] [PubMed] [Google Scholar]
- 12.Tang Y, Xu X, Song H, Yang S, Shi S, Wei J, et al. Early diagnostic and prognostic significance of a specific Th1/Th2 cytokine pattern in children with haemophagocytic syndrome. British journal of haematology. 2008;143:84–91. doi: 10.1111/j.1365-2141.2008.07298.x. [DOI] [PubMed] [Google Scholar]
- 13.Tamura K, Kanazawa T, Tsukada S, Kobayashi T, Kawamura M, Morikawa A. Increased serum monocyte chemoattractant protein-1, macrophage inflammatory protein-1beta, and interleukin-8 concentrations in hemophagocytic lymphohistiocytosis. Pediatric blood & cancer. 2008;51:662–8. doi: 10.1002/pbc.21660. [DOI] [PubMed] [Google Scholar]
- 14.Takada H, Takahata Y, Nomura A, Ohga S, Mizuno Y, Hara T. Increased serum levels of interferon-gamma-inducible protein 10 and monokine induced by gamma interferon in patients with haemophagocytic lymphohistiocytosis. Clin Exp Immunol. 2003;133:448–53. doi: 10.1046/j.1365-2249.2003.02237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Osugi Y, Hara J, Tagawa S, Takai K, Hosoi G, Matsuda Y, et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood. 1997;89:4100–3. [PubMed] [Google Scholar]
- 16.Henter JI, Elinder G, Soder O, Hansson M, Andersson B, Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 1991;78:2918–22. [PubMed] [Google Scholar]
- 17.Bhattacharyya M, Ghosh MK. Hemophagoctic lymphohistiocytosis–recent concept. J Assoc Physicians India. 2008;56:453–7. [PubMed] [Google Scholar]
- 18.Koehne G, Hasan A, Doubrovina E, Prockop S, Tyler E, Wasilewski G, et al. Immunotherapy with Donor T Cells Sensitized with Overlapping Pentadecapeptides for Treatment of Persistent Cytomegalovirus Infection or Viremia. Biol Blood Marrow Transplant. 2015;21:1663–78. doi: 10.1016/j.bbmt.2015.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Briso EM, Dienz O, Rincon M. Cutting edge: soluble IL-6R is produced by IL-6R ectodomain shedding in activated CD4 T cells. Journal of immunology. 2008;180:7102–6. doi: 10.4049/jimmunol.180.11.7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calabrese LH, Rose-John S. IL-6 biology: implications for clinical targeting in rheumatic disease. Nat Rev Rheumatol. 2014;10:720–7. doi: 10.1038/nrrheum.2014.127. [DOI] [PubMed] [Google Scholar]
- 21.Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, Kakehi T. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood. 2008;112:3959–64. doi: 10.1182/blood-2008-05-155846. [DOI] [PubMed] [Google Scholar]
- 22.Garbers C, Aparicio-Siegmund S, Rose-John S. The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr Opin Immunol. 2015;34:75–82. doi: 10.1016/j.coi.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 23.Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. International journal of biological sciences. 2012;8:1237–47. doi: 10.7150/ijbs.4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rose-John S, Heinrich PC. Soluble receptors for cytokines and growth factors: generation and biological function. The Biochemical journal. 1994;300(Pt 2):281–90. doi: 10.1042/bj3000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uno K, Yoshizaki K, Iwahashi M, Yamana J, Yamana S, Tanigawa M, et al. Pretreatment Prediction of Individual Rheumatoid Arthritis Patients’ Response to Anti-Cytokine Therapy Using Serum Cytokine/Chemokine/Soluble Receptor Biomarkers. PLoS ONE. 2015;10:e0132055. doi: 10.1371/journal.pone.0132055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maney SK, McIlwain DR, Polz R, Pandyra AA, Sundaram B, Wolff D, et al. Deletions in the cytoplasmic domain of iRhom1 and iRhom2 promote shedding of the TNF receptor by the protease ADAM17. Sci Signal. 2015;8:ra109. doi: 10.1126/scisignal.aac5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burska A, Boissinot M, Ponchel F. Cytokines as biomarkers in rheumatoid arthritis. Mediators Inflamm. 2014;2014:545493. doi: 10.1155/2014/545493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–95. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borowitz MJ, Devidas M, Hunger SP, Bowman WP, Carroll AJ, Carroll WL, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children’s Oncology Group study. Blood. 2008;111:5477–85. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weir EG, Cowan K, LeBeau P, Borowitz MJ. A limited antibody panel can distinguish B-precursor acute lymphoblastic leukemia from normal B precursors with four color flow cytometry: implications for residual disease detection. Leukemia. 1999;13:558–67. doi: 10.1038/sj.leu.2401364. [DOI] [PubMed] [Google Scholar]
- 32.Holm S. A simple sequentially rejective multiple test procedure. Scandinavian Journal of Statistics. 1979;6:70. [Google Scholar]
- 33.Berdat PA, Wehrle TJ, Kung A, Achermann F, Sutter M, Carrel TP, et al. Age-specific analysis of normal cytokine levels in healthy infants. Clin Chem Lab Med. 2003;41:1335–9. doi: 10.1515/CCLM.2003.204. [DOI] [PubMed] [Google Scholar]
- 34.Lilic D, Cant AJ, Abinun M, Calvert JE, Spickett GP. Cytokine production differs in children and adults. Pediatr Res. 1997;42:237–40. doi: 10.1203/00006450-199708000-00018. [DOI] [PubMed] [Google Scholar]
- 35.Kleiner G, Marcuzzi A, Zanin V, Monasta L, Zauli G. Cytokine levels in the serum of healthy subjects. Mediators Inflamm. 2013;2013:434010. doi: 10.1155/2013/434010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.