

Abstract
Rationale: Idiopathic pulmonary fibrosis (IPF) is a chronic pulmonary disorder of unknown etiology with few treatment options. Although tetraspanins are involved in various diseases, their roles in fibrosis have not been determined.
Objectives: To investigate the role of tetraspanin CD151 in pulmonary fibrosis.
Methods: CD151 knockout (KO) mice were studied by histological, biochemical, and physiological analyses and compared with wild-type mice and CD9 KO mice. Further mechanistic analyses were performed in vitro, in vivo, and on samples from patients with IPF.
Measurements and Main Results: A microarray study identified an enrichment of genes involved in connective tissue disorders in the lungs of CD151 KO mice, but not in CD9 KO mice. Consistent with this, CD151 KO mice spontaneously exhibited age-related pulmonary fibrosis. Deletion of CD151 did not affect pulmonary fibroblast functions but instead degraded epithelial integrity via attenuated adhesion strength on the basement membrane; CD151-deleted alveolar epithelial cells exhibited increased α-SMA expression with activation of p-Smad2, leading to fibrotic changes in the lungs. This loss of epithelial integrity in CD151 KO lungs was further exacerbated by intratracheal bleomycin exposure, resulting in severe fibrosis with increased mortality. We also observed decreased numbers of CD151-positive alveolar epithelial cells in patients with IPF.
Conclusions: CD151 is essential for normal function of alveolar epithelial cells; loss of CD151 causes pulmonary fibrosis as a result of epithelial disintegrity. Given that CD151 may protect against fibrosis, this protein represents a novel target for the treatment of fibrotic diseases.
Keywords: alveolar epithelial cell, adhesion strength, epithelial-to-mesenchymal transition, Smad2, CD9
At a Glance Commentary
Scientific Knowledge on the Subject
Although tetraspanin CD151 is abundantly expressed in the lung and plays a critical role in epithelial cells, its role in pulmonary fibrosis has not been determined.
What This Study Adds to the Field
Deletion of CD151 in mice results in progressive pulmonary fibrosis. The underlying mechanism could involve loss of epithelial integrity: deletion of CD151 in alveolar epithelial cells attenuates the strength of adhesion to the basement membrane, resulting in mesenchymal-like changes and increased p-Smad2 signaling.
Idiopathic pulmonary fibrosis (IPF) is a chronic pulmonary disorder of unknown etiology; the condition has a poor prognosis, and there are no effective treatments. IPF is characterized by progressive deposition of collagen and other extracellular matrix (ECM) molecules (1). Previously, IPF was viewed as a “smoldering” inflammatory response that ultimately led to chronic lung injury with subsequent fibrosis. However, inflammation is now not regarded as crucial to the etiology, largely because current antiinflammatory therapies for IPF have provided little benefit for patients (2). Instead, it has become clear that abnormal behavior of alveolar epithelial cells (AECs) is a primary source of the development of pulmonary fibrosis (3, 4). The disease process is initiated through repetitive injury of AECs, causing AEC activation, which in turn leads to recruitment of immune cells and fibroblasts within the lung microenvironment. Aberrant activated AECs, in cooperation with migrated immune cells and fibroblasts, secrete and activate latent transforming growth factor (TGF)-β1, as well as other profibrotic factors, which promote the differentiation of fibroblasts and AECs to myofibroblasts, resulting in overproduction of ECM in the lung.
The tetraspanins are a protein family comprising at least 33 members in mammals. Tetraspanins contain four transmembrane regions and associate with a variety of membrane proteins, including integrins, growth factors, and intracellular signaling molecules (5). Tetraspanins facilitate the formation of these multimolecular complexes, thereby regulating cell activation, fusion, motility, and signaling (6). Thus, tetraspanins are involved in a number of normal and pathological processes, such as tumor progression, angiogenesis, and infectious diseases (7). For example, tetraspanins CD9 and CD151 regulate tumor progression, either negatively (CD9) or positively (CD151), in multiple tumor types (7, 8). Specifically regarding lung diseases, CD9 plays protective roles in lung inflammation and emphysema, which we have studied using genetic models in mice (9, 10).
In contrast to CD9, which is ubiquitously expressed at the apical and lateral sides of the cell, CD151 are predominantly expressed at the basolateral surface of epithelial and endothelial cells. Moreover, CD151 and a homologous tetraspanin, TSP-15, are essential for maintenance of epithelial characteristics (11, 12), which is crucial for the development and maintenance of lung structure. However, the involvement of CD151 in lung diseases has remained unclear. Recently, close attention has been paid to β1 integrins in AECs and fibroblasts, focusing on their potential roles in pulmonary fibrosis (13, 14). Therefore, we investigated whether CD151, the most robust partner of laminin-binding integrins among tetraspanins, is involved in lung diseases.
In this study, we found that CD151 is essential in order for AECs to maintain epithelial integrity via firm adhesion to the basement membrane. The deletion of CD151 promotes mesenchymal-like changes and activation of TGF-β signaling in AECs and ultimately leads to pulmonary fibrosis.
Methods
See the online supplemental for additional information.
Animal Model
CD151 knockout (KO) mice were generated as previously described (15). CD9 KO mice were kindly provided by Eisuke Mekada (16). The mice were backcrossed to C57BL/6 mice for more than eight generations. All experiments comparing CD151 KO and WT mice were performed with age- and sex-matched littermates.
Microarrays
mRNA was extracted from WT (n = 4), CD151 KO (n = 5), and CD9 KO (n = 5) lungs at 20 weeks of age. After reverse transcription, cDNA was hybridized to GeneChip Mouse Genome 430 2.0 Arrays (Affymetrix, Santa Clara, CA). Array data were analyzed using Ingenuity Pathway Analysis (Ingenuity Systems, www.ingenuity.com), comparing the gene expression lists of CD151 KO/CD9 KO lungs and WT lungs; significantly differentially regulated genes were classified into subcategories by gene functions.
Bleomycin-induced Pulmonary Fibrosis Model
Eight- to 10-week-old mice were anesthetized with 13 μl/g of 4% Avertin. Intratracheal instillation of saline or 1.2 U/kg of bleomycin (BLM) (Nippon Kayaku Co., Tokyo, Japan) was performed after cervical incision. Mice were killed 7 to003F21 days after instillation, and lungs were removed for further analysis.
Assessment of Pulmonary Fibrosis
Hydroxyproline was measured as previously described (17). Lung compliance of mice was measured using a whole-body plethysmograph as part of the Pulmonary Maneuvers System (Buxco Electronics, Wilmington, NC), as previously described (10).
Isolation and Culture of Primary Alveolar Epithelial Type II Cells
Mouse AECs were isolated as previously described (18) with minor modifications. Because it was difficult to maintain the phenotype of primary epithelial cells in vitro, we used AECs isolated from H-2Kb-tsA58 transgenic mice (Immortomouse, Charles River, Wilmington, MA) (19).
Human Lung Tissues
Lung tissue samples from patients with IPF were obtained from diagnostic surgical lung biopsies, surgical lung resection from patients with lung cancer, or explanted lung obtained during lung transplantation procedures performed at the Kinki-Chuo Chest Medical Center or Osaka University Hospital. All cases were diagnosed as IPF/usual interstitial pneumonia according to the American Thoracic Society/European Respiratory Society Consensus Conference (20). Normal controls were obtained from healthy tissue derived from surgical lung resection from patients with lung cancer. We performed quantitative polymerase chain reaction using homogenates from 5 IPF and 6 healthy lung tissue samples and immunohistochemical analyses using sections from 10 IPF and 3 healthy lungs. The protocol was approved by the Institutional Review Board of both institutes; written informed consent was obtained from all subjects.
Statistical Analysis
All results are expressed as mean ± SEM. The significance of differences between two sample means was determined by two-tailed Student t-test. Analyses of survival were conducted using the Kaplan-Meier method; statistical differences between two groups were calculated using a log-rank test. P < 0.05 was considered statistically significant. All statistical analyses were performed using SPSS 11.0 for Windows (SPSS, Inc., Chicago, IL).
Results
DNA Microarray Analyses Identify Genes Associated with Connective Tissue Disorders in CD151 KO Lungs
To determine the expression level of CD151 in human lung tissues, we first performed immunohistochemical analyses. In normal human lungs, CD151 was expressed abundantly in AECs, alveolar macrophages, and peripheral bronchial epithelial cells (see Figure E1 in the online supplement). mRNA expression of CD151 was confirmed in the lungs of WT mice; in contrast, in the lungs of CD151 KO mice, expression of CD151 was clearly absent, but there was no effect on expression of other tetraspanins or associated integrins (Figure E2).
To investigate the changes in mRNA expression levels in CD151 KO lungs in an unbiased and comprehensive manner, we performed DNA microarray analyses on mouse lungs. We analyzed the list of genes differentially expressed between WT lungs and CD151/CD9 KO lungs using Ingenuity Pathway Analysis and identified enriched functional categories. “Cancer” was ranked at the top of the functional categories in both CD151 KO and CD9 KO lungs (Figure 1A), consistent with previous reports (7, 8). Unexpectedly, two categories were ranked highly in CD151 KO lungs but not in CD9 KO lungs: “connective tissue disorders” and “connective tissue development and function” (Figure 1A). In the category “connective tissue disorders,” there were 13 genes (detected by 16 probe sets) differentially expressed between WT lungs and CD151 KO lungs; most of these were collagen genes, such as collagen-1 and -3. As shown in Figure 1B, all of these genes were up-regulated in CD151 KO lungs. Moreover, expression levels of other ECM component genes, such as elastin and laminin, were also increased in CD151 KO lungs (data not shown). These data suggest that deletion of CD151 could result in fibrotic changes in the lung.
Figure 1.
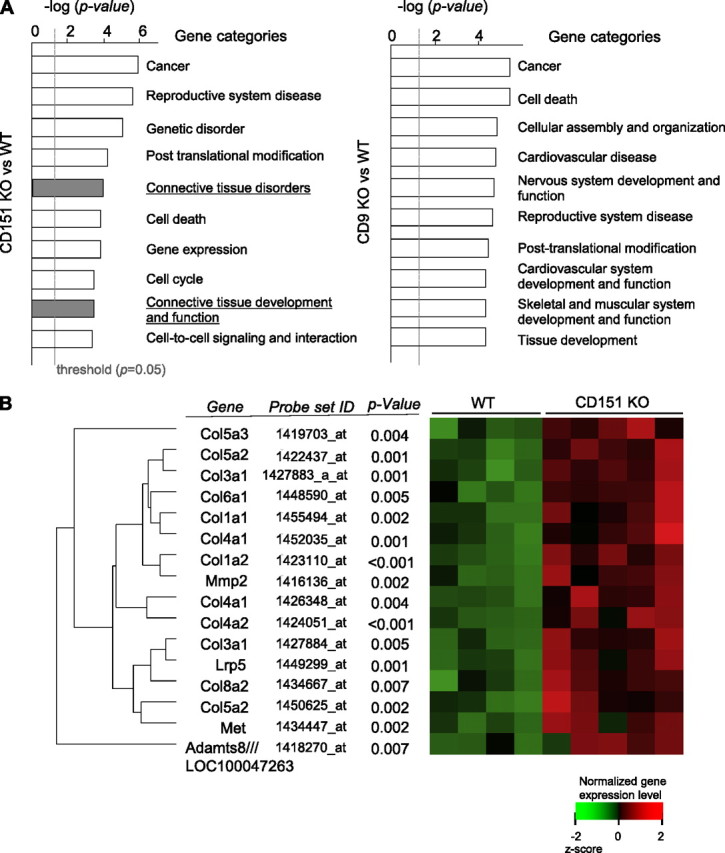
Microarray analysis indicated that genes associated with connective tissue disorders are differentially expressed in CD151 knockout (KO) lungs. (A) The list of genes differentially expressed between wild-type (WT) and CD151/CD9 KO lungs was analyzed using Ingenuity Pathway Analysis (IPA), and enriched functional gene categories were identified; results shown are the top 10 gene categories differentially expressed between CD151 KO and WT lungs (left) and between CD9 KO and WT lungs (right). Common logarithm of P value calculated by IPA (the length of the bar) reflects the significance of differential expression in each category. (B) Differentially expressed genes in the category “connective tissue disorders” are shown. A heat map shows the expression level of each gene (red = higher than mean value; green = lower than mean value). Adamts8 /// LOC100047263 = a disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 8; Lrp5 = low-density lipoprotein receptor-related protein 5; Met = met protooncogene; MMP2 = matrix metallopeptidase 2.
CD151 KO Mice Spontaneously Develop Age-related Pulmonary Fibrosis
No lung phenotype has ever been reported in CD151 KO mice, but these novel findings from microarrays led us to investigate the phenotype of CD151 KO mice in more detail. Consistent with our array results, Azan staining revealed slightly increased collagen depositions in the alveolar wall of CD151 KO lungs, although hematoxylin and eosin staining revealed minimal changes to alveolar structures (Figure 2A). Furthermore, collagen-1 expression in CD151 KO lungs was significantly increased relative to WT and CD9 KO lungs (Figure 2B). Likewise, hydroxyproline content of CD151 KO lungs was significantly increased compared with WT lungs (Figure 2C). Additionally, CD151 heterozygous mice also exhibited increased collagen-1 expression in their lungs. In lung function analyses, the lung compliance (C chord value) of CD151 KO mice was significantly lower than that of WT, CD9 KO, and CD151 heterozygous mice (Figure 2D), indicating that restrictive respiratory dysfunction occurred in CD151 KO lungs. These changes were obvious at 30 weeks of age but not at 10 weeks of age, suggesting that developmental abnormalities were only minimally involved. On the other hand, total cell numbers in bronchoalveolar lavage fluid were comparable between CD151 KO and WT lungs (Figure 2E). These data indicate that CD151 KO mice spontaneously develop age-related fibrotic phenotypes in the lungs.
Figure 2.
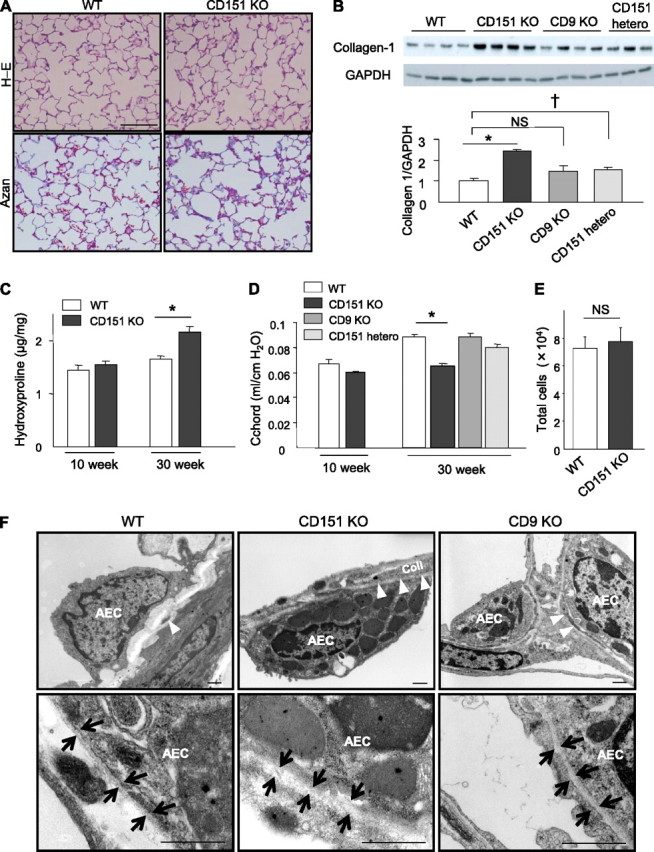
CD151 knockout (KO) mice spontaneously develop age-related pulmonary fibrosis. (A) Representative lung sections from wild-type (WT) and CD151 KO mice at 30 weeks of age stained with hematoxylin-eosin (H-E) and Azan. Bar = 100 μm. (B) Immunoblotting of total lung homogenate revealed increased expression of collagen-1 in CD151 KO and heterozygous (hetero) mice but not in CD9 KO mice compared with WT mice at 30 weeks of age. *P < 0.001 (WT vs. CD151 KO), †P < 0.05 (WT vs. CD151 hetero). (C) Hydroxyproline contents of total lungs were measured and normalized against lung weight (μg/mg, n = 5 per group). *P < 0.05. (D) Lung compliance was measured by a quasistatic pressure/volume maneuver, and is shown as C chord values (n = 4−5 per group). *P < 0.001. (E) Total cell number in bronchoalveolar lavage fluids was comparable between CD151 KO and WT mice at 30 weeks of age (n = 4 per group). (F) Electron microscopy of CD151 KO lung at 16 weeks of age revealed increased collagen (coll) deposition in alveolar walls and hypertrophied type II alveolar epithelial cells (AECs) with many lamellar body−like structures. Basement membranes (BM) under these AECs were irregularly thickened. The number of lamellar body−like structures was also increased in CD9 KO lungs but not as prominently as in CD151 KO lungs. Arrowheads indicate coll in alveolar walls; arrows indicate BM. Bar = 1 μm.
As expected, transmission electron microscopy revealed increased collagen deposition in the interstitium of CD151 KO lungs (Figure 2F). Of note, type II AECs of CD151 KO lungs exhibited alterations: these cells were hypertrophied and contained many dense cytoplasmic, lamellar body−like structures (Figure 2F). Furthermore, the basement membranes (BM) adjacent to these AECs were irregularly thickened (Figure 2F). No obvious changes were detected in macrophages, fibroblasts, or endothelial cells of CD151 KO lungs.
We next examined whether similar fibrotic changes could be observed in other organs. In CD151 KO mice, Azan staining revealed mild fibrotic changes not only in the glomeruli of kidneys but also around the intrahepatic bile ducts (Figure E3). However, fibrotic changes could not be detected in the heart (data not shown). These findings implicate CD151 in fibrosis not only in the lung but also in the liver and kidney.
Phosphorylation of Smad2 Is Enhanced in AECs in CD151 KO Mice
Because TGF-β signaling is crucial for inducing tissue fibrosis (21), we investigated TGF-β and related signaling pathways in CD151 KO lungs. Among TGF-β targets, phosphorylated Smad 2 (p-Smad2) was augmented in CD151 KO lungs. This finding became evident in CD151 KO mice at 30 weeks of age, but the tendency was already detectable at 10 weeks of age (Figure 3A), suggesting that this change is crucial in the development of fibrosis. However, no significant changes were observed in other signals, including Smad-7, p-Akt, p-Erk1/2, p-JNK, and p-p38 (Figure 3B and data not shown). To determine the main source of the elevated p-Smad2, we performed immunofluorescence analyses. As shown in Figure 3C, increased p-Smad2 was predominantly observed in nuclei of AECs but could scarcely be detected in alveolar macrophages (Figure 3D). In contrast, levels of activated TGF-β1 in whole lungs were comparable between WT and CD151 KO (Figure 3E). Combined with the abnormal morphology of AECs observed by electron microscopy, these findings suggest that increased p-Smad2 signals arising from aberrant AECs underlie the fibrotic phenotype of CD151 KO lungs.
Figure 3.
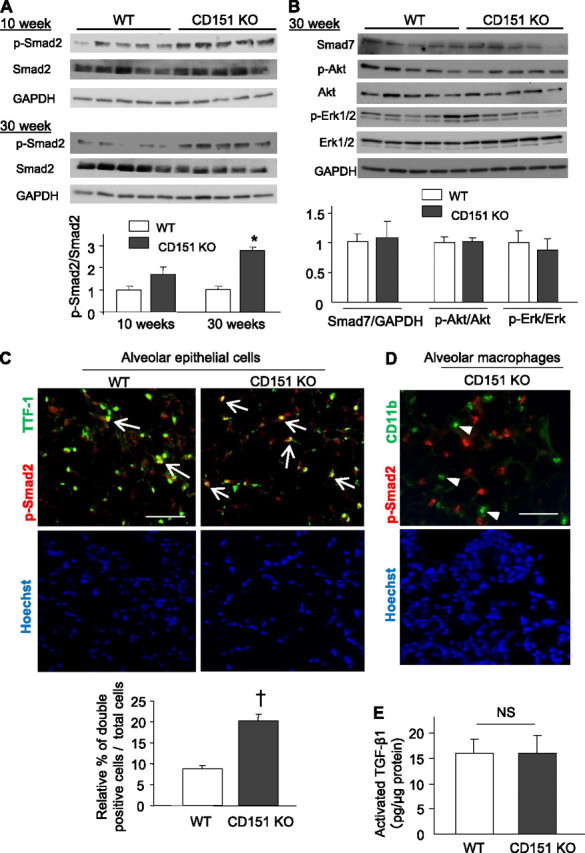
Enhanced p-Smad2 signaling in the alveolar epithelial cells (AECs) of CD151 knockout (KO) mice. (A) Western blot analysis and densitometric quantification of p-Smad2 in total protein homogenates from wild-type (WT) and CD151 KO lungs. Values of p-Smad2 were normalized against total Smad2. *P < 0.001. (B) Western blot analysis and densitometric quantification of Smad7 and p-Akt in total protein homogenates from WT and CD151 KO lungs. Values of Smad7, p-Akt, and p-Erk1/2 were normalized against GAPDH, Akt, and Erk, respectively. (C, D) Immunofluorescence of mouse lungs stained for p-Smad2 and TTF-1 or CD11b. The numbers of p-Smad2 and TTF-1 double-positive AECs (arrows) were significantly larger in CD151 KO lungs (C, n = 4 per group, †P < 0.05). p-Smad2−positive signals were scarcely seen in CD11b-positive alveolar macrophages (D, arrowheads). Bar = 100 μm. (E) Activated TGF-β1 in whole lung was measured by ELISA and normalized against total protein. Levels of activated TGF-β1 were comparable between WT and CD151 KO lungs (n = 3 per group).
CD151 Is Required for Stable Adhesion of AECs on BM
To evaluate the role of CD151 in AECs, we knocked down the CD151 gene in AECs isolated from H-2Kb-tsA58 transgenic mice. The CD151 gene was knocked down specifically, without affecting other tetraspanins and associated integrins (Figure 4A). Because tight attachment to the BM is essential for epithelia to maintain their characteristics, we initially analyzed the role of CD151 on cell binding to Matrigel, which contains ECM components similar to those in BM (e.g., laminin, collagen). To investigate the ECM-dependent function of CD151, we also conducted the same analyses on fibronectin, which is not present in Matrigel/BM. CD151-deleted AECs adhered onto ECM similarly to control cells (Figure 4B). However, they were easily detached by chemical and mechanical forces when grown on Matrigel, but not fibronectin (Figures 4C and 4D), suggesting that the adhesion strengthening of CD151-deleted AECs on Matrigel was impaired. On the other hand, deletion of CD151 in AECs did not affect random migration, wound closing assays, proliferation, or exogenous TGF-β1−induced signaling (Figure E4). Collectively, the data suggest that the presence of CD151 in AECs is required for firm adhesion to BM.
Figure 4.
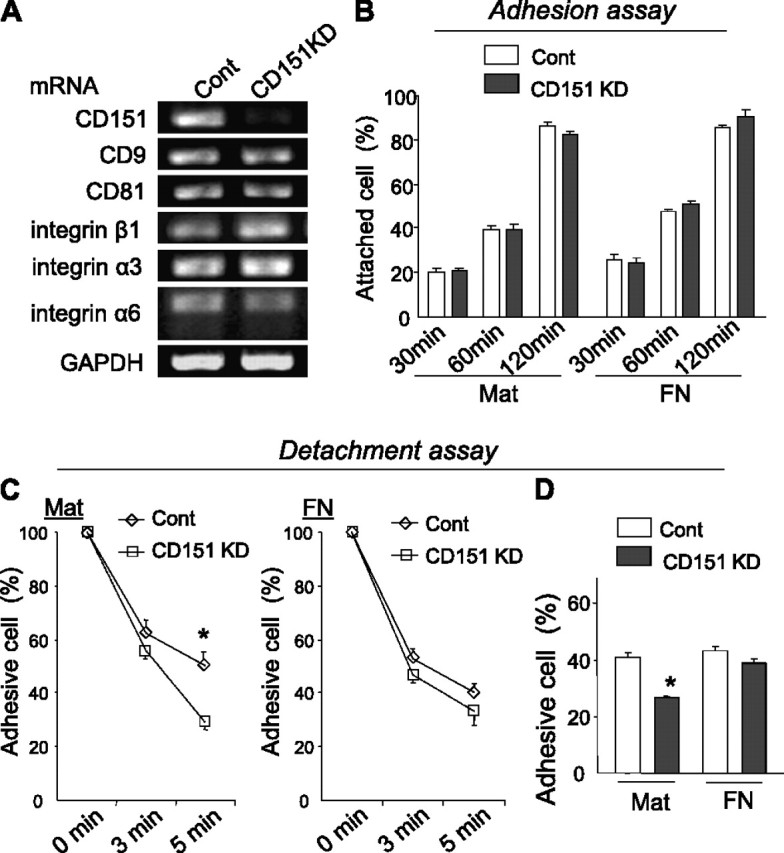
Deletion of CD151 results in attenuated adhesion strength of alveolar epithelial cells (AECs) on Matrigel. (A) The expression of CD151 was significantly down-regulated in CD151 knockdown (KD) AECs compared with cells treated with control siRNAs (cont). No significant change was observed in the expression of other tetraspanins (CD9, CD81) and associated integrins (β1, α3, and α6). (B) AECs were seeded on Matrigel (Mat)- or fibronectin (FN)-coated 96-well plates for 30, 60, and 120 minutes. After medium was gently changed, the attached cells were counted. The number of attached cells is shown in relation to total seeded cells. CD151 KD cells adhered to Mat and FN similarly to cont. (C, D) Adhesion strength of AECs on Mat is attenuated by CD151 knockdown. Trypsin/EDTA-induced (C) or mechanical vibration–induced (D) detachment was enhanced in CD151 KD cells cultured on Mat but not on FN. *P < 0.05. Data are representative of three independent experiments, which yielded similar results.
CD151 Is Necessary for AECs to Maintain Epithelial Characteristics on BM
Adhesion to BM was indispensable in order for epithelial cells to maintain their epithelial integrity (22, 23). Indeed, in the presence of serum, AECs maintained epithelial characteristics on Matrigel; in contrast, on collagen-1−, fibronectin-, or non-ECM−coated dishes, they exhibited elevation of p-Smad2 and mesenchymal-like changes, namely, increased α-SMA and decreased E-cadherin (Figure 5A). Again, these findings indicate that adhesion to Matrigel, but not collagen/fibronectin, protects AECs against mesenchymal changes and up-regulation of TGF-β signaling. However, these protective roles of Matrigel were overcome in the presence of a large amount of TGF-β1 (Figure 5A). Of note, CD151-deleted AECs lost epithelial integrity on Matrigel; they exhibited an enlarged morphology and increased α-SMA expression concomitant with upregulation of p-Smad2 levels, similar to cells treated with TGF-β1 (Figures 5B and 5C). These effects of CD151 deletion were not obvious on fibronectin-coated dishes, indicating that they might result from attenuated interaction with Matrigel. Consistent with this, α-SMA and pro-SPC double-positive cells were observed in CD151 KO lungs, but not in WT lungs, suggesting that these cells were in a state of epithelial-to-mesenchymal transition (EMT; Figure 5D). Furthermore, AECs isolated from CD151 KO mice exhibited relatively higher α-SMA expression (Figure 5E). Taken together, our data suggest that CD151 is required in order for AECs to preserve their integrity by maintaining firm adhesion to BM.
Figure 5.
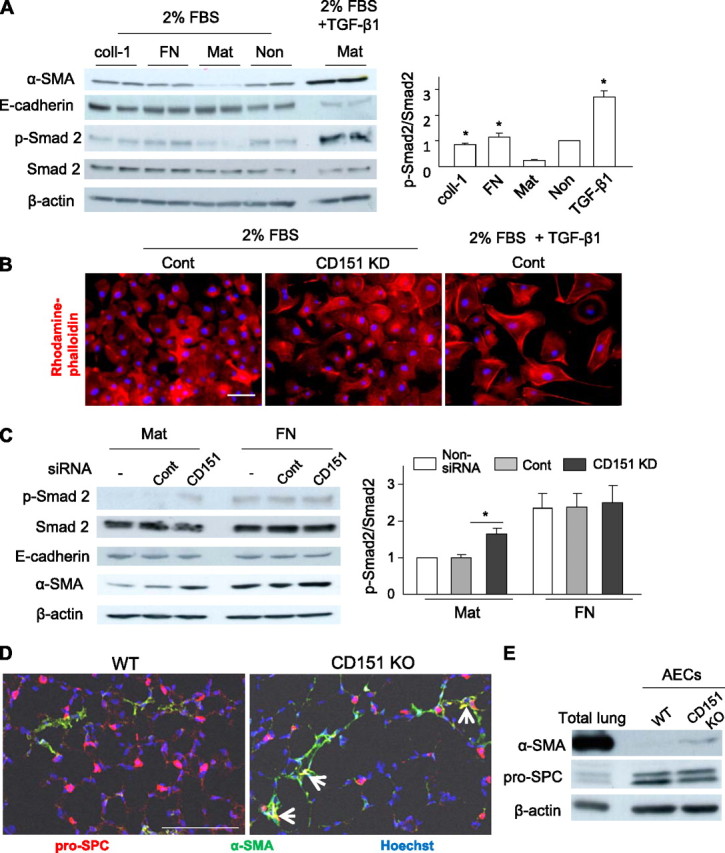
Deletion of CD151 causes disintegrity of alveolar epithelial cells (AECs) on basement membrane, with increased α-SMA expression and transforming growth factor (TGF)-β signaling. (A) AECs isolated from H-2Kb-tsA58 transgenic mice were cultured on collagen-1 (coll-1)–coated, fibronectin (FN)-coated, Matrigel (Mat)-coated, and noncoated (Non) dishes. After incubation for 72 hours in Dulbecco’s modified Eagle medium with 2% fetal bovine serum, only AECs cultured on Mat maintained an epithelial character and lower p-Smad2 signaling. As a control, AECs were stimulated with 10 ng/ml of TGF-β1 for 48 hours on Mat. In the densitometric quantitation, values of p-Smad2 were normalized against Smad2 and divided by values obtained on noncoated dishes (n = 4 per group). *P < 0.01 (vs. Mat). (B) The morphology of AECs cultured on Mat was analyzed by rhodamine-phalloidin staining. Compared with AECs treated by control siRNAs (cont), CD151 knockdown (KD) cells exhibited enlarged morphologies similar to those observed in cells treated by TGF-β1. After 48 hours of treatment with TGF-β1 (10 ng/ml), AECs lost their epithelial integrity and exhibited mesenchymal-like changes; they became enlarged and spindle-shaped, and actin filaments formed stress fibers. Bar = 20 μm. (C) Deletion of CD151 caused up-regulation of p-Smad2 levels, with increased α-SMA expression, in AECs on Mat, whereas these effects of CD151 deletion were not obvious on FN. *P < 0.05. Data are representative of three independent experiments, which yielded similar results. (D) In immunofluorescence analyses of frozen lung sections, epithelial cells expressing both pro-SPC and α-SMA were observed in CD151 knockout (KO) lungs, suggesting that they were in a state of epithelial-to-mesenchymal transition. However, no epithelial cells in the lung tissues of wild-type (WT) mice exhibited α-SMA expression. Bar = 100 μm. (E) AECs isolated from CD151 KO lungs exhibited higher expression of α-SMA than ones from WT lungs. Representatives of at least three similar results were shown.
Deletion of CD151 Does Not Affect Lung Fibroblast Function
Because resident fibroblasts are believed to be the main source of collagen-secreting myofibroblasts in pulmonary fibrosis (24), we investigated the functional role of CD151 in primary lung fibroblasts. Although fibroblasts from CD151 KO mice exhibited increased migration on Matrigel, the effect did not reach statistical significance. Otherwise, we could not see any differences between primary fibroblasts isolated from WT and CD151 KO lungs regarding chemotaxis, proliferation, differentiation into myofibroblasts, or collagen production (Figure E5).
BLM Injury Exacerbates AEC Disintegrity in CD151 KO Mice, Followed by Severe Pulmonary Fibrosis with Increased Mortality
We next examined the significance of CD151 under intratracheal BLM exposure, which initially causes direct injury to AECs and ultimately leads to pulmonary fibrosis (25). Low-dose intratracheal BLM induced a moderate decrease in CD151 expression in WT lungs and a gradual increase in collagen-1 expression (Figure E6). In CD151 KO lungs, BLM treatment caused enlargement of AECs, and their α-SMA expression was dramatically increased (Figure 6A), suggesting that disintegrity of AECs was accelerated by BLM injury. On the other hand, the number of inflammatory cells in bronchoalveolar lavage fluid of CD151 KO lungs was only mildly increased (Figure 6B). Accelerated epithelial disintegrity was followed by severe fibrosis in CD151 KO lungs, as was evident from histopathology, CT scan, increased hydroxyproline content, and decreased lung compliance (Figures 6C−6F). Furthermore, these changes resulted in increased and prolonged body weight loss (Figure 6G) and severe mortality (Figure 6H). Nevertheless, BLM-induced fibrosis in CD9 KO mice was not increased compared with that in WT mice, although the number of lung inflammatory cells increased (Figure E7).
Figure 6.
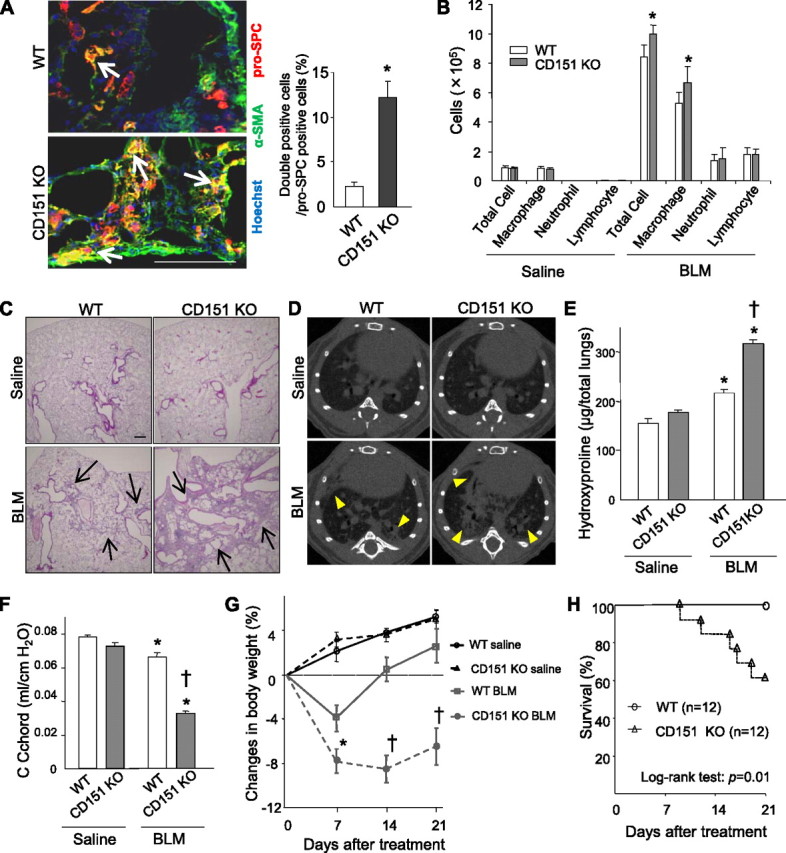
Low-dose bleomycin (BLM) injury accelerates epithelial disintegrity, followed by exacerbated pulmonary fibrosis in CD151 knockout (KO) mice. (A) Fourteen days after BLM injury (1.2 U/kg), alveolar epithelial cells (AECs) were deformed, and the proportion of AECs expressing both α-SMA and pro-SPC was significantly increased in CD151 KO lungs compared with wild-type (WT) (n = 4 per group, *P < 0.01). Bar = 100 μm. (B) Cell numbers in bronchoalveolar lavage fluids peaked 7 days after saline/BLM treatment. CD151 KO mice exhibited mildly increased lung inflammatory cells relative to WT mice after BLM injury (n = 4−6 per group). *P < 0.05 (vs. WT). (C, D) Twenty-one days after BLM injury, increased lung fibrosis was observed in both WT mice and CD151 KO mice compared with their saline controls, but this phenomenon was much more severe in CD151 KO mice, as indicated by histopathology (hematoxylin-eosin staining, C, arrows; bar = 100 μm) and computed tomography (D, arrowheads). (E) Hydroxyproline assays confirmed increased collagen content in total lungs after BLM treatment in CD151 KO mice relative to WT mice (n = 5−6 per group). *P < 0.01 (vs. saline), †P < 0.001 (vs. WT). (F) Twenty-one days after BLM injury, compliance (C chord) of CD151 KO lungs was significantly decreased to around half the WT level (n = 5−6 per group). *P < 0.01 (vs. saline), †P < 0.001 (vs. WT). (G) Changes in body weight after BLM injury. From 14 to 21 days after treatment, WT mice regained their weight, whereas the weight loss of CD151 KO mice continued. During the whole period of the experiment, saline control mice gradually gained weight (n = 4−6 per group). *P < 0.05 (vs. WT), †P < 0.01 (vs. WT). (H) After BLM injury, the mortality of CD151 KO mice was larger than that of WT mice (log-rank test: P = 0.01).
In summary, disintegrity of AECs in CD151 KO lungs was further exacerbated by BLM exposure, ultimately resulting in severe fibrosis.
CD151 Expression Is Significantly Decreased in Human AECs from Patients with IPF
Finally, to investigate the possible involvement of CD151 in human pulmonary fibrosis, we analyzed lung sections from patients with IPF/usual interstitial pneumonia. P-Smad2 staining from lung sections from patients with IPF revealed increased positive signals relative to healthy lungs (Figures 7A and 7D). These signals in IPF lungs were enriched in nuclei of AECs (Figures 7A–7F, Figure E8), implying that TGF-β1 signaling in AECs might play an important role in the development of human pulmonary fibrosis.
Figure 7.
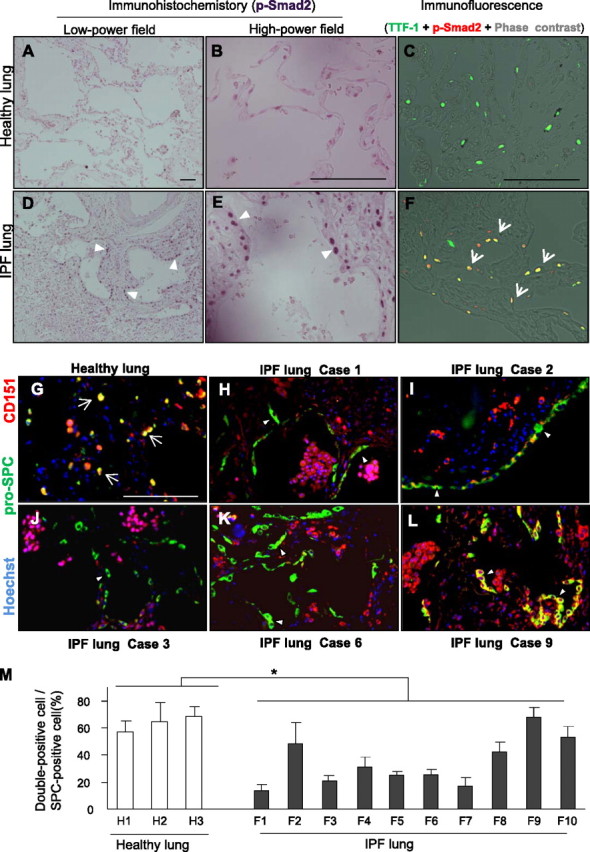
CD151 expression is significantly decreased in alveolar epithelial cells (AECs) from patients with idiopathic pulmonary fibrosis (IPF). (A−F) IPF lungs contained more p-Smad2 signals than healthy lungs (A, D, arrowheads). P-Smad2−positive signals of IPF lungs were preferentially observed in nuclei of AECs (B, C, E, and F, arrowheads and arrows). Representative IPF and healthy lung sections were shown. Bar = 100 μm. TTF-1 = specific marker for type II AECs. (G−L) Representative immunostaining of pro-SPC and CD151 in healthy (n = 3) and IPF (n = 10) lungs. Most AECs expressed CD151 in healthy lungs (G, arrows), whereas CD151-negative AECs were prominent in the lungs of IPF cases 1, 2, 3, and 6 (H−K, arrowheads). Notably, clear and diffuse expression of CD151 was observed in AECs in the lung of case 9 (L, arrowheads). Bar = 100 μm. (M) In histological quantification, at least three fields from each lung were randomly chosen, and an average of 50 pro-SPC–stained AECs per field were quantitated. The number of CD151-positive AECs was significantly decreased in the lungs of patients with IPF (F1−10) compared with healthy lungs (H1−3, *P < 0.05).
In our quantitative polymerase chain reaction analysis using human lung homogenates, we detected no significant difference in CD151 expression levels between healthy and IPF lungs (Figure E9). In immunofluorescence, however, most AECs in healthy lung tissues expressed CD151 (Figure 7G), whereas CD151-negative AECs could be focally observed in IPF lungs (Figures 7H−7L). Histological quantitation revealed fewer CD151-positive AECs in lung samples from patients with IPF compared with healthy lungs (Figure 7M), although CD151 expression levels fluctuated. In contrast, CD151 expression in alveolar macrophages was comparable between the two groups. Taken together, these observations indicate that CD151 in AECs may play a crucial role in the pathogenesis of pulmonary fibrosis in humans as well as mice.
Discussion
Here, we provide new evidence that CD151 KO mice spontaneously develop age-related pulmonary fibrosis, which is exacerbated by BLM exposure. To date, it has been reported that CD151 KO mice have renal dysfunction (in the FVB strain) (26–28), impaired pathologic angiogenesis (15), delayed wound closure (29), and deficiencies in platelet aggregation (30). However, the involvement of CD151 in lung diseases remains unclear; this is the first report implicating CD151 in protection against pulmonary fibrosis. We also observed focal fibrotic changes in the kidneys and liver of CD151 KO mice, further supporting the critical role played by CD151 in fibrosis.
Accumulating evidence suggests that in addition to intrinsic factors in host lungs (i.e., genetic predisposition), extrinsic factors that accelerate injury of AECs also play an etiologic role in IPF. These factors include environmental exposure, transbronchial infection, and aging (2, 3, 31, 32). Our results fit this idea well because the fibrotic phenotype and epithelial disintegrity of CD151 KO mice became more evident with increased age and was exacerbated by low-dose BLM exposure. In contrast to CD151 KO mice, however, patients with IPF do not commonly present with kidney and liver symptoms. In some IPF lungs, we found that CD151 expression is focally decreased in AECs but not in alveolar macrophages, suggesting that CD151 down-regulation in AECs is the result of an extrinsic factor, rather than a genetic factor. Consistent with this idea, intratracheal BLM decreased CD151 expression in the lung. Recently, hypoxic stress was shown to down-regulate CD151 expression in colon cancer cells through the up-regulation of hypoxia-inducible factor-1 (HIF-1) (33). Likewise, in AECs, we showed that hypoxic stress down-regulates CD151 expression (Figure E10). It has become clear that HIF-1 is significantly up-regulated in AECs of IPF and BLM-induced fibrotic mouse lungs (34). In this context, hypoxic stress and subsequent up-regulation of HIF-1 are one possible cause of focal decrease of CD151 in IPF lungs. However, the expression levels of CD151 appeared to differ among lungs from different patients with IPF. Fujishima and colleagues previously reported that CD151 expression in epithelial cells and macrophages is relevant to the activation of matrilysin-1 in IPF lungs (35). It thus appears that expression of CD151 in cases of pulmonary fibrosis depends on the site, clinical phase, and possible complications involving infection. Further investigation will be required to address the clinical significance of CD151 expression in IPF lungs.
Despite the increased number of lung inflammatory cells, CD9 KO mice did not exhibit severe fibrotic changes compared with WT mice, even after BLM exposure. These findings are in line with the idea that increased inflammation does not necessarily lead to augmented fibrosis. Double deletion of CD9 and a homologous tetraspanin, CD81, spontaneously leads to pulmonary emphysema in mice (10). Determination of the shared and distinctive pathways/molecules associated with CD151 and CD9 might help us to understand the pathogenesis of pulmonary fibrosis and emphysema.
We found that deletion of CD151 resulted in disintegrity of AECs, activated p-Smad2, and EMT-like changes, which might causally contribute to the development of pulmonary fibrosis. First, activation of TGF-β signaling in AECs may be a primary source of the development of pulmonary fibrosis, because mice with epithelial cell−specific loss of TGF-β receptor are protected from BLM-induced pulmonary fibrosis (36, 37). Consistent with this, in our analyses of IPF lungs, activated p-Smad2 signals were predominantly observed in AECs. Second, myofibroblasts that arise from EMT could themselves contribute to the development of pulmonary fibrosis (38–40). Changes in CD151-deleted AECs were mild, but similar to those observed in cells treated with TGF-β1, suggesting that CD151 deletion promotes EMT. In line with our study, CD151 deficiency in epithelial cells results in increased actin stress fibers at the basal cell surface and perturbs cell polarity (11, 41). In contrast, in hepatocellular carcinoma cells, the complex containing CD151 and α6β1 integrin promotes EMT (42). Further investigation will be needed to address the role of CD151 in EMT in normal and malignant cells.
We found that CD151 is essential for firm adhesion of AECs on BM. Among several ECM molecules contained in BM, laminin may be the most crucially involved in this adhesion, because CD151 determines the strength of cell−ECM interactions by forming complexes with laminin-binding integrins such as α3β1, α6β4, and α6β1 (43–45). We also found that this firm adhesion to BM protects AECs against EMT-like changes and up-regulation of p-Smad2. Likewise, during embryonic development, BM protects against EMT in primitive ectoderm (22), further supporting our results. However, we could not determine how loss of adhesion to BM leads to elevation of p-Smad2 in serum. Possible explanations include response to the low level of TGF-β1 contained in serum or an increase in autocrine/paracrine TGF-β signaling from activated AECs exhibiting mesenchymal-like changes. In a previous study of breast cancer cells, CD151 was shown to regulate TGF-β1−induced cell scattering and proliferation via activation of p38 (46). Additional studies will be required to elucidate how CD151 is involved in TGF-β1 signaling in normal and malignant cells.
However, there may be alternative explanations for fibrotic changes, because CD151 is implicated in a variety of cell functions through its association with multiple proteins. First, CD151 modulates various integrin-dependent cell functions. Recently, mice were generated with lung epithelium–specific loss of α3 integrin expression (14). In contrast to CD151 KO mice, these mutant mice exhibited a blunted response to BLM injury, in which a CD151-independent function of α3β1 integrin could be involved. However, similar to our CD151 KO mice, the mutants exhibited increased collagen deposition in their lungs, suggesting that some functions mediated by CD151-α3β1 integrin complexes may be involved in the fibrotic phenotype of CD151 KO mice. Further study is warranted to determine how CD151 deletion affects the function of associated integrins. Second, other cells, such as resident fibroblasts, could contribute to the progression of fibrosis. However, as far as we could determine, CD151 is minimally involved in lung fibroblast functions related to fibrosis.
Consistent with a crucial role for CD151 in mouse pulmonary fibrosis, CD151 expression in patients with IPF is focally diminished in type II AECs. In addition, type II AECs in CD151 KO mice are hypertrophied and contain increased numbers of lamellar body−like structures, similar to what is observed in familial human IPF (47). These findings suggest that CD151 in AECs plays critical roles in the pathogenesis of pulmonary fibrosis, in humans as well as mice. Recently, a mutation in CD151 was reported in a few patients with Alport syndrome, who exhibit sensorineural deafness and pretibial epidermolysis bullosa (48). Our results suggest that these patients should also be assayed for fibrotic changes in the lungs.
In conclusion, we here provide evidence that CD151 is essential for AECs, and deletion of CD151 results in pulmonary fibrosis. To date, numerous transgenic mice overexpressing profibrogenic cytokines exhibit pulmonary fibrosis (25), whereas only a few molecules that play protective roles in pulmonary fibrosis have been identified in mouse models or in humans: SPC (47), caveolin-1 (49, 50), relaxin (51), and connexin (52). Most therapies for IPF targeted against profibrogenic cytokines have not been effective in clinical trials (53). Therefore, it may be reasonable to focus efforts on protective factors, such as CD151, which provide a clue about the pathogenesis of pulmonary fibrosis and represent novel targets for development of therapies.
Acknowledgments
The authors thank Dr. Yasuyuki Yokosaki (Hiroshima University, Japan) for helpful discussions and critical reading of the manuscript, and Dr. Hideshi Kaneko (Teijin Pharmaceutical Company, Japan) for helpful comments on the electron microscopy studies.
Footnotes
Supported by a Grant-in-Aid for Scientific Research (C) No. 21590992, the Takeda Science Foundation, the Okamoto Satoshi Memorial Fund for Pulmonary Fibrosis Research, the Kato Memorial Trust For Nambyo Research (Y.T.), National Institutes of Health grant CA42368 (M.E.H.), the Funding Program for Next Generation World-Leading Researchers (NEXT Program), and Special Coordination Funds for Promoting Science and Technology (A.K.).
Author Contributions: Conception and design, K. Tsujino, Y.T., I.T., A.K.; analysis and interpretation, K. Tsujino, Y.T., R.I., H.S., T.I., S.T., Y.J., S.I., T.M., M.S., I.N., K.I., H.K., T.K., M.I., K. Takeda; collection of human samples, K. Tsujino, T.A., Y.S., M.K., Y.I., M.O.; preparing the manuscript, K. Tsujino, Y.T., M.E.H., A.K.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201201-0117OC on May 16, 2012
References
- 1.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 2001;134:136–151. [DOI] [PubMed] [Google Scholar]
- 2.Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med 2001;345:517–525. [DOI] [PubMed] [Google Scholar]
- 3.King TE, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011;378:1949–1961. [DOI] [PubMed] [Google Scholar]
- 4.Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol 2010;298:L715–L731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy S, Shoham T. Protein-protein interactions in the tetraspanin web. Physiology (Bethesda) 2005;20:218–224. [DOI] [PubMed] [Google Scholar]
- 6.Hemler ME. Tetraspanin proteins mediate cellular penetration, invasion, and fusion events and define a novel type of membrane microdomain. Annu Rev Cell Dev Biol 2003;19:397–422. [DOI] [PubMed] [Google Scholar]
- 7.Hemler ME. Targeting of tetraspanin proteins–potential benefits and strategies. Nat Rev Drug Discov 2008;7:747–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang HX, Li Q, Sharma C, Knoblich K, Hemler ME. Tetraspanin protein contributions to cancer. Biochem Soc Trans 2011;39:547–552. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki M, Tachibana I, Takeda Y, He P, Minami S, Iwasaki T, Kida H, Goya S, Kijima T, Yoshida M, et al. Tetraspanin CD9 negatively regulates lipopolysaccharide-induced macrophage activation and lung inflammation. J Immunol 2009;182:6485–6493. [DOI] [PubMed] [Google Scholar]
- 10.Takeda Y, He P, Tachibana I, Zhou B, Miyado K, Kaneko H, Suzuki M, Minami S, Iwasaki T, Goya S, et al. Double deficiency of tetraspanins CD9 and CD81 alters cell motility and protease production of macrophages and causes chronic obstructive pulmonary disease-like phenotype in mice. J Biol Chem 2008;283:26089–26097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shigeta M, Sanzen N, Ozawa M, Gu J, Hasegawa H, Sekiguchi K. CD151 regulates epithelial cell-cell adhesion through PKC- and Cdc42-dependent actin cytoskeletal reorganization. J Cell Biol 2003;163:165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moribe H, Yochem J, Yamada H, Tabuse Y, Fujimoto T, Mekada E. Tetraspanin protein (TSP-15) is required for epidermal integrity in Caenorhabditis elegans. J Cell Sci 2004;117:5209–5220. [DOI] [PubMed] [Google Scholar]
- 13.Xia H, Diebold D, Nho R, Perlman D, Kleidon J, Kahm J, Avdulov S, Peterson M, Nerva J, Bitterman P, et al. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med 2008;205:1659–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, Frank JA, Brumwell AN, Wheeler SE, Kreidberg JA, et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest 2009;119:213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeda Y, Kazarov AR, Butterfield CE, Hopkins BD, Benjamin LE, Kaipainen A, Hemler ME. Deletion of tetraspanin Cd151 results in decreased pathologic angiogenesis in vivo and in vitro. Blood 2007;109:1524–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyado K, Yamada G, Yamada S, Hasuwa H, Nakamura Y, Ryu F, Suzuki K, Kosai K, Inoue K, Ogura A, et al. Requirement of CD9 on the egg plasma membrane for fertilization. Science 2000;287:321–324. [DOI] [PubMed] [Google Scholar]
- 17.Oku H, Shimizu T, Kawabata T, Nagira M, Hikita I, Ueyama A, Matsushima S, Torii M, Arimura A. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol 2008;590:400–408. [DOI] [PubMed] [Google Scholar]
- 18.Rice WR, Conkright JJ, Na CL, Ikegami M, Shannon JM, Weaver TE. Maintenance of the mouse type II cell phenotype in vitro. Am J Physiol Lung Cell Mol Physiol 2002;283:L256–L264. [DOI] [PubMed] [Google Scholar]
- 19.Saiga H, Nishimura J, Kuwata H, Okuyama M, Matsumoto S, Sato S, Matsumoto M, Akira S, Yoshikai Y, Honda K, et al. Lipocalin 2-dependent inhibition of mycobacterial growth in alveolar epithelium. J Immunol 2008;181:8521–8527. [DOI] [PubMed] [Google Scholar]
- 20.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med 1994;331:1286–1292. [DOI] [PubMed] [Google Scholar]
- 22.Fujiwara H, Hayashi Y, Sanzen N, Kobayashi R, Weber CN, Emoto T, Futaki S, Niwa H, Murray P, Edgar D, et al. Regulation of mesodermal differentiation of mouse embryonic stem cells by basement membranes. J Biol Chem 2007;282:29701–29711. [DOI] [PubMed] [Google Scholar]
- 23.Shintani Y, Maeda M, Chaika N, Johnson KR, Wheelock MJ. Collagen I promotes epithelial-to-mesenchymal transition in lung cancer cells via transforming growth factor-beta signaling. Am J Respir Cell Mol Biol 2008;38:95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kisseleva T, Brenner DA. Fibrogenesis of parenchymal organs. Proc Am Thorac Soc 2008;5:338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2008;294:L152–L160. [DOI] [PubMed] [Google Scholar]
- 26.Sachs N, Kreft M, van den Bergh Weerman MA, Beynon AJ, Peters TA, Weening JJ, Sonnenberg A. Kidney failure in mice lacking the tetraspanin CD151. J Cell Biol 2006;175:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baleato RM, Guthrie PL, Gubler MC, Ashman LK, Roselli S. Deletion of CD151 results in a strain-dependent glomerular disease due to severe alterations of the glomerular basement membrane. Am J Pathol 2008;173:927–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sachs N, Claessen N, Aten J, Kreft M, Teske GJ, Koeman A, Zuurbier CJ, Janssen H, Sonnenberg A. Blood pressure influences end-stage renal disease of Cd151 knockout mice. J Clin Invest 2012;122:348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cowin AJ, Adams D, Geary SM, Wright MD, Jones JC, Ashman LK. Wound healing is defective in mice lacking tetraspanin CD151. J Invest Dermatol 2006;126:680–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lau LM, Wee JL, Wright MD, Moseley GW, Hogarth PM, Ashman LK, Jackson DE. The tetraspanin superfamily member CD151 regulates outside-in integrin alphaIIbbeta3 signaling and platelet function. Blood 2004;104:2368–2375. [DOI] [PubMed] [Google Scholar]
- 31.Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006;3:293–298. [DOI] [PubMed] [Google Scholar]
- 32.du Bois RM. Strategies for treating idiopathic pulmonary fibrosis. Nat Rev Drug Discov 2010;9:129–140. [DOI] [PubMed] [Google Scholar]
- 33.Chien CW, Lin SC, Lai YY, Lin BW, Lee JC, Tsai SJ. Regulation of CD151 by hypoxia controls cell adhesion and metastasis in colorectal cancer. Clin Cancer Res 2008;14:8043–8051. [DOI] [PubMed] [Google Scholar]
- 34.Tzouvelekis A, Harokopos V, Paparountas T, Oikonomou N, Chatziioannou A, Vilaras G, Tsiambas E, Karameris A, Bouros D, Aidinis V. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis. Am J Respir Crit Care Med 2007;176:1108–1119. [DOI] [PubMed] [Google Scholar]
- 35.Fujishima S, Shiomi T, Yamashita S, Yogo Y, Nakano Y, Inoue T, Nakamura M, Tasaka S, Hasegawa N, Aikawa N, et al. Production and activation of matrix metalloproteinase 7 (matrilysin 1) in the lungs of patients with idiopathic pulmonary fibrosis. Arch Pathol Lab Med 2010;134:1136–1142. [DOI] [PubMed] [Google Scholar]
- 36.Li M, Krishnaveni MS, Li C, Zhou B, Xing Y, Banfalvi A, Li A, Lombardi V, Akbari O, Borok Z, et al. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest 2011;121:277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Degryse AL, Tanjore H, Xu XC, Polosukhin VV, Jones BR, Boomershine CS, Ortiz C, Sherrill TP, McMahon FB, Gleaves LA, et al. TGFbeta signaling in lung epithelium regulates bleomycin-induced alveolar injury and fibroblast recruitment. Am J Physiol Lung Cell Mol Physiol 2011;300:L887–L897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol 2005;166:1321–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest 2007;132:1311–1321. [DOI] [PubMed] [Google Scholar]
- 40.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA 2006;103:13180–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson JL, Winterwood N, DeMali KA, Stipp CS. Tetraspanin CD151 regulates RhoA activation and the dynamic stability of carcinoma cell-cell contacts. J Cell Sci 2009;122:2263–2273. [DOI] [PubMed] [Google Scholar]
- 42.Ke AW, Shi GM, Zhou J, Huang XY, Shi YH, Ding ZB, Wang XY, Devbhandari RP, Fan J. CD151 amplifies signaling by integrin α6β1 to PI3K and induces the epithelial-mesenchymal transition in HCC cells. Gastroenterology 2011;140:1629–1641.e15. [DOI] [PubMed] [Google Scholar]
- 43.Lammerding J, Kazarov AR, Huang H, Lee RT, Hemler ME. Tetraspanin CD151 regulates alpha6beta1 integrin adhesion strengthening. Proc Natl Acad Sci USA 2003;100:7616–7621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sterk LM, Geuijen CA, Oomen LC, Calafat J, Janssen H, Sonnenberg A. The tetraspan molecule CD151, a novel constituent of hemidesmosomes, associates with the integrin alpha6beta4 and may regulate the spatial organization of hemidesmosomes. J Cell Biol 2000;149:969–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishiuchi R, Sanzen N, Nada S, Sumida Y, Wada Y, Okada M, Takagi J, Hasegawa H, Sekiguchi K. Potentiation of the ligand-binding activity of integrin alpha3beta1 via association with tetraspanin CD151. Proc Natl Acad Sci USA 2005;102:1939–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sadej R, Romanska H, Kavanagh D, Baldwin G, Takahashi T, Kalia N, Berditchevski F. Tetraspanin CD151 regulates transforming growth factor beta signaling: implication in tumor metastasis. Cancer Res 2010;70:6059–6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas AQ, Lane K, Phillips J, Prince M, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med 2002;165:1322–1328. [DOI] [PubMed] [Google Scholar]
- 48.Karamatic Crew V, Burton N, Kagan A, Green CA, Levene C, Flinter F, Brady RL, Daniels G, Anstee DJ. CD151, the first member of the tetraspanin (TM4) superfamily detected on erythrocytes, is essential for the correct assembly of human basement membranes in kidney and skin. Blood 2004;104:2217–2223. [DOI] [PubMed] [Google Scholar]
- 49.Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001;293:2449–2452. [DOI] [PubMed] [Google Scholar]
- 50.Wang XM, Zhang Y, Kim HP, Zhou Z, Feghali-Bostwick CA, Liu F, Ifedigbo E, Xu X, Oury TD, Kaminski N, et al. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med 2006;203:2895–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Samuel CS, Zhao C, Bathgate RA, Bond CP, Burton MD, Parry LJ, Summers RJ, Tang ML, Amento EP, Tregear GW. Relaxin deficiency in mice is associated with an age-related progression of pulmonary fibrosis. FASEB J 2003;17:121–123. [DOI] [PubMed] [Google Scholar]
- 52.Koval M, Billaud M, Straub AC, Johnstone SR, Zarbock A, Duling BR, Isakson BE. Spontaneous lung dysfunction and fibrosis in mice lacking connexin 40 and endothelial cell connexin 43. Am J Pathol 2011;178:2536–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bouros D, Antoniou KM. Current and future therapeutic approaches in idiopathic pulmonary fibrosis. Eur Respir J 2005;26:693–702. [DOI] [PubMed] [Google Scholar]