

Abstract
The epithelial–mesenchymal transition (EMT) is a crucial morphological event that occurs during progression of epithelial tumors. We reported previously that levels of the δ‐crystallin/E2‐box factor 1 (δEF1) family proteins (Zinc finger E‐box binding homeobox 1 [ZEB1]/δEF1 and ZEB2/ Smad‐interacting protein 1), key regulators of the EMT, are positively correlated with EMT phenotypes and aggressiveness of breast cancer. Here, we show that Ets1 induces ZEB expression and activates the ZEB1 promoter, independently of its threonine 38 phosphorylation status. In the basal‐like subtype of breast cancer cells, siRNAs targeting Ets1 repressed expression of ZEBs and partially restored their epithelial phenotypes and sensitivity to antitumor drugs. Epithelium‐specific ETS transcription factor 1 (ESE1), a member of the Ets transcription factor family, was originally characterized as being expressed in an epithelial‐restricted pattern, placing it within the epithelium‐specific ETS subfamily. ESE1, highly expressed in the luminal subtype of breast cancer cells, was repressed by activation of the MEK–ERK pathway, resulting in induction of ZEBs through Ets1 upregulation. Conversely, Ets1, highly expressed in the basal‐like subtype, was repressed by inactivation of MEK–ERK pathway, resulting in reduction of ZEBs through ESE1 upregulation. These findings suggest that ESE1 and Ets1, whose expressions are reciprocally regulated by the MEK–ERK pathway, define the EMT phenotype through controlling expression of ZEBs in each subtype of breast cancer cells.
Keywords: EMT, ESE1, Ets1, signal transduction, ZEB
Breast cancer is a heterogeneous disease, and treatment must be adapted to the clinical, histologic, cellular, and molecular characteristics of individual cases. Molecular profiling of breast cancers revealed two distinct molecular subtypes, in which gene expression patterns are precisely compatible with pathological and clinical features.1 These subtypes, luminal and basal‐like, are generally considered to be exclusively composed of cells with epithelial and mesenchymal phenotypes, respectively.2, 3 These opposing features are attributed to modulation of the EMT. Notably, the basal‐like subtype represents 10–20% of all breast carcinoma and almost matches a clinical subtype known as triple‐negative breast cancer, defined as a tumor that lacks expression of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2. These tumors are highly progressive, drug‐resistant, and frequently recurrent. Clinical treatment of patients with triple‐negative breast cancer has not yet been successful.4
The process of cancer cell invasion involves the loss of cell–cell interactions along with acquisition of motile properties, and is often associated with the EMT. Formation of tight cell–cell adhesions depends primarily on the E‐cadherin system, and a hallmark of the EMT is downregulation of E‐cadherin. Although this can occur as a consequence of various interventions, transcriptional repression of E‐cadherin, frequently observed in malignant tumors, is mediated by the δEF1 family of two‐handed zinc‐finger factors (ZEB1/δEF1 and ZEB2/SIP1), the Snail family (Snail, Slug, and Smuc), and basic helix–loop–helix factors (Twist and E12/E47). These factors, which are overexpressed in cancer cells, induce the EMT and promote development of metastatic properties such as migration and invasion. The ZEBs repress E‐cadherin expression through direct binding to the E‐box elements in the E‐cadherin promoter region. The ZEBs are highly expressed in basal‐like breast cancers, but hardly expressed in luminal‐type breast cancers.5 The molecular mechanisms underlying induction of ZEB expression remain to be fully elucidated.
The ETS family of transcription factors, which has 28 members in the human genome, regulate many different biological processes, including cell proliferation, cell differentiation, embryonic development, neoplasia, hematopoiesis, angiogenesis, and inflammation.6 Ets1, a prototypic member of this family, regulates the EMT during chicken embryo development.7 Additionally, Ets1 enhances ZEB1 promoter activity in murine mammary gland epithelial NMuMG cells to elicit transforming growth factor‐β‐induced EMT.8 In addition, expression levels of Ets1 and Ets2 in breast cancer are correlated with poor prognosis and high rates of tumor recurrence, supporting the findings that inactive mutation of Ets2 inhibits mammary tumor progression.9, 10 Ets2 functions redundantly with Ets1 to regulate various cellular phenomena.11 Although many ETS family members are expressed in non‐epithelial cells, such as hematopoietic and endothelial cells, ESE1 belongs to the ESE subfamily of ETS transcription factors, which are expressed only in epithelial‐rich tissues.12 Also known as ESX, ELF3, ERT, and JEN, ESE1 is amplified in 50% of ductal carcinoma in situ (an early stage breast cancer),13 whereas ESE1 suppresses cell invasion in cancer cells.14 Moreover, ESE2, also known as ELF5, suppresses the EMT through transcriptional repression of Slug (also known as Snail2).15
Zinc‐finger E‐box binding homeobox 1 is highly expressed in the basal‐like subtype of breast cancer cells,5 and ZEB1 promoter reporter activity is promoted by Ets1 in NMuMG cells.8 Based on a previous report that Ets1 is a predictive marker for poor prognosis of breast cancer,10 we hypothesized that Ets1 regulates expression of ZEB proteins in these tumors. In this study, we found that Ets1 siRNAs repressed expression of ZEBs in basal‐like breast cancer cells and partially altered EMT phenotypes including expression of epithelial/mesenchymal marker proteins and sensitivity to antitumor drugs. A MEK1/2 inhibitor, U0126, suppressed expression of ZEB proteins by repressing the Ets1 protein level, but not by repressing phosphorylation of Ets1 at threonine 38. Thus, low activation status of the MEK–ERK pathway, widely observed in the luminal subtype, upregulates ESE1 and downregulates Ets1, leading to acquiring the MET phenotype through reduction of ZEBs. By contrast, high activation status of the MEK–ERK pathway, widely observed in the basal‐like subtype, downregulates ESE1 and upregulates Ets1, leading to acquiring the EMT phenotype through induction of ZEBs. Enhancement of ZEB1 expression by Ets1 required putative Ets‐binding sites in the ZEB1 promoter region. Therefore, Ets1 and ESE1 appear to reciprocally control EMT by regulating expression of the ZEB proteins, which promote breast cancer aggressiveness.
Materials and Methods
Cell culture
All cells used in this study were from the ATCC (Manassas, VA, USA). They were cultured in DMEM (Nacalai Tesque, Kyoto, Japan) supplemented with 4.5 g/L glucose, 10% FBS, 50 U/mL penicillin, and 50 μg/mL streptomycin, in a 5% CO2 atmosphere at 37°C.
Reagents and antibodies
U0126, SP600125, and SB203580 were purchased from Millipore (Billerica, MA, USA). Rabbit polyclonal anti‐ZEB1 and anti‐ZEB2 antibodies were from Novus Biologicals (Littleton, CO, USA). Rabbit polyclonal anti–Ets1 (C20) was from Santa Cruz Biotechnology (Dallas, TX, USA). Rabbit polyclonal anti‐phospho‐Ets1 (phospho‐T38) antibodies and monoclonal anti‐ESE1 antibody were from Abcam (Cambridge, UK). Rabbit polyclonal anti‐ERK1/2 and anti‐phospho‐ERK1/2 antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). Mouse monoclonal anti‐E‐cadherin and anti‐N‐cadherin antibodies were from BD Transduction Laboratories (Lexington, KY, USA). Mouse monoclonal anti‐α‐tubulin and anti‐Flag (M2) antibodies were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Rhodamine‐conjugated phalloidin was purchased from Cytoskeleton (Denver, CO, USA).
RNA extraction and quantitative RT‐PCR
Total RNA was purified using Isogen (Nippon Gene, Tokyo, Japan). cDNAs were synthesized using the PrimeScript First Strand cDNA synthesis kit (TaKaRa, Otsu, Japan). Quantitative RT‐PCR was carried out using Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Values in each sample were normalized to the corresponding level of the mRNA encoding GAPDH. Polymerase chain reactions were carried out using the following primers: ZEB1, forward, 5′‐CAATGATCAGCCTCAATCTGCA‐3′, reverse, 5′‐CCATTGGTGGTTGATCCCA‐3′; ZEB2, forward, 5′‐AAGCCCCATCAACCCATACAAG‐3′ reverse, 5′‐AAATTCCTGAGGAAGGCCCA‐3′; Ets1, forward, 5′‐CCCGTACGTCCCCCACTCCT‐3′, reverse, 5′‐TGGGACATCTGCACATTCCA‐3′; GAPDH, forward, 5′‐CGACCACTTTGTCAAGCTCA‐3′, reverse, 5′‐CCCTGTTGCTGTAGCCAAAT‐3′; Ets1, forward, 5′‐CCCGTACGTCCCCCACTCCT‐3′, reverse, 5′‐TGGGACATCTGCACATTCCA‐3′; and ESE1, forward, 5′‐CAACTATGGGGCCAAAAGAA‐3′, reverse, 5′‐TCCAGGATCTCCCGTTTGTA‐3′.
DNA construction
The human ZEB1 promoter reporter construct (hZEB1‐Luc) was previously described.8 Three truncated versions of hZEB1‐Luc were constructed by a PCR‐based strategy. hZEB1‐Luc with point mutations and human Ets1 with a point mutation at threonine 38 were constructed by PCR‐based mutagenesis. Human Ets1 cDNA was as previously described,8 and human ESE1 cDNA was PCR‐amplified using cDNA prepared from MCF7 cells. All constructs were confirmed by sequencing.
RNA interference
Transfection of siRNAs (Stealth RNAi; Invitrogen, Carlsbad, CA, USA, Ets1 [103402, 103403, 103404], ESE1 [103187, 176434], and control [12935300]) was carried out in six‐well tissue culture plates using Lipofectamine RNAiMax (Invitrogen). Final concentration of siRNA was 10 nM.
Immunoblot and immunofluorescence analyses
Cells were seeded at a density of 2 × 105 cells/well in six‐well tissue culture plates. Cells were lysed in lysis buffer (20 mM Tris–HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Nonidet P‐40, and protease and phosphatase inhibitors). After measurement of protein concentration with a BCA protein assay kit (Pierce, Rockford, IL, USA), equal amounts of total proteins per lane were subjected to SDS‐PAGE, followed by semi‐dry transfer of proteins to Fluoro Trans W membrane (Pall, Glen Cove, NY, USA). Immunodetection was undertaken with the ECL blotting system (Amersham Bioscience, Piscataway, NJ) on a Luminescent Image Analyzer (LAS400; Fujifilm, Tokyo, Japan). For immunofluorescence labeling, cells were fixed in 3.7% formaldehyde in PBS for 15 min, permealized with 0.2% Triton X‐100 in PBS for 5 min, and incubated with primary antibodies diluted with Blocking One solution (Nacalai Tesque) overnight at 4°C. The cells were then incubated with secondary antibodies for 1 h and TOPRO (Invitrogen) for 5 min. Fluorescence was examined by a confocal laser scanning microscopy (Olympus, Tokyo Japan).
Generation and infection of lentiviruses
We used a lentiviral expression system to establish MCF7 cells stably expressing H‐Ras (G12V). Complementary DNA encoding Ras (G12V) with an N‐terminal FLAG epitope tag was inserted into the multicloning site of lentiviral pCSII‐EF/CMV‐RfA vectors using Gateway Technology (Invitrogen). For production of lentiviral vectors, 293FT cells were transfected by Lipofectamine 2000 (Invitrogen) with pCAG‐HIVgp and pCMV‐VSV‐G‐RSV‐Rev vectors. The culture media were collected 72 h after transfection and used for infection into MCF7 cells.
Luciferase assay
HeLa cells were seeded in duplicate in 24‐well tissue culture plates, followed by transient transfection with hZEB1‐Luc, pTKRenilla (Promega, San Luis Obispo, CA, USA), and the indicated expression plasmids using X‐treme Gene HP DNA transfection reagent (Roche, Indianapolis, IN, USA). Twenty‐four hours later, luciferase activity was determined using the Dual Luciferase Reporter Assay (Promega) on a luminometer (SpectraMax L Microplate Reader; Molecular Devices, Sunnyvale, CA, USA). Firefly luciferase activity was normalized to sea‐pansy luciferase activity from cotransfected pTKRenilla.
Cell proliferation assay
Twenty‐four hours after transfection with the siRNAs, cells were seeded in triplicate in 96‐well tissue culture plates at the cell density of 2 × 104 cells/well. Twenty‐four hours later, cells were exposed to adriamycin (Sigma‐Aldrich) or cisplatin (Sigma‐Aldrich) in serum‐free media at a concentration of 0, 20, 30, or 40 μM. After cultured for 24 h, cell count assays were carried out using WST‐8 (Nacalai Tesque).
Results
Expression of both ZEB1 and ZEB2 is repressed by Ets1 siRNAs
Ets1 mRNA and protein are expressed at higher levels in the basal‐like subtype than the luminal subtype of breast cancer cells.16 Because we previously reported that ZEB1 and ZEB2 are also highly expressed in the basal‐like subtype,5, 17 we examined expression of Ets1 along with ZEB1 and ZEB2 in the basal‐like subtype (MDA‐MB‐231) and the luminal subtype (MCF7 and T47D). As with previous findings, protein levels of Ets1, ZEB1, and ZEB2 were higher in MDA‐MB‐231 cells than in MCF7 and T47D cells (Fig. 1a). To determine whether Ets1 regulates expression of ZEB1 and ZEB2, we transfected three different siRNAs against human Ets1 into MDA‐MB‐231 cells. All siRNAs successfully silenced endogenous Ets1 at both the protein and mRNA levels (Fig. 1a,b). Notably, protein as well as mRNA expression of ZEBs were also suppressed by all three Ets1 siRNAs (Fig. 1a,b). The effects of the siRNAs were confirmed in two other basal‐like subtype cell lines, BT549 and Hs578T (Fig. 1c). Expression of E‐cadherin, a representative epithelial maker, was examined in both MDA‐MB‐231 and BT549 cells, because E‐cadherin in Hs578T cells is epigenetically regulated and not affected by transfection with ZEB siRNA alone.17 E‐cadherin was upregulated by siRNA transfection in both cell lines (Fig. 1d). By contrast, N‐cadherin, a representative mesenchymal maker, was downregulated in MDA‐MB‐231 following transfection of siRNA, whereas it was expressed below the detectable level in BT549 cells (Fig. 1d). Immunofluorescence analysis also showed upregulation of E‐cadherin and downregulation of N‐cadherin in MDA‐MB‐231 transfected with Ets1 siRNAs, which were accompanied by a change in mRNA levels (Figs 1e,S1). Actin stress fiber formation determined by phalloidin staining and cell morphology were insufficiently changed by Ets1 siRNA (Fig. S1a and data not shown). Vimentin mRNA was moderately downregulated by Ets1 siRNA (Fig. S1b), whereas mRNA levels of fibronectin and α‐smooth muscle actin were low and unchanged dramatically by the siRNA (Fig. S1b). These findings suggest that Ets1 positively regulates expressions of ZEB and some EMT marker molecules in the basal‐like subtype of breast cancer cells.
Figure 1.
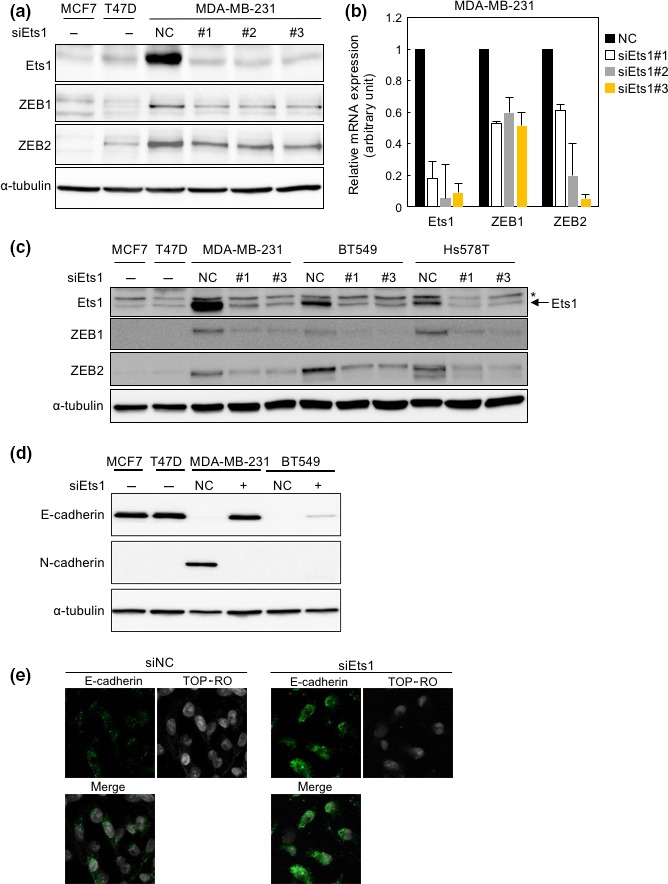
Repression of zinc‐finger E‐box binding homeobox (ZEB)1 and ZEB2 by Ets1 siRNAs. (a–c) Forty‐eight hours after transfection of breast cancer cells with control siRNA (NC) or three different kinds of siRNAs targeting human Ets1 (#1, #2, and #3), expression of the indicated factors was examined at the protein and mRNA levels by immunoblot (a,c) and quantitative RT‐PCR (b) analyses, respectively. *Non‐specific bands (c). (d,e) Forty‐eight hours after transfection with human control siRNA (NC) and Ets1 siRNA (siEts1#1), expression of the indicated proteins was evaluated by immunoblot analyses (d). MDA‐MB‐231 cells transfected with control siRNA (siNC) and Ets1 siRNA (#1) were analyzed by immunofluorescence analyses with anti‐E‐cadherin antibody (green), and TO‐PRO (white) to detect nuclei (e). α‐Tubulin levels were monitored as a loading control (a,c,d).
Ets1 activates the ZEB1 promoter through putative Ets1‐binding sites at the distal region from the transcriptional start site
To determine how Ets1 upregulates expression of ZEBs, we transfected the ZEB1 promoter reporter construct (hZEB1‐Luc) into HeLa cells and measured luciferase activities. As in a previous report using NMuMG cells,8 Ets1 activated hZEB1‐Luc (WT containing −1129 to +76 of the human ZEB1 promoter region) in the cells (Fig. 2a). Next, we constructed three truncated mutants by a PCR‐based strategy (Fig. 2a) and measured luciferase activities following transfection with Flag‐tagged Ets1 (Fig. 2a). Although Ets1 could activate hZEB1‐Luc (WT), it failed to activate the shorter constructs, suggesting that the region spanning from −1129 to −820 is involved in transactivation by Ets1. In this region, we identified two typical Ets1‐binding sites (5′‐A/GGGAAG/C‐3′) and two putative Ets1‐binding sites (5′‐GGAT‐3′) (Fig. 2b).18 hZEB1‐Luc containing mutation in the two typical Ets1‐binding sites (Mutation C in Fig. 2b) was activated by Ets1 to a similar extent as the wild‐type construct (Fig. 2b). However, hZEB1‐Luc constructs harboring mutations in either or both of the two putative binding sites were not activated by Ets1 (Fig. 2b). Responses of various hZEB1‐Luc constructs to Ets1 were also confirmed by breast cancer MCF7 cells (Fig. S2a). These findings indicate that both putative binding sites in the region spanning from −1129 to −820 are indispensable for activation by Ets1.
Figure 2.
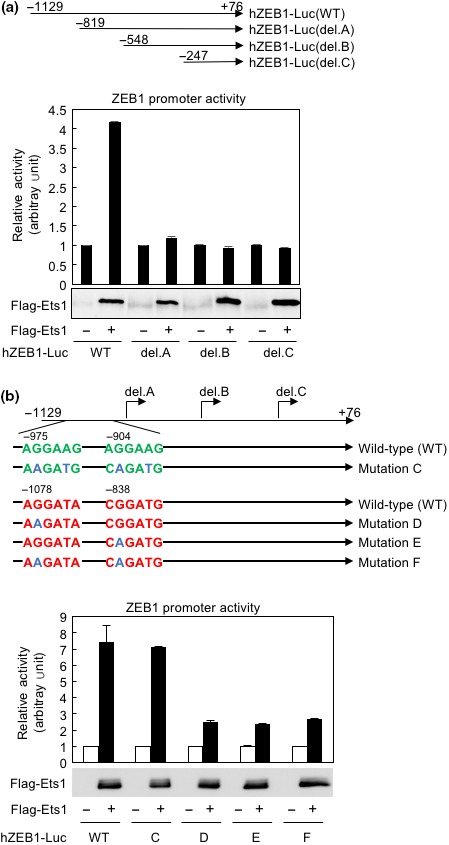
Activation of the zinc‐finger E‐box binding homeobox (ZEB)1 promoter by Ets1. (a) Schematic illustration of the human ZEB1 promoter reporter construct (hZEB1‐Luc, WT containing −1129 to +76 of the human ZEB1 promoter region) and three truncated constructs (del. A, del. B, and del. C) (top panel). The reporter constructs were cotransfected with Flag‐tagged Ets1 expression plasmid (Flag‐Ets1) in HeLa cells, and luciferase activities were measured. Expression of transfected Ets1 was confirmed by immunoblot analyses using anti‐Flag antibody (bottom). (b) Two typical Ets1‐binding sites (A/GGGAAG/C) and two putative Ets1‐binding sites (GGAT) in the region spanning from −1129 to −820 are shown in green and red, respectively. Point mutations in each reporter construct are shown in blue. Arrows indicate the starting sites of each deletion construct. After transfection with the indicated plasmids, luciferase activities were measured. Expression of transfected Ets1 (Flag‐Ets1) was confirmed by immunoblot analyses using anti‐Flag antibody (bottom).
Effect of MEK1/2 inhibitor U0126 on expression of ZEB1 and ZEB2
Ets1 is phosphorylated at threonine 38 by activated ERK1/2.19 Despite the rarity of Ras mutation in breast cancers (<2%),20 high levels of ERK1/2 phosphorylation are observed in the basal‐like subtype of breast cancer cells.21 To examine the effect of Ets1 phosphorylation on induction of ZEBs, we treated MDA‐MB‐231 cells with a MEK1/2 inhibitor (U0126) as well as other MAPK inhibitors, a JNK inhibitor (SP600125), and a p38 inhibitor (SB203580), and then monitored expression of ZEBs and Ets1. Treatment with U0126 for 24 h dramatically suppressed phosphorylation of both ERK1/2 and Ets1. Notably, the levels of Ets1 protein were considerably decreased by U0126 (Figs 3a,S2b), accompanied by repression of ZEB proteins, whereas SP600125 and SB203580 had no dramatic effect (Fig. 3a). The effect of U0126 was also observed in other breast cancer cells (Fig. 3b). Although ZEB mRNA expression was repressed by U0126 in a time‐dependent manner, similar to that of Ets1 (Fig. 3c), we could not exclude the involvement of Ets1 phosphorylation in repression of ZEBs. To explore this issue, we mutated threonine 38 of Ets1, a phosphorylation site by ERK1/2, to alanine. This mutant was not recognized by anti‐phospho‐Ets1 (threonine 38) antibody (Fig. 3d, top panel), and it enhanced ZEB1 promoter activity as efficiently as the wild type (Flag‐Ets1) in HeLa cells (Fig. 3d) and MCF7 cells (Fig. S2a), suggesting that phosphorylation of threonine 38 is less important for activation of the ZEB1 promoter by Ets1. Therefore, ZEB expression appears to be regulated by the Ets1 protein itself or by post‐translational modifications of Ets1, including phosphorylation at residues other than threonine 38.
Figure 3.
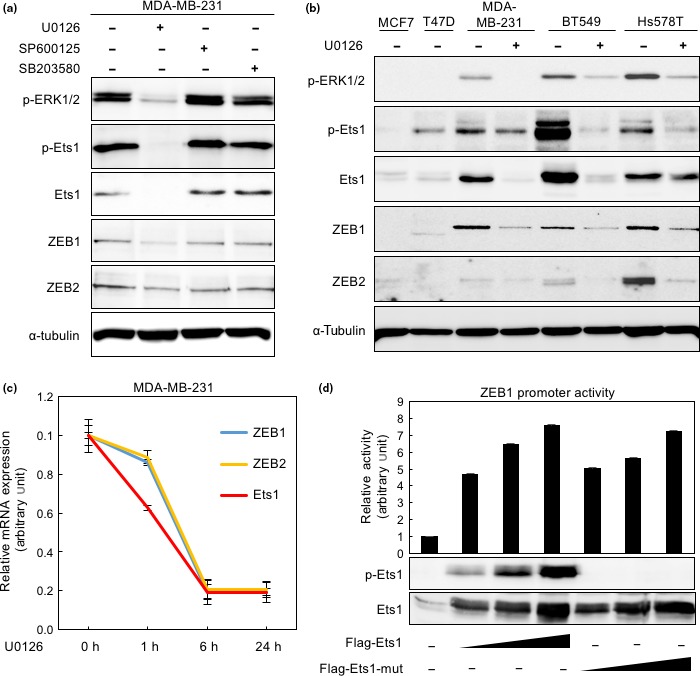
Effects of MEK1/2 inhibitor U0126 on expression of zinc‐finger E‐box binding homeobox (ZEB)1 and ZEB2. (a) MDA‐MB‐231 cells were treated with U0126, a JNK inhibitor (SP600125), and a p38 inhibitor (SB203580) at a concentration of 10 μM for 24 h.30, 31, 32, 33 Immunoblot analyses were carried out using the indicated antibodies. (b) Protein levels in breast cancer cells treated with U0126 (10 μM) for 24 h were determined by immunoblot analysis. (c) Relative levels of mRNA expression were determined by quantitative RT‐PCR analyses in MDA‐MB‐231 cells treated with U0126 (10 μM) for 0, 1, 6, or 24 h. Each value represents the mean ± SD of triplicate determinations from a representative experiment. Similar results were obtained in at least three independent experiments. The value at time point 0 is indicated as “1”. (d) hZEB1‐Luc was cotransfected into HeLa cells along with Flag‐tagged wild‐type Ets1 (Flag‐Ets1) or Flag‐tagged mutated Ets1 expression plasmid (Flag‐Ets1‐mut, threonine 38 converted to alanine), followed by measurement of luciferase activities. Expression levels of phosphorylated Ets1 (threonine 38) and total Ets1 were confirmed by immunoblot analyses using anti‐phospho‐Ets1 (threonine 38) and anti‐Flag antibodies (bottom). α‐Tubulin levels were monitored as a loading control (a,b). p‐, phospho‐.
Ets1‐enhanced ZEB1 promoter activity is inhibited by ESE1
Epithelium‐specific ETS transcription factor 1 is a member of the Ets transcription factors, which are preferentially expressed in epithelial cells.12 Both ESE1 and Ets1 are expressed reciprocally in breast cancer cells (Fig. 4a,b, see Fig. 1c).16 Because the MEK1/2 inhibitor, U0126, suppressed Ets1 expression in MDA‐MB‐231 cells (Fig. 3b), we examined whether U0126 upregulates ESE1 in the cells. Following U0126 treatment, ESE1 expression was moderately increased in MDA‐MB‐231 cells in a fashion reciprocal to ERK1/2 phosphorylation status (Fig. 4c). Conversely, when ERK1/2 was continuously and intensely activated by infection with active H‐Ras (RasG12V) in MCF7 cells, ESE1 expression was suppressed and Ets1 was upregulated, which were not observed in transient activation of ERK1/2 by stimulation with hepatocyte growth factor (Fig. 4d). These findings suggested that sustained MEK–ERK activation upregulates Ets1 and downregulates ESE1 in breast cancer cells.
Figure 4.
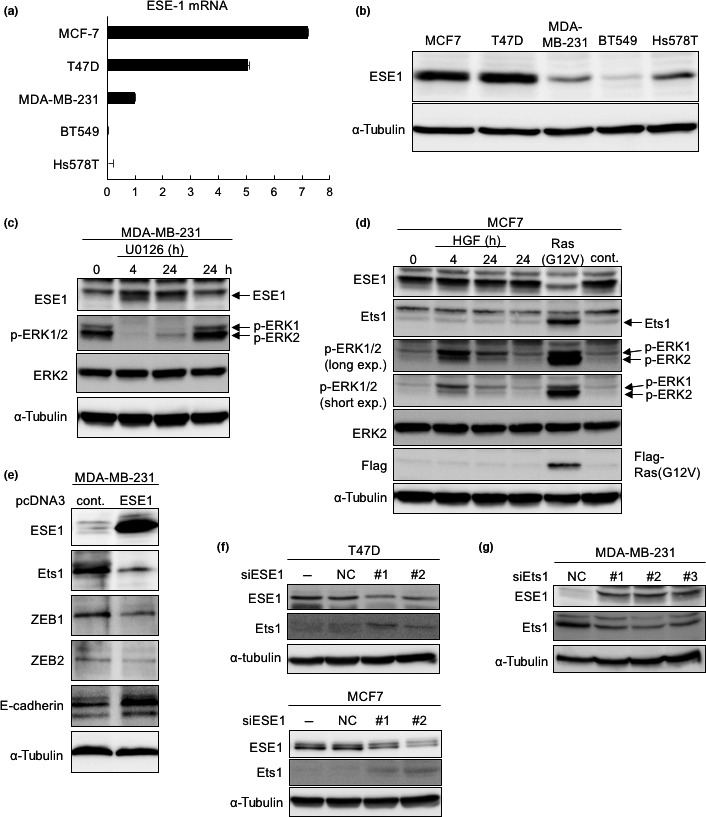
Role of epithelium‐specific E26 transformation‐specific transcription factor 1 (ESE1) in the luminal subtype of breast cancer cells (a,b). Expression of endogenous ESE1 was examined by quantitative RT‐PCR (a) and immunoblot (b) analyses. (c) After MDA‐MB‐231 cells were treated with U0126 (10 μM) for 4 or 24 h, immunoblot analyses were carried out using the indicated antibodies. (d) MCF7 cells were treated with hepatocyte growth factor (HGF; 100 ng/mL) for 4 or 24 h, or infected with control (cont.) or Flag‐H‐Ras (G12V) lentiviruses, followed by immunoblot analyses using the indicated antibodies. (e) Forty‐eight hours after transfection with expression plasmids encoding control (cont.) and ESE1 in MDA‐MB‐231 cells, levels of the indicated proteins were evaluated by immunoblot analyses. (f) Thirty‐six hours after transfection with control siRNA (NC) and two different kinds of ESE1 siRNAs (#1 and #2) in T47 (top) and MCF7 (bottom) cells, levels of the indicated proteins were evaluated by immunoblot analyses. (g) Thirty‐six hours after transfection with control siRNA (NC) and three different kinds of Ets1 siRNAs (#1, #2 and #3) in MDA‐MB‐231 cells, levels of the indicated proteins were evaluated by immunoblot analyses. α‐Tubulin levels were monitored as a loading control (b–g). p‐, phospho‐.
Next, we examined whether ESE1 represses expression of ZEBs. To investigate this, we prepared ESE1 cDNA using RNA isolated from MCF7 cells and cloned it into an expression vector. When ESE1 was transfected into MDA‐MB‐231 cells, the expression of ZEBs was suppressed, leading to partial acquisition of MET (Figs 4e,S2c), suggesting that ESE1 represses expression of ZEB1 and ZEB2. Interestingly, ESE1 also downregulated Ets1 expression in MDA‐MB‐231 cells; consistent with this, siRNAs targeting ESE1 restored ETS1 expression in the luminal‐subtype MCF7 and T47D cells (Fig. 4f). Reciprocally, Ets1 siRNA increased ESE1 expression in MDA‐MB‐231 cells (Fig. 4g). These findings suggested that ESE represses Ets1 expression, and in turn expression of ZEBs. Therefore, ESE1 and Ets1 reciprocally regulate expression of ZEBs, dependent on MEK–ERK activation, and ESE1 itself suppresses Ets1 expression in breast cancer cells.
Silencing of Ets1 sensitizes basal‐like breast cancer cells to antitumor drugs
Previous studies reported significant correlations between EMT and drug resistance.22 The basal‐like subtype of breast cancer cells transfected with Ets1 siRNAs expressed lower levels of ZEBs and showed partial changes in EMT marker protein expression (see Fig. 1d), suggesting that Ets1 siRNA partially converts cellular phenotypes from mesenchymal to epithelial. Thus, we investigated whether Ets1 siRNA could sensitize the basal‐like subtype of breast cancer cells to antitumor drugs. In both MDA‐MB‐231 and BT549 cells, transfection with Ets1 siRNA increased sensitivity to cisplatin and adriamycin (Figs 5,S3). Therefore, these findings suggest that Ets1 plays pivotal roles in maintaining EMT phenotypes by regulating ZEB expression in the basal‐like subtype of breast cancer cells, and that silencing of Ets1 sensitizes cells to antitumor drugs.
Figure 5.
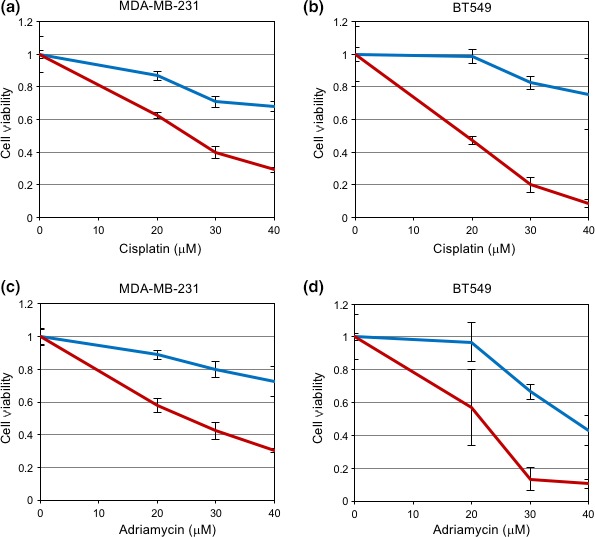
Silencing of Ets1 sensitizes breast cancer cells to antitumor drugs. (a–d) After transfection with control siRNA (blue) and Ets1 siRNA (red), MDA‐MB‐231 (a,c) and BT549 (b,d) cells were exposed to cisplatin (a,b) or adriamycin (c,d) at the indicated concentrations. Twenty‐four hours later, cell viability was evaluated by MTT assay. Each value represents the mean ± SD of triplicate determinations from a representative experiment. Similar results were obtained in at least three independent experiments using cells transfected with NC or Ets1 siRNAs.
Discussion
The mechanism of transcriptional regulation of ZEB1 and ZEB2 by Ets1 has not been fully elucidated. In this study, we found that phosphorylation of Ets1 at threonine 38 does not affect its ability to transactivate the ZEB1 promoter, and that U0126 downregulates expression of Ets1 and ZEBs at both the mRNA and protein levels (Figs 2, 3). The Ets1 promoter contains two consensus AP‐1 binding sites.23 Activator protein‐1 is a homo‐ or heterodimeric transcription factor composed of proteins of the Jun (c‐Jun, JunB, and JunD), Fos (c‐Fos, FosB, Fra1, and Fra2), or closely related activating factor (ATF‐2, ATF‐3, and B‐ATF) families, some of which are activated by ERK1/2. In addition, the Ets1 promoter also contains a typical Ets binding site,23 suggesting that ERK1/2 signals activate both AP‐1 and Ets1 (or Ets family/Ets‐like) pathways to upregulate Ets1 expression, possibly forming a positive‐feedback loop. In addition to its effect on transcription of Ets1, U0126 may affect the stability of Ets1 protein. The effect of threonine 38 phosphorylation on protein stability has not been fully elucidated. However, we found that U0126 downregulated Ets1 protein overexpressed from a transgene (data not shown). To date, several phosphorylation sites of Ets1 other than threonine 38 have been reported. For example, phosphorylation at serines 251, 257, and 285 by Ca2+/calmodulin‐dependent kinase II facilitates degradation of Ets1 protein.24 Phosphorylation of Ets1 at serines 276 and 282 facilitates binding of Ets1 to COP1 (ubiquitin ligase complex), thereby promoting ubiquitination and degradation of Ets1. Additionally, phosphorylation of tyrosine 138 in Ets1 by Src family kinases disrupts the interaction with COP1, increasing the stability of Ets1.25 Thus, it is possible that U0126 transcriptionally represses Ets1 by disrupting a positive‐feedback loop with AP‐1, and also destabilizes Ets1 protein by affecting its post‐translational modifications, independent of phosphorylation of threonine 38.
The ETS family of transcription factors comprises 28 members in humans, including Ets1 and Ets2. Ets1 and Ets2 share high sequence identity and conserved ERK1/2 phosphorylation sites. Because Ets family proteins act as homo‐ or heterodimers to regulate transcription, Ets1 can interact with Ets2.26 Overexpression of Ets2 in HeLa cells also activates the ZEB1 promoter, although siRNAs targeting Ets2 did not effectively repress ZEB1 expression (data not shown), suggesting that Ets1, rather than Ets2, is a major regulator of ZEB expression in breast cancer cells.
We cannot exclude the possibility that Ets family members other than Ets1 and Ets2 are involved in regulating ZEB expression in breast cancer cells. We found that ESE1 expression is regulated by the activation status of ERK1/2 in breast cancer cells (Fig. 4c,d); that is, most of the cell lines with high levels of ESE1 expression, and low levels of expression of ZEBs and Ets1 and phospho‐ERK1/2 status, were categorized into the “luminal” subtype of breast cancer cells. When the MEK–ERK pathway is activated by infection with active Ras, ESE1 and Ets1 are downregulated and upregulated, respectively (Fig. 4d). Conversely, most of the cell lines with low level of ESE1 expression, and high levels of expression of ZEBs and Ets1 and phospho‐ERK1/2 status were categorized into the “basal‐like” subtype of breast cancer cells. When the MEK–ERK pathway is inactivated by certain chemical compounds, such as U0126, ESE1 and Ets1 are upregulated and downregulated, respectively (Figs 3b,4c). These findings suggest that ESE1 and Ets1 control MET and EMT, respectively, through regulating ZEB expression in each subtype of breast cancer cells.
Moreover, ESE1 siRNAs increased Ets1 expression (Fig. 4f), whereas transfection with both ESE1 siRNA and Ets1 siRNA failed to induce expression of ZEBs (data not shown), suggesting that loss of ESE1 alone is not sufficient to regulate ZEB expression, and that upregulated Ets1 is important for upregulation of ZEBs. From loss of function experiments using either ESE1 siRNA or Ets1 siRNA, both proteins were shown to regulate each other (Fig. 4f,g). However, when Ets1 was ectopically overexpressed in MCF7 cells, expression of ESE1 was not dramatically repressed (data not shown), resulting in coexistence of both ESE1 and Ets1 in the cells. When both ESE1 and Ets1 were simultaneously expressed, ESE1 inhibited the Ets1‐promoted activity of hZEB1‐Luc without affecting protein levels of transfected Ets1 (Fig. S4). The underlying mechanism remains to be elucidated, because an interaction between Ets1 and ESE1 was not clearly detected (data not shown). Thus, Ets1 overexpression in MCF7 cells failed to induce ZEBs (data not shown).
Both ESE1 and Ets1 are dominantly expressed in the luminal and basal‐like subtypes of breast cancer cells (Figs 1c,4b), respectively.16 In prostate cancer cells and bronchial epithelial cells, ESE1 expression is induced by interleukin‐1β through nuclear factor‐κB.27, 28 By contrast, nuclear factor‐κB signals are activated in the basal‐like subtype of breast cancer cells,29 in which ESE1 is only poorly expressed. Thus, the molecular mechanism of ESE1 upregulation observed in the luminal subtype of breast cancer cells and normal epithelial cells remains largely unclear. In the present study, we found that reciprocal expression between ESE1 and Ets1 in breast cancer cells is largely dependent on activation status of the MEK–ERK pathway. In addition, activation of ERK2, rather than ERK1, appears to play a more dominant role in regulation of Ets family proteins (Fig. 4c,d), but the mechanism underlying reciprocal status of ERK1/2 phosphorylation in each subtype of breast cancer cells has not yet been elucidated.
The results of this study suggest that ESE1 is expressed in the luminal subtype of breast cancer cells and represses expression levels of Ets1 and ZEBs, resulting in low malignancy and favorable prognosis. Conversely, the basal‐like subtype of breast cancer cells, which is characterized by aggressive behavior and poor prognosis, expresses high levels of Ets1 and ZEBs and low levels of ESE1. Thus, Ets1 and ESE1, whose expressions are dependent on activation of the MEK–ERK pathway, act reciprocally by regulating ZEB expression during cancer progression in breast cancer.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- AP‐1
activator protein‐1
- δEF1
δ‐crystallin/E2‐box factor 1
- EMT
epithelial–mesenchymal transition
- ESE1
epithelium‐specific ETS transcription factor 1
- ETS
E26 transformation‐specific
- MET
mesenchymal–epithelial transition
- SIP1
Smad‐interacting protein 1
- ZEB
zinc‐finger E‐box binding homeobox
Supporting information
Fig. S1. Repression of zinc‐finger E‐box binding homeobox (ZEB)1 and ZEB2 by Ets1 siRNAs.
Fig. S2. Roles of epithelium‐specific E26 transformation‐specific transcription factor 1 (ESE1) and Ets1 in breast cancer cells.
Fig. S3. Silencing of Ets1 sensitizes breast cancer cells to antitumor drugs.
Fig. S4. Inhibition of Ets1‐promoted activity by epithelium‐specific E26 transformation‐specific transcription factor 1 (ESE1).
Acknowledgments
We thank Dr. T. Shirakihara for providing expression plasmids and for valuable discussions. This work was supported by the Japan Society for the Promotion of Science (JSPS KAKENHI Grant Nos. JP24390419 and JP15H05018), the research program of the Project for Development of Innovative Research on Cancer Therapeutics (P‐Direct) of the Ministry of Education, Culture, Sports, Science and Technology of Japan, and the JSPS Core‐to‐Core Program “Cooperative International Framework in TGF‐β Family Signaling”.
Cancer Sci 108 (2017) 952–960
Funding Information
Japan Society for the Promotion of Science (JP24390419 and JP15H05018); Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Perou CM, Sorlie T, Eisen MB et al Molecular portraits of human breast tumours. Nature 2000; 406: 747–52. [DOI] [PubMed] [Google Scholar]
- 2. Neve RM, Chin K, Fridlyand J et al A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10: 515–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Charafe‐Jauffret E, Ginestier C, Monville F et al Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 2006; 25: 2273–84. [DOI] [PubMed] [Google Scholar]
- 4. Foulkes WD, Smith IE, Reis‐Filho JS. Triple‐negative breast cancer. N Engl J Med 2010; 363: 1938–48. [DOI] [PubMed] [Google Scholar]
- 5. Horiguchi K, Sakamoto K, Koinuma D et al TGF‐β drives epithelial‐mesenchymal transition through deltaEF1‐mediated downregulation of ESRP. Oncogene 2012; 31: 3190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garrett‐Sinha LA. Review of Ets1 structure, function, and roles in immunity. Cell Mol Life Sci 2013; 70: 3375–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fafeur V, Tulasne D, Queva C et al The ETS1 transcription factor is expressed during epithelial‐mesenchymal transitions in the chick embryo and is activated in scatter factor‐stimulated MDCK epithelial cells. Cell Growth Differ 1997; 8: 655–65. [PubMed] [Google Scholar]
- 8. Shirakihara T, Saitoh M, Miyazono K. Differential regulation of epithelial and mesenchymal markers by δEF1 proteins in epithelial mesenchymal transition induced by TGF‐β. Mol Biol Cell 2007; 18: 3533–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Man AK, Young LJ, Tynan JA et al Ets2‐dependent stromal regulation of mouse mammary tumors. Mol Cell Biol 2003; 23: 8614–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Myers E, Hill AD, Kelly G et al Associations and interactions between Ets‐1 and Ets‐2 and coregulatory proteins, SRC‐1, AIB1, and NCoR in breast cancer. Clin Cancer Res 2005; 11: 2111–22. [DOI] [PubMed] [Google Scholar]
- 11. Wei G, Srinivasan R, Cantemir‐Stone CZ et al Ets1 and Ets2 are required for endothelial cell survival during embryonic angiogenesis. Blood 2009; 114: 1123–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oliver JR, Kushwah R, Hu J. Multiple roles of the epithelium‐specific ETS transcription factor, ESE‐1, in development and disease. Lab Invest 2012; 92: 320–30. [DOI] [PubMed] [Google Scholar]
- 13. Eckel KL, Tentler JJ, Cappetta GJ, Diamond SE, Gutierrez‐Hartmann A. The epithelial‐specific ETS transcription factor ESX/ESE‐1/Elf‐3 modulates breast cancer‐associated gene expression. DNA Cell Biol 2003; 22: 79–94. [DOI] [PubMed] [Google Scholar]
- 14. Iwai S, Amekawa S, Yomogida K et al ESE‐1 inhibits the invasion of oral squamous cell carcinoma in conjunction with MMP‐9 suppression. Oral Dis 2008; 14: 144–9. [DOI] [PubMed] [Google Scholar]
- 15. Chakrabarti R, Hwang J, Andres Blanco M et al Elf5 inhibits the epithelial‐mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nat Cell Biol 2012; 14: 1212–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. He J, Pan Y, Hu J, Albarracin C, Wu Y, Dai JL. Profile of Ets gene expression in human breast carcinoma. Cancer Biol Ther 2007; 6: 76–82. [DOI] [PubMed] [Google Scholar]
- 17. Fukagawa A, Ishii H, Miyazawa K, Saitoh M. deltaEF1 associates with DNMT1 and maintains DNA methylation of the E‐cadherin promoter in breast cancer cells. Cancer Med 2015; 4: 125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wei GH, Badis G, Berger MF et al Genome‐wide analysis of ETS‐family DNA‐binding in vitro and in vivo. EMBO J 2010; 29: 2147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wasylyk C, Bradford AP, Gutierrez‐Hartmann A, Wasylyk B. Conserved mechanisms of Ras regulation of evolutionary related transcription factors, Ets1 and Pointed P2. Oncogene 1997; 14: 899–913. [DOI] [PubMed] [Google Scholar]
- 20. Rochlitz CF, Scott GK, Dodson JM et al Incidence of activating ras oncogene mutations associated with primary and metastatic human breast cancer. Cancer Res 1989; 49: 357–60. [PubMed] [Google Scholar]
- 21. Shirakihara T, Kawasaki T, Fukagawa A et al Identification of integrin α3 as a molecular marker of cells undergoing epithelial‐mesenchymal transition and of cancer cells with aggressive phenotypes. Cancer Sci 2013; 104: 1189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell 2016; 166: 21–45. [DOI] [PubMed] [Google Scholar]
- 23. Seth A, Papas TS. The c‐ets‐1 proto‐oncogene has oncogenic activity and is positively autoregulated. Oncogene 1990; 5: 1761–7. [PubMed] [Google Scholar]
- 24. Dittmer J. The biology of the Ets1 proto‐oncogene. Mol Cancer 2003; 2: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu G, Zhang Q, Huang Y et al Phosphorylation of ETS1 by Src family kinases prevents its recognition by the COP1 tumor suppressor. Cancer Cell 2014; 26: 222–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Basuyaux JP, Ferreira E, Stehelin D, Buttice G. The Ets transcription factors interact with each other and with the c‐Fos/c‐Jun complex via distinct protein domains in a DNA‐dependent and ‐independent manner. J Biol Chem 1997; 272: 26188–95. [DOI] [PubMed] [Google Scholar]
- 27. Longoni N, Sarti M, Albino D et al ETS transcription factor ESE1/ELF3 orchestrates a positive feedback loop that constitutively activates NF‐κB and drives prostate cancer progression. Cancer Res 2013; 73: 4533–47. [DOI] [PubMed] [Google Scholar]
- 28. Wu J, Duan R, Cao H et al Regulation of epithelium‐specific Ets‐like factors ESE‐1 and ESE‐3 in airway epithelial cells: potential roles in airway inflammation. Cell Res 2008; 18: 649–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamaguchi N, Ito T, Azuma S et al Constitutive activation of nuclear factor‐κB is preferentially involved in the proliferation of basal‐like subtype breast cancer cell lines. Cancer Sci 2009; 100: 1668–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu Y, Mueller BM. Protease‐activated receptor‐2 regulates vascular endothelial growth factor expression in MDA‐MB‐231 cells via MAPK pathways. Biochem Biophys Res Commun 2006; 344: 1263–70. [DOI] [PubMed] [Google Scholar]
- 31. Park EJ, Kwon TK. Rottlerin enhances IL‐1beta‐induced COX‐2 expression through sustained p38 MAPK activation in MDA‐MB‐231 human breast cancer cells. Exp Mol Med 2011; 43: 669–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Palit S, Kar S, Sharma G, Das PK. Hesperetin induces apoptosis in breast carcinoma by triggering accumulation of ROS and activation of ASK1/JNK pathway. J Cell Physiol 2015; 230: 1729–39. [DOI] [PubMed] [Google Scholar]
- 33. Mingo‐Sion AM, Marietta PM, Koller E, Wolf DM, Van Den Berg CL. Inhibition of JNK reduces G2/M transit independent of p53, leading to endoreduplication, decreased proliferation, and apoptosis in breast cancer cells. Oncogene 2004; 23: 596–604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Repression of zinc‐finger E‐box binding homeobox (ZEB)1 and ZEB2 by Ets1 siRNAs.
Fig. S2. Roles of epithelium‐specific E26 transformation‐specific transcription factor 1 (ESE1) and Ets1 in breast cancer cells.
Fig. S3. Silencing of Ets1 sensitizes breast cancer cells to antitumor drugs.
Fig. S4. Inhibition of Ets1‐promoted activity by epithelium‐specific E26 transformation‐specific transcription factor 1 (ESE1).