

Abstract
Zinc‐finger protein 143 (ZNF143) is a transcription factor that is involved in anticancer drug resistance and cancer cell survival. In the present study, we identified a novel small molecule N‐(5‐bromo‐2‐methoxyphenyl)‐3‐(pyridine‐3‐yl) propiolamide (YPC‐21661) that inhibited ZNF143 promoter activity and down‐regulated the expression of the ZNF143‐regulated genes, RAD51, PLK1, and Survivin, by inhibiting the binding of ZNF143 to DNA. In addition, YPC‐21661 was cytotoxic and induced apoptosis in the human colon cancer cell line, HCT116 and human prostate cancer cell line, PC‐3. 2‐(pyridine‐3‐ylethynyl)‐5‐(2‐(trifluoromethoxy)phenyl)‐1,3,4‐oxadiazole (YPC‐22026), a metabolically stable derivative of YPC‐21661, induced tumor regression accompanied by the suppression of ZNF143‐regulated genes in a mouse xenograft model. The present study revealed that the inhibition of ZNF143 activity by small molecules induced tumor regression in vitro and in vivo; therefore, ZNF143 is a promising target of cancer therapeutics.
Keywords: anticancer drug, apoptosis, cell cycle, transcription factor, ZNF143
Zinc‐finger protein 143 (ZNF143) is a transcription factor that has been identified as a human homolog of Xenopus Staf.1 In humans, ZNF143 has been suggested to play roles in various activities.2 Previous studies demonstrated that ZNF143 is involved in the cell cycle, cell viability, and drug resistance.3, 4, 5, 6, 7, 8, 9, 10, 11 Furthermore, the knockdown of ZNF143 expression by small interfering RNA (siRNA) downregulated the expression of various cell cycle/DNA replication‐associated genes and inhibited the progression of the cell cycle and tumor cell growth.3 ZNF143 was also more strongly expressed in solid tumors than in adjacent non‐tumor samples.3 In clinical specimens, ZNF143 protein levels have been correlated with clinical outcomes in lung adenocarcinoma.12 Therefore, ZNF143 is considered to be a promising and unique cancer drug target. However, molecules that inhibit the activity of ZNF143 have not yet been identified. We herein identified a novel small molecule N‐(5‐bromo‐2‐methoxyphenyl)‐3‐(pyridine‐3‐yl) propiolamide (YPC‐21661) by screening in‐house chemical libraries for ZNF143 inhibitors. We demonstrated that YPC‐21661 down‐regulated the expression of ZNF143‐regulated genes. We also showed that YPC‐21661 and its derivative 2‐(pyridine‐3‐ylethynyl)‐5‐(2‐(trifluoromethoxy)phenyl)‐1,3,4‐oxadiazole (YPC‐22026) exhibited anti‐tumor activities in vitro and in vivo.
Materials and Methods
Reagents
YPC‐21661 and YPC‐22026 (Fig. 1) were chemically synthesized by Yakult Honsha (Tokyo, Japan). In in vitro assays, these drugs were dissolved in dimethyl sulfoxide (DMSO). The final concentration of DMSO in all treatments was adjusted to 0.1%. In the in vivo study, YPC‐22026 was dissolved in 5% glucose containing 10% (w/v) Tween80 and 5% (w/v) propylene glycol.
Figure 1.
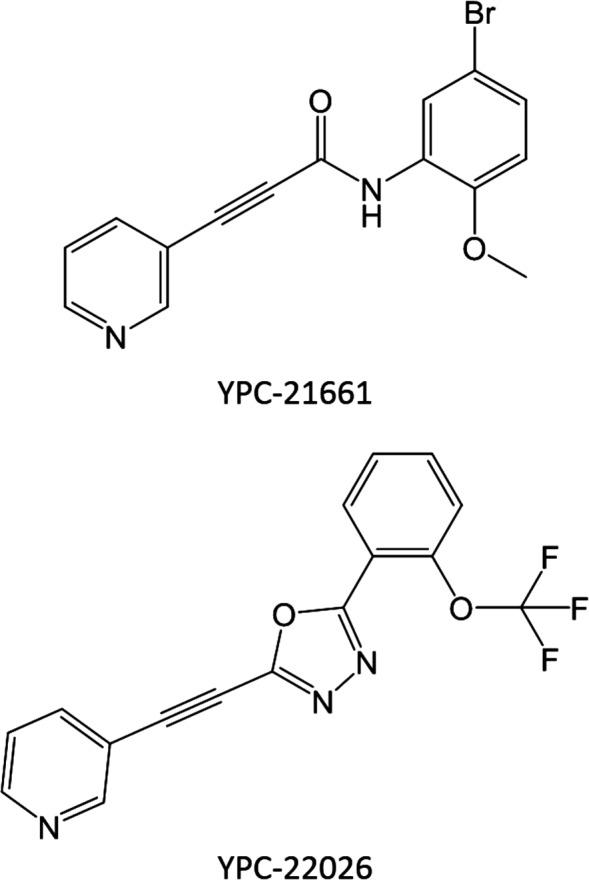
Chemical structures of YPC‐21661 and YPC‐22026.
Cell cultures
The human colon cancer cell lines HCT116 and HT‐29 were purchased from the American Type Culture Collection (Manassas, VA, USA). The human colon cancer cell line DLD‐1, human non‐small cell lung cancer cell line PC‐9, and human endometrial adenocarcinoma cell line Ishikawa were purchased from DS Pharma Biomedical (Osaka, Japan). The human prostate cancer cell line, PC‐3 and PC‐3 cells stably expressing 3xFlag‐tagged ZNF143 (PC‐3/3xFlag‐ZNF143) were obtained from the University of Occupational and Environmental Health, Japan.3 All cell lines were cultured in RPMI medium 1640 containing 10% (v/v) fetal bovine serum and maintained at 37°C in a 5% CO2 atmosphere.
Plasmid construct
In order to prepare the tandem repeat sequence of the Staf‐binding site, human genomic DNA was amplified by PCR with the following primer pair: 5′‐ACGCGTACTACGCCTCCCAGCGTGCTTTGCGGCGGGC‐3′ and 5′‐ CGGCCACTACGCCTCCCAGCGTGCTTTGCGGCTCGAG‐3′. Restriction enzyme sites are underlined. This PCR product was ligated into the MluI/XhoI site of the pGL3‐basic vector (Promega, Madison, WI, USA). This plasmid was named pGL3‐SBSx2‐Luc. The pGL3 control vector, which contains the SV40 promoter upstream of the luciferase gene, was purchased from Promega.
Reporter assays
PC‐3 cells that were stably transfected with the pGL3‐SBSx2‐Luc or pGL3 control vector were seeded on 96‐well white plates. YPC‐21661 or YPC‐22026 was added to each well the next day. After a 16‐h incubation, luciferase activity was detected using the Bright‐Glo luciferase assay system (Promega). The results obtained were normalized for a viable cell count measured by the MTT assay.
Quantitative RT‐PCR
Total RNA was isolated from cancer cells or resected xenograft tumors using an RNAspin mini kit (GE Healthcare Biosciences, Piscataway, NJ, USA), according to the manufacturer's protocol. First‐strand cDNA was synthesized from isolated total RNA using the GoScript Reverse Transcription System (Promega). Quantitative RT‐PCR was performed using the 7500 Fast real‐time PCR system (Thermo Fisher Scientific, Waltham, MA, USA) with the GoTaq qPCR Master Mix (Promega). The results obtained were normalized to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) as an endogenous control. The primer sequences used were as follows: human RAD51, 5′‐TGCTGATCCCAAAAAACCTATTG‐3′ (forward) and 5′‐CCCTCTTCCTTTCCTCAGATACAA‐3′ (reverse): human PLK1, 5′‐CCACCAGCACGTCGTAGGAT‐3′ (forward) and 5′‐CTTGACATGCTGCGGTGTTT‐3′ (reverse): human Survivin, 5′‐TGACGACCCCATAGAGGAACAT‐3′ (forward) and 5′‐TTCACCAAGGGTTAATTCTTCAAAC‐3′ (reverse): human GAPDH 5′‐CTCTCTGCTCCTGTTCGA‐3′ (forward) and 5′‐ACCTTCCCCATGGTGTCTGA‐3′ (reverse).
Western blotting
Cancer cells were lysed in solubilization buffer (10 mmol/L Tris‐HCl, pH 7.4, 0.1% [w/v] Nonidet P‐40, 0.1% [w/v] sodium deoxycholate, 0.1% [w/v] SDS, 150 mmol/L NaCl, 1 mmol/L EDTA, and 10 μg/mL aprotinin). Lysates were subjected to SDS‐PAGE using 10% (w/v) gels under reducing conditions. The separated proteins were electrotransferred to polyvinylidene difluoride membranes using a semi‐dry blotter. Membranes were treated with 5% (w/v) skimmed milk in 10 mmol/L Tris‐HCl, pH 7.4, 0.1% (v/v) Tween20, and 150 mmol/L NaCl for 1 h. Each membrane was then reacted with an anti‐Survivin antibody (AF‐886 R&D systems, Minneapolis, MN, USA), anti‐RAD51 antibody (sc‐8349 Santa Cruz, St. Louis, MO, USA), anti‐PLK1 antibody (37‐7000 Thermo Fisher Scientific), anti‐ZNF143 antibody (H00007702‐M01 Abnova, Taipei, Taiwan), anti‐Poly (ADP‐ribose) polymerase (PARP) antibody (9542S Cell Signaling Technologies, Danvers, MA, USA), anti‐Flag antibody (F1804 Sigma‐Aldrich, St. Louis, MO, USA), and anti‐ β‐actin antibody (A5316 Sigma‐Aldrich). After washing the membranes with T‐TBS, they were reacted with a horseradish peroxidase‐conjugated secondary antibody. They were then treated with an ECL kit (GE Healthcare Biosciences) and exposed to LAS‐3000 (GE Healthcare Biosciences).
Chromatin immunoprecipitation (ChIP) assays
PC‐3/3xFlag‐ZNF143 cells were treated with or without YPC‐21661 or YPC‐22026 for 16 hours. Protein–DNA crosslinking was performed by incubating cells with formaldehyde. Cells were lysed in radioimmunoprecipitation assay buffer (50 mmol/L Tris‐HCl [pH 7.5], 1 mmol/L EDTA, 150 mmol/L NaCl, 0.1% [w/v] SDS, 0.5% [w/v] sodium deoxycholate, 1% [v/v] Nonidet P‐40, and 1 mmol/L phenylmethylsulfonyl fluoride) and the lysates were sonicated. Soluble chromatin was incubated with an anti‐Flag (M2) affinity gel or anti‐mouse immunoglobulin G (IgG) with protein A/G agarose (Santa Cruz) by rotation at 4°C for 16 h. Immune complexes were collected by centrifugation. They were then treated with 0.2 mol/L NaCl to break protein–DNA crosslinking, and were digested with proteinase K and RNase A. Purified DNA was dissolved with H2O. DNA was then used in a PCR analysis with the following primer pairs for the PLK1 promoter region: 5′‐CCGCATCCACGCCGGGTTTGG‐3′ forward primer and 5′‐GCGCTGCAGACCTCGATCCGAGC‐3′ reverse primer. PCR products were separated by electrophoresis on 2% agarose gels and stained with ethidium bromide.
Cell viability assays
Cell viability was assayed in a 96‐well plate using TetraColor ONE (Kishida chemical, Osaka, Japan), according to the manufacturer's protocol. Briefly, exponentially growing cells were seeded at a density of 5 × 102 cells/well. Serially diluted YPC‐21661 or YPC‐22026 were added to each well the next day. After a 96‐h incubation, the 2‐(2‐methoxy‐4‐nitrophenyl)‐3‐(4‐nitrophenyl)‐5‐(2,4‐disulfophenyl)‐2H‐tetrazolium monosodium salt, WST‐8, was added to each well and the plates were incubated for 1 h at 37°C. Absorbance was then measured at 450 nm using a SPECTRA Max PLUS384 (Molecular Devices, Sunnyvale, CA, USA).
Evaluation of nuclear morphology
Cells were seeded in a 24‐well plate and treated with YPC‐21661. After a 24‐h incubation, cells were stained in 0.5 μg/mL Hoechst 33342 in culture medium for 30 min. Nuclear morphology was visually evaluated using a fluorescence microscope (Biozero BZ‐8100, Keyence, Osaka, Japan).
DNA fragmentation assays
DNA fragmentation, which is characteristic of apoptosis, was quantitatively evaluated using Cell Death Detection ELISA PLUS (Roche Life Science, Indianapolis, IN, USA), according to the manual instructions. Briefly, the cytoplasmic fractions of untreated control and YPC‐21661‐treated cells were transferred onto a streptavidin‐coated plate and incubated at room temperature for 2 h with a mixture of peroxidase‐conjugated anti‐DNA and biotin‐labeled anti‐histone. The plate was washed thoroughly and incubated with ABTS solution. Absorbance was then measured at 405 nm with a reference wavelength at 490 nm.
Analysis of cell cycle distribution
HCT116 cells were treated with or without YPC‐21661 or YPC‐22026. The attached cells were fixed and stained with propidium iodide using a cell cycle phase determination kit (Cayman Chemical, Ann Arbor, MI, USA), according to the manual instructions. Cell cycle distribution was analyzed by flow cytometry using a GUAVA EasyCyte Plus System (Merck Millipore, Darmstadt, Germany).
Metabolic stability in mouse microsomes
BALB/c mouse hepatic microsomes were purchased from XenoTech, LLC (Kansas City, KS, USA). A total of 0.5 mL of the reaction mixture, containing 0.5 mg/mL microsomal protein and 1 μmol/L of the test compound in 100 mmol/L phosphate buffer (pH 7.4), was preincubated at 37°C for 5 min, and the reaction was initiated by adding 30 μL of NADPH‐regenerating system solution (Corning Gentest, Woburn, MA, USA). The reaction mixture (50 μL) was sampled, and the reaction was terminated with 150 μL of 1% formic acid at 0, 10, and 30 min. All incubations were performed in duplicate. The test compound in the reaction mixture was measured by LC‐MS/MS using a Waters ACQUITY UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm). The column temperature and flow rate were 40°C and 0.4 mL/min, respectively. Mobile phases A and B were acetonitrile and 0.1% formic acid, respectively. Gradient elution was as follows: mobile phase A was linearly increased from 5% to 95% over a period of 2 min, maintained at 95% for 1 min, and then equilibrated at 5% for 2 min. Metabolic stability (half‐life, T 1/2) was calculated as follows.
Evaluation of antitumor activity in vivo
Six‐week‐old male BALB/c nude mice were purchased from Japan SLC, Inc. (Shizuoka, Japan). HCT116 cells were suspended in saline (2 × 107 cells/mL) and 0.1 mL of the cell suspension was injected subcutaneously into the flank of each mouse. When the tumor volume (length × width × width × 0.5) reached 50 to 120 mm3 (day 1), mice were randomly divided into three groups of five mice per group. YPC‐22026 was administered intraperitoneally at 50 and 100 mg/kg on days 1, 2, 3, 8, 9, 12, and 16 and days 1, 2, 3, 12, and 16, respectively. Tumor size was measured using calipers twice a week and the body weight of each mouse was monitored twice a week in order to assess toxicity. On day 16, antitumor activity was evaluated by weighing tumor tissues. The tumor growth inhibition rate, IR (%), was calculated as (1 – [average tumor weight of each treated group]/[average tumor weight of the control group]) × 100. The weight loss rate was calculated as (1 – [average body weight on Day 16]/[average body weight on Day 1]) × 100.
In quantitative RT‐PCR of xenograft tumors, 6‐week‐old male BALB/c nude mice were purchased from Japan SLC, Inc. PC‐3 cells were suspended in saline (2 × 107 cells/mL) containing 50% matrigel (Becton Dickinson, Franklin Lakes, NJ, USA) and 0.1 mL of the cell suspension was injected subcutaneously into the flank of each mouse. When tumor volumes reached 110–180 mm3 (day 1), mice were randomly divided into four groups of three mice per group. YPC‐22026 was administered intraperitoneally at 75 mg/kg on days 1, 2, and 3. After 4, 8, and 24 h from the final administration, tumors were resected and frozen in RNAlater (Thermo Fisher Scientific).
All experiments with animals were conducted in accordance with the Guidelines of the Yakult Central Institute and protocols approved by the Animal Experimental Committee of the Yakult Central Institute.
Statistical analysis
Statistical analyses were performed using Dunnett's test. P‐values <0.05 were considered significant.
Results
YPC‐21661 inhibits ZNF143 transcriptional activity and down‐regulates the expression of ZNF143 target genes
We first evaluated the effects of YPC‐21661 on the transcriptional activity of ZNF143 using a luciferase reporter gene assay. We found that YPC‐21661 inhibited ZNF143 promoter activity, but did not significantly inhibit SV40 promoter activity at concentrations up to 3.0 μmol/L (Fig. 2). ZNF143 plays important roles in the expression of DNA repair genes, cell cycle‐related genes, and anti‐apoptotic genes. Therefore, we examined the effects of YPC‐21661 on the expression of these genes using real‐time PCR and Western blotting. As shown in Figure 3, YPC‐21661 decreased the expression of ZNF143 target gene mRNAs (RAD51, PLK1 and Survivin) (Fig. 3a) and their respective proteins (Fig. 3b). Collectively, these results show that YPC‐21661 inhibits ZNF143 activity and down‐regulates the expression of its target genes.
Figure 2.
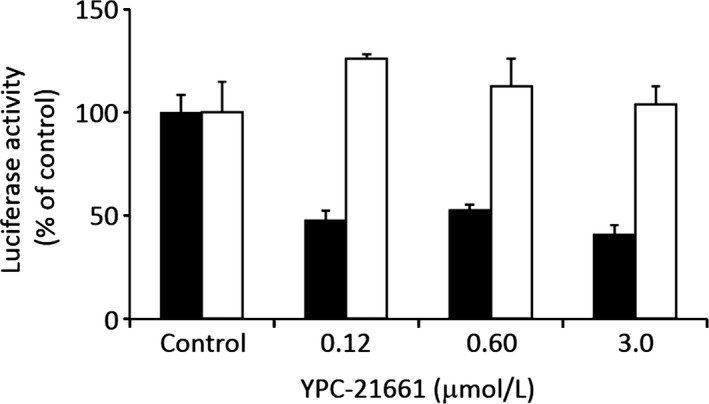
Effects of YPC‐21661 on ZNF143‐dependent luciferase gene activity. PC‐3 cells stably transfected with the pGL3‐SBSx2‐Luc (black bars) or pGL3 control vector (white bars) were treated with YPC‐21661 at the indicated concentrations. After 16 h, luciferase activity was measured. Data are the mean ± SD of triplicate determinations.
Figure 3.
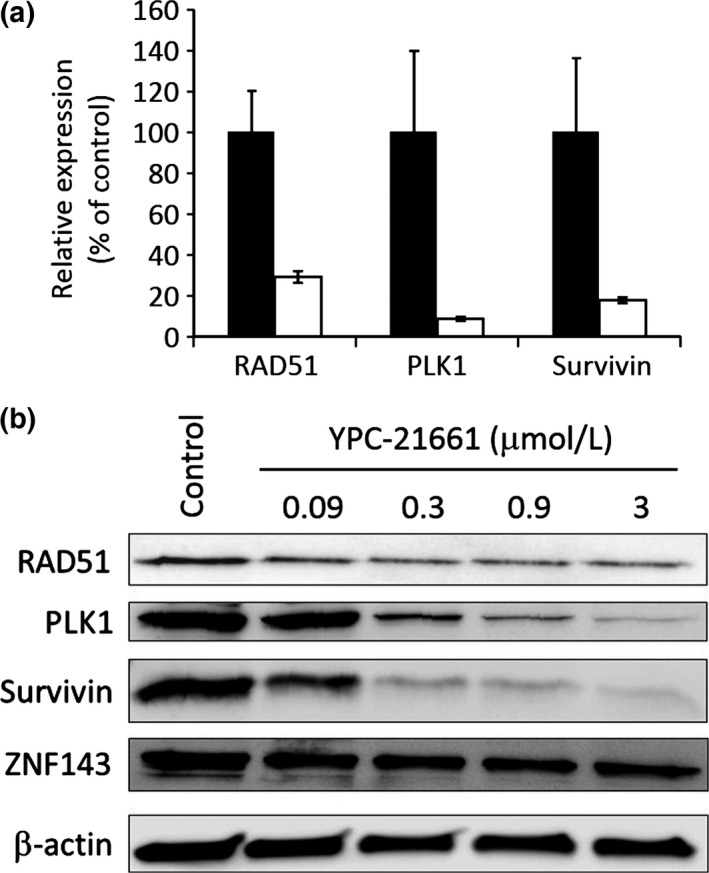
Effects of YPC‐21661 on expression levels of ZNF143 target genes. (a) PC‐3 cells were treated with (white bars) or without (black bars) YPC‐21661 (1.5 μmol/L) for 16 h. Cells were analyzed to detect ZNF143 target gene mRNAs by real‐time PCR. mRNA expression levels were normalized to that of GAPDH and expressed as relative expression levels (% of control). Data are the mean ± SD of triplicate determinations. (b) HCT116 cells were treated with YPC‐21661 for 16 h at the indicated concentrations. Cells were analyzed to detect ZNF143 target gene proteins and the ZNF143 protein by Western blotting.
YPC‐21661 inhibits the DNA binding of ZNF143
In order to investigate the effects of YPC‐21661 on ZNF143 binding at the STAF binding site in the promoter region of ZNF143 target genes, ChIP assays were performed after PC‐3/3xFlag‐ZNF143 was treated with YPC‐21661 for 24 h. As shown in Figure 4, YPC‐21661 decreased the binding of ZNF143 to the STAF binding site in a dose‐dependent manner (Fig. 4a,b).
Figure 4.
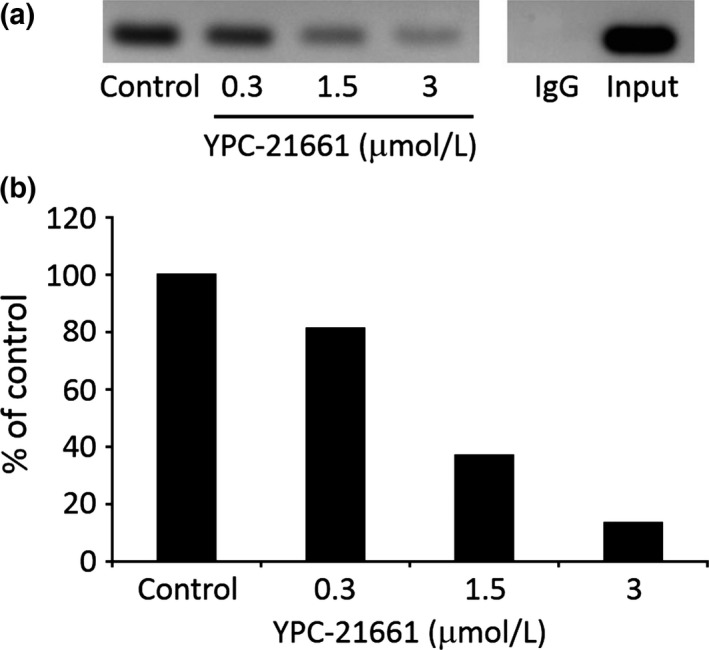
Effects of YPC‐21661 on DNA binding of ZNF143. (a) PC‐3/3xFlag‐ZNF143 was treated with YPC‐21661 for 16 h at the indicated concentrations. Soluble chromatin was incubated with an anti‐Flag (M2) affinity gel or anti‐mouse IgG with protein A/G agarose by rotation at 4°C for 16 h. Purified DNA was amplified using primer pairs for the PLK1 promoter region. (b) The amounts of PCR products were quantified by a densitometric analysis.
YPC‐21661 is cytotoxic to tumor cells and inhibits cell cycle progression by inducing apoptosis
The cytotoxic effects of YPC‐21661 were examined in HCT116 and PC‐3 cells. YPC‐21661 was potently cytotoxic to HCT116 and PC‐3 cells with 50% inhibitory concentration (IC50) values of 6.0 and 6.8 nmol/L, respectively (Fig. 5a). We then analyzed the effects of YPC‐21661 on cell cycle distribution because ZNF143 regulates the expression of cell cycle‐related genes.3, 7, 8 Histograms were shown in Figure 5(b). Increases in the numbers of G2/M phase cells and subG1 cells were observed when HCT116 cells were treated with YPC‐21661 at 0.1 μmol/L (Fig. 5b). Increases in the numbers of subG1 cells suggest that YPC‐21661 induces apoptosis; therefore, we investigated whether the cytotoxic effects of YPC‐21661 are caused by apoptosis. PC‐3 and HCT116 cells were treated with YPC‐21661 and stained with Hoechst 33342. Both cells showed nuclear morphological changes and chromatin condensation, consistent with apoptosis in response to YPC‐21661 (Fig. 6a). As another confirmatory test for YPC‐21661‐mediated apoptosis, we performed an ELISA assay to detect fragmented DNA and Western blotting for cleaved poly (ADP‐ribose) polymerase (PARP). As shown in Figure 6(b, c), the treatment with YPC‐21661 led to the accumulation of fragmented DNA at 0.3 μmol/L (Fig. 6b) and induced PARP cleavage in a dose‐dependent manner (Fig. 6c).
Figure 5.
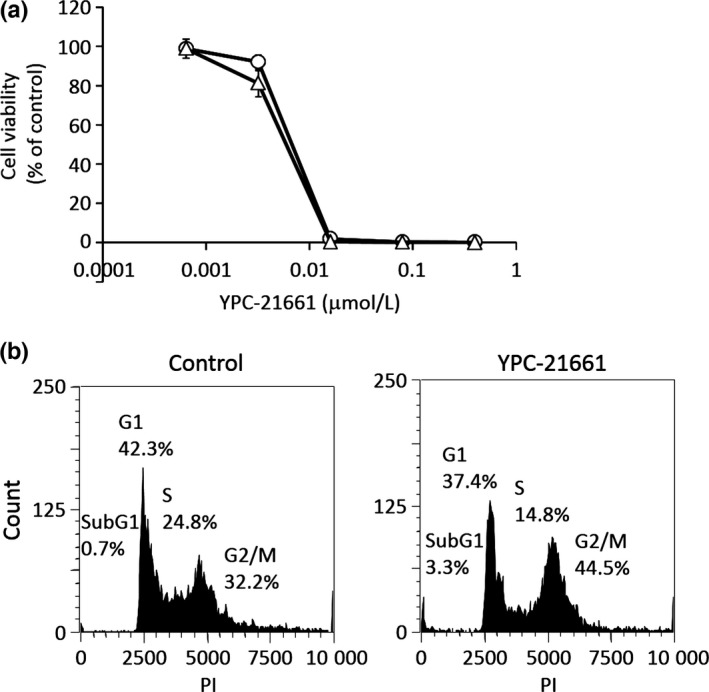
Effects of YPC‐21661 on cell viability and the cell cycle. (a) PC‐3 (open circles) and HCT116 (open triangles) cells were treated with YPC‐21661 for 96 h. Cell viability was assessed by WST‐8 and presented as a percentage of the value for the control culture. (b) HCT116 cells were treated with or without YPC‐21661 at 0.1 μmol/L for 24 h. Cells were stained with propidium iodide and the DNA content in a single cell was measured using a flow cytometer.
Figure 6.
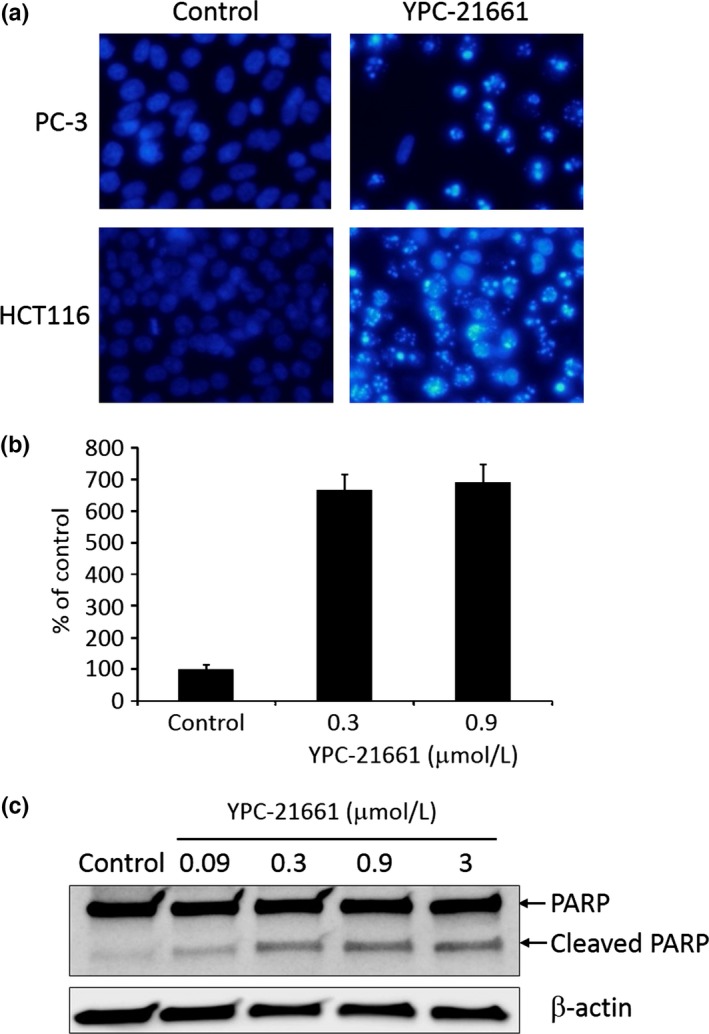
Effects of YPC‐21661 on apoptosis. (a) PC‐3 and HCT116 cells treated with YPC‐21661 for 24 h at 0.3 μmol/L were stained with the DNA binding dye Hoechst 33342. Condensed and fragmented nuclei were observed under a fluorescent microscope. (b) HCT116 cells were treated with YPC‐21661 at the indicated concentrations for 24 h. Fragmented DNA was quantified, according to the manual instructions. The amount of fragmented DNA was expressed as a relative value (% of control). Data are the means ± SD of triplicate determinations. (c) Western blotting for PARP and cleaved PARP expressed in HCT116 cells treated with YPC‐21661 for 16 h at the indicated concentrations.
These results demonstrate that YPC‐21661 is cytotoxic to tumor cells by inducing G2/M phase cell cycle arrest and apoptosis.
YPC‐22026 inhibits the growth of human xenograft tumors by inhibiting the expression of ZNF143‐regulated genes
Since YPC‐21661 is unstable in mouse liver microsomes, we synthesized YPC‐22026 in order to improve stability. The in vitro activity of YPC‐22026 was evaluated using a luciferase reporter assay and MTT assay. YPC‐22026 inhibited ZNF143 activity with an IC50 value of 9.0 μmol/L (Fig. 7a), and was cytotoxic to HCT116 and PC‐3 with IC50 values of 0.33 and 0.66 μmol/L, respectively (Fig. 7b). In addition, YPC‐22026 induced G2/M arrest (Fig. S1A) and decreased the binding of ZNF143 to the STAF binding site (Fig. S1B). The in vitro activity of YPC‐22026 was weaker than that of YPC‐21661, whereas YPC‐22026 exhibited cytotoxicity by inhibiting ZNF143 activity. Therefore, we evaluated the in vivo antitumor activity of YPC‐22026. In the HCT116 xenograft model, the treatment with YPC‐22026 at 50 or 100 mg/kg significantly inhibited the growth of tumors, with IR values of 40.8% and 56.1%, respectively, on day 22 (Fig. 7c). In order to confirm whether YPC‐22026 alters ZNF143 activity in tumors during the drug treatment, we evaluated the effects of YPC‐22026 on the intratumoral expression of the ZNF143 target genes, RAD51, PLK1, and Survivin. Mice with xenografted PC‐3 tumors received a 3‐day continuous infusion of YPC‐22026 at 75 mg/kg. Animals treated with YPC‐22026 showed a significant decrease in the intratumoral expression levels of all ZNF143 target genes analyzed after 8 and 24 h from the last administration (Fig. 7d). These results show that YPC‐22026 inhibits the growth of tumors by decreasing the intratumoral expression levels of ZNF143 target genes.
Figure 7.
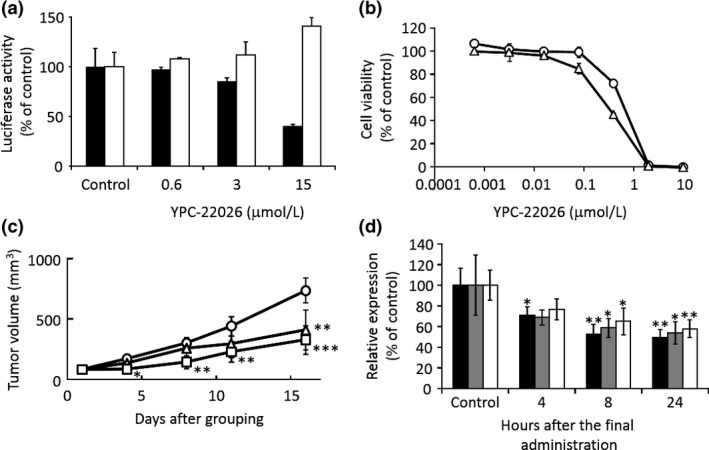
Effects of YPC‐22026 on tumor growth and ZNF143 activity in vitro and in vivo. (a) PC‐3 cells stably transfected with the pGL3‐SBSx2‐Luc (black bars) or pGL3 control vector (white bars) were treated with YPC‐22026 at the indicated concentrations. After 16 h, luciferase activity was measured. Data are the means ± SD of triplicate determinations. (b) PC‐3 (open circles) and HCT116 (open triangles) cells were treated with YPC‐22026 for 96 h. Cell viability was assessed by WST‐8 and presented as a percentage of the value for the control culture. (c) BALB/c nude mice (n = 5/group) were inoculated subcutaneously with HCT116 cells. After tumors had formed, mice were treated intraperitoneally with 50 (open triangles) or 100 (open squares) mg/kg YPC‐22026 or with vehicle only (open circles). Data are expressed as the mean ± SD *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle control. (d) Mice bearing PC‐3 xenograft tumors (n = 3/group) were treated intraperitoneally with YPC‐22026 at 75 mg/kg. The treatment was given on days 1–3. After the indicated hours from the last administration, tumors were resected and the expression of the ZNF143 target genes, RAD51 (black bars), Survivin (gray bars), and PLK1 (white bars), was analyzed. Data are expressed as the mean ± SD *P < 0.05, **P < 0.01 versus vehicle control.
Discussion
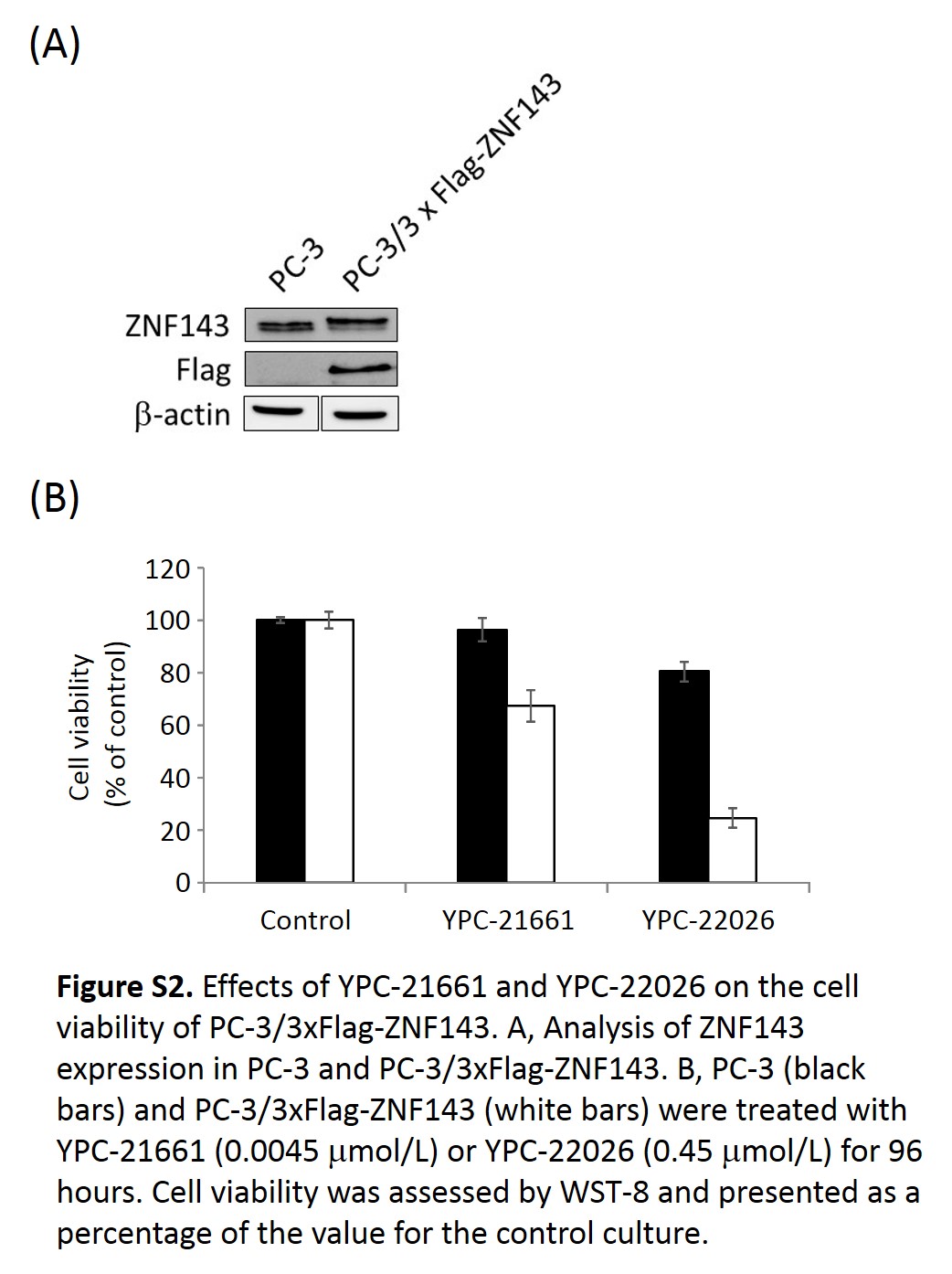
YPC‐21661 is a novel ZNF143 inhibitor that has been identified using a luciferase reporter assay. YPC‐21661 downregulated the expression of ZNF143 target genes by inhibiting the binding of ZNF143 to DNA. In addition, YPC‐21661 was cytotoxic to cancer cells through its induction of G2/M phase cell cycle arrest and apoptosis. These results were consistent with previous findings obtained from ZNF143 knockdown experiments.3 This agreement confirmed that YPC‐21661 inhibits ZNF143 activity. In addition, the ZNF143‐over‐expressing cell line, PC‐3/3xFlag‐ZNF143, was more vulnerable to YPC‐21661 and YPC‐22026 than parent cells (Fig. S2A,B), suggesting that the efficacy of YPC compounds depend on ZNF143 expression levels. These results also indicate that YPC compounds are potent inhibitors of ZNF143. On the other hand, the efficacy of YPC compounds on cancer cell lines that intrinsically express ZNF143 was not related to ZNF143 expression levels (Fig. S3 and Table S1). This discordance may be attributed to differences in genetic backgrounds. PC‐3 and PC‐3/3xFlag‐ZNF143 have similar genetic backgrounds; therefore, the efficacy of YPC compounds is relevant to ZNF143 expression levels. However, the cancer cells used in the present study have various genetic backgrounds, and their growth is not decided based only on the expression levels of ZNF143; therefore, the efficacies of YPC compounds are not relevant to ZNF143 expression levels. YPC‐21661‐induced apoptosis may be caused by the down‐regulation of the anti‐apoptotic factor Survivin (Fig. 3b). However, previous studies reported that the down‐regulation of Survivin reduces the number of G2/M phase cells.13, 14 Therefore, G2/M arrest induced by YPC‐21661 does not appear to be essential to apoptosis.
YPC‐22026, designed to improve stability in human liver microsomes, exhibited anti‐tumor activity in the HCT116 xenograft model by inhibiting the expression of ZNF143 target genes. This in vivo anti‐tumor activity suggests that ZNF143 inhibitors are promising anti‐cancer drugs.
ZNF143 is a transcription factor related to the cell cycle. E2F and c‐Myc are well‐known transcription factors that are involved in cell cycle progression and cancer malignancy. Small molecules that inhibit E2F or c‐Myc transcriptional activity induce cell cycle arrest at the G0/G1 phase.15, 16, 17, 18, 19, 20 On the other hand, the inhibition of ZNF143 by YPC‐21661 induced G2/M arrest (Fig. 5b) in accordance with the findings of a previous knockdown analysis.3 These results indicate that the role of ZNF143 in cancer cell proliferation is distinct from those of E2F and c‐Myc.
In the in vivo study, the amide bond included in YPC‐21661 is metabolically unstable. Therefore, we converted the amide bond for 1,3,4‐oxadiazole, which is a bioisostere of the amide bond, in order to improve the elimination half‐life of YPC‐22026 in mouse microsomes over that of YPC‐21661 (34.4 and <1.7 min, respectively). ZNF143 controls the expression of various genes related to cell survival,3, 7, 8, 9, 10 and ZNF143 is essential for normal development in zebrafish.21 Therefore, we anticipated that ZNF143 inhibitors exhibit severe toxicity against mice. However, the toxicity of YPC‐22026 against mice only led to slight weight loss (<20%) at therapeutically effective doses (data not shown).
The expression of ZNF143 is activated by DNA damage reagents such as etoposide, cisplatin, and adriamycin.6 In addition, ZNF143 binds to cisplatin‐modified DNA and is involved in cisplatin resistance;4, 5, 6 therefore, YPC‐21661 and 22026 may augment the efficacy of cisplatin. Further extensive studies on this matter are needed.
In the present study, we demonstrated that YPC‐21661 inhibited the activity of ZNF143; however, the mechanisms responsible for its inhibitory effects have not yet been elucidated. Therefore, further studies are needed in order to develop YPC‐22026 or its derivatives as novel anti‐cancer drugs. In conclusion, YPC‐21661 and YPC‐22026 appear to be the first ZNF143 inhibitors to exhibit anti‐cancer activities in vitro and in vivo. These results indicate that YPC‐21661 and 22026 are promising first‐in‐class drug seeds.
Disclosure Statement
Hirotaka Haibara, Ryuta Yamazaki, Yukiko Nishiyama, Masahiro Ono, Tsuneyuki Kobayashi, Atsuko Hokkyo‐Itagaki, Fukiko Nishisaka, Hiroyuki Nishiyama, Akinobu Kurita, and Takeshi Matsuzaki are employees of Yakult Honsha Co., Ltd., Tokyo, Japan.
Supporting information
Fig. S1. Effects of YPC‐22026 on the cell cycle and DNA binding of ZNF143.
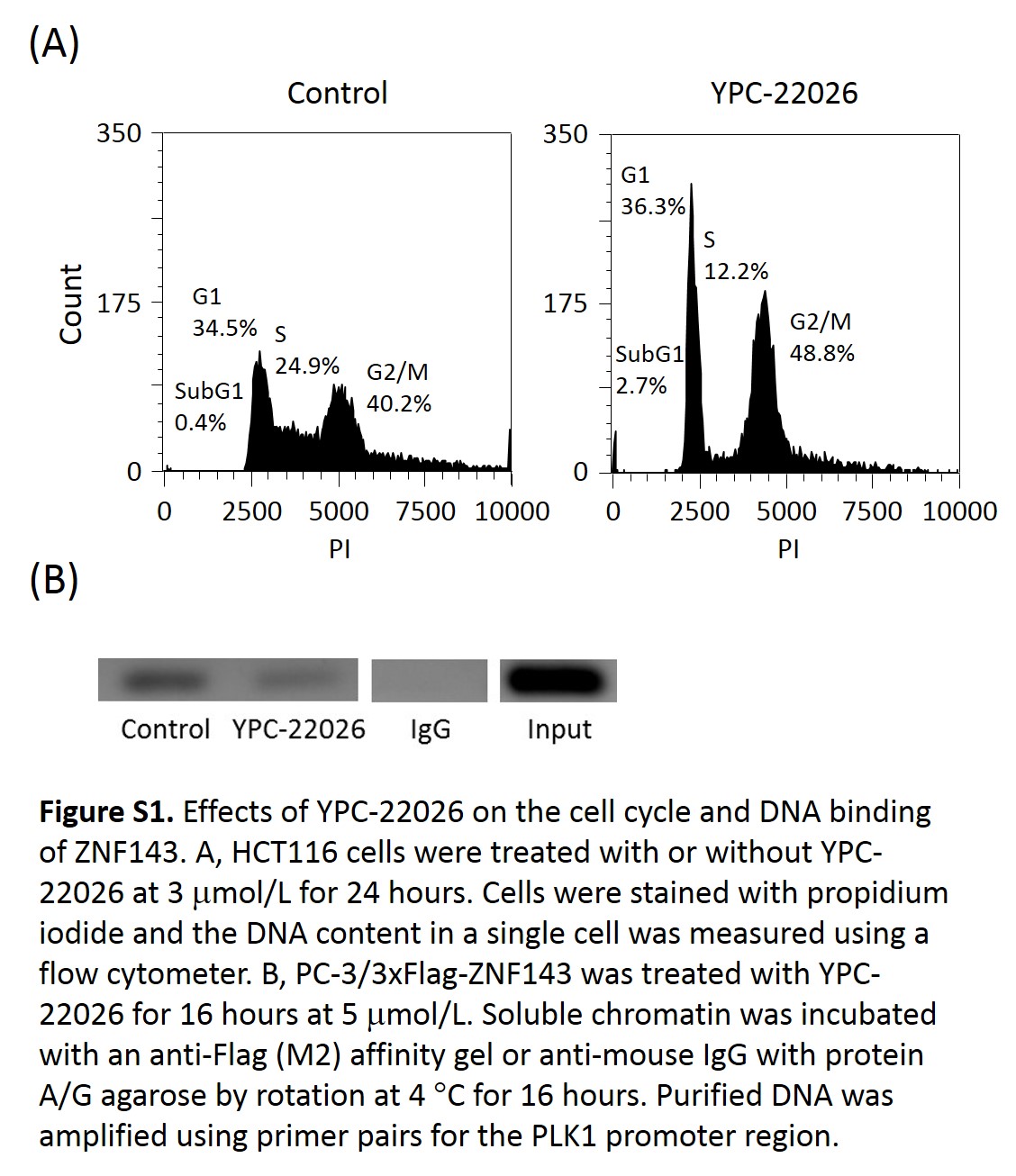{kind=link}
Fig. S2. Effects of YPC‐21661 and YPC‐22026 on the cell viability of PC‐3/3xFlag‐ZNF143.
{kind=link}
Fig. S3. Analysis of ZNF143 expression in cancer cells.
{kind=link}
Table S1. IC50 values by the 96‐h treatment of YPC‐21661 and YPC‐22026 in human cancer cell lines.
Cancer Sci 108 (2017) 1042–1048
References
- 1. Myslinski E, Krol A, Carbon P. ZNF76 and ZNF143 are two human homologs of the transcriptional activator Staf. J Biol Chem 1998; 273: 21998–2006. [DOI] [PubMed] [Google Scholar]
- 2. Myslinski E, Gérard MA, Krol A, Carbon P. A genome scale location analysis of human Staf/ZNF143‐binding sites suggests a widespread role for human Staf/ZNF143 in mammalian promoters. J Biol Chem 2006; 281: 39953–62. [DOI] [PubMed] [Google Scholar]
- 3. Izumi H, Wakasugi T, Shimajiri S et al Role of ZNF143 in tumor growth through transcriptional regulation of DNA replication and cell‐cycle‐associated genes. Cancer Sci 2010; 101: 2538–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wakasugi T, Izumi H, Uchiumi T et al ZNF143 interacts with p73 and is involved in cisplatin resistance through the transcriptional regulation of DNA repair genes. Oncogene 2007; 26: 5194–203. [DOI] [PubMed] [Google Scholar]
- 5. Torigoe T, Izumi H, Ishiguchi H et al Cisplatin resistance and transcription factors. Curr Med Chem Anticancer Agents 2005; 5: 15–27. [DOI] [PubMed] [Google Scholar]
- 6. Ishiguchi H, Izumi H, Torigoe T et al ZNF143 activates gene expression in response to DNA damage and binds to cisplatin‐modified DNA. Int J Cancer 2004; 111: 900–9. [DOI] [PubMed] [Google Scholar]
- 7. Izumi H, Yasuniwa Y, Akiyama M et al Forced expression of ZNF143 restrains cancer cell growth. Cancers (Basel) 2011; 3: 3909–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Myslinski E, Gérard MA, Krol A, Carbon P. Transcription of the human cell cycle regulated BUB1B gene requires hStaf/ZNF143. Nucleic Acids Res 2007; 35: 3453–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu W, Chen Z, Zhang H, Wang Y, Luo Y, Huang P. ZNF143 transcription factor mediates cell survival through upregulation of the GPX1 activity in the mitochondrial respiratory dysfunction. Cell Death Dis 2012; 3: e422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van der Lee R, Szklarczyk R, Smeitink J, Smeets HJ, Huynen MA, Vogel R. Transcriptome analysis of complex I‐deficient patients reveals distinct expression programs for subunits and assembly factors of the oxidative phosphorylation system. BMC Genom 2015; 16: 691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Paek AR, Kim SH, Kim SS, Kim KT, You HJ. IGF‐1 induces expression of zinc‐finger protein 143 in colon cancer cells through phosphatidylinositide 3‐kinase and reactive oxygen species. Exp Mol Med 2010; 42: 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kawatsu Y, Kitada S, Uramoto H et al The combination of strong expression of ZNF143 and high MIB‐1 labelling index independently predicts shorter disease‐specific survival in lung adenocarcinoma. Br J Cancer 2014; 110: 2583–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu W, Zhu F, Jiang Y, Sun D, Yang B, Yan H. siRNA targeting survivin inhibits the growth and enhances the chemosensitivity of hepatocellular carcinoma cells. Oncol Rep 2013; 29: 1183–8. [DOI] [PubMed] [Google Scholar]
- 14. Qin Q, Cheng H, Lu J et al Small‐molecule survivin inhibitor YM155 enhances radiosensitization in esophageal squamous cell carcinoma by the abrogation of G2 checkpoint and suppression of homologous recombination repair. J Hematol Oncol 2014; 7: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hahm ER, Singh SV. Honokiol causes G0‐G1 phase cell cycle arrest in human prostate cancer cells in association with suppression of retinoblastoma protein level/phosphorylation and inhibition of E2F1 transcriptional activity. Mol Cancer Ther 2007; 6: 2686–95. [DOI] [PubMed] [Google Scholar]
- 16. Oliveira AR, Beyer G, Chugh R et al Triptolide abrogates growth of colon cancer and induces cell cycle arrest by inhibiting transcriptional activation of E2F. Lab Invest 2015; 95: 648–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luo X, Peng JM, Su LD, Wang DY, Yu YJ. Fangchinoline inhibits the proliferation of SPC‐A‐1 lung cancer cells by blocking cell cycle progression. Exp Ther Med 2016; 11: 613–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin CP, Liu JD, Chow JM, Liu CR, Liu HE. Small‐molecule c‐Myc inhibitor, 10058‐F4, inhibits proliferation, downregulates human telomerase reverse transcriptase and enhances chemosensitivity in human hepatocellular carcinoma cells. Anticancer Drugs 2007; 18: 161–70. [DOI] [PubMed] [Google Scholar]
- 19. Zhang M, Fan HY, Li SC. Inhibition of c‐Myc by 10058‐F4 induces growth arrest and chemosensitivity in pancreatic ductal adenocarcinoma. Biomed Pharmacother 2015; 73: 123–8. [DOI] [PubMed] [Google Scholar]
- 20. Fry DW, Harvey PJ, Keller PR et al Specific inhibition of cyclin‐dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 2004; 3: 1427–38. [PubMed] [Google Scholar]
- 21. Halbig KM, Lekven AC, Kunkel GR. The transcriptional activator ZNF143 is essential for normal development in zebrafish. BMC Mol Biol 2012; 13: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Effects of YPC‐22026 on the cell cycle and DNA binding of ZNF143.
Fig. S2. Effects of YPC‐21661 and YPC‐22026 on the cell viability of PC‐3/3xFlag‐ZNF143.
Fig. S3. Analysis of ZNF143 expression in cancer cells.
Table S1. IC50 values by the 96‐h treatment of YPC‐21661 and YPC‐22026 in human cancer cell lines.