

Abstract
Objective
Therapeutic hypothermia is an established treatment for perinatal asphyxia. Yet, many term infants continue to die or suffer from neurodevelopmental disability. Several experimental studies have demonstrated a beneficial effect of mild-to-moderate hypothermia after hypoxic injury, but the understanding of hypothermia-induced neuroprotection remains incomplete. In general, global protein synthesis is attenuated by hypothermia, but a small group of RNA-binding proteins including the RNA-binding motif 3 (RBM3) is upregulated in response to cooling. The aim of this study was to establish an in vitro model to investigate the effects of hypoxia and hypothermia on neuronal cell survival, as well as to examine the kinetics of concurrent cold-shock protein RBM3 gene expression.
Methods
Experiments were performed by using human SK-N-SH neurons exposed to different oxygen concentrations (21%, 8%, or 0.2% O2) for 24 hours followed by moderate hypothermia (33.5°C) or normothermia for 24, 48, or 72 hours. Cell death was determined by quantification of lactate dehydrogenase and neuron-specific enolase releases into the cell cultured medium, and cell morphology was assessed by using immunofluorescence staining. The regulation of RBM3 gene expression was assessed by reverse transcriptase-quantitative polymerase chain reaction and Western blot analysis.
Results
Exposure to hypoxia (0.2% O2) for 24 hours resulted in significantly increased cell death in SK-N-SH neurons, whereas exposure to 8% O2 had no significant impact on cell viability. Post-hypoxia treatment with moderate hypothermia for 48 or 72 hours rescued the neurons from hypoxia-induced cell death. Moreover, exposure to severe hypoxia led to observable cell swelling, which was also attenuated by moderate hypothermia. Finally, moderate hypothermia but not hypoxia led to the induction of RBM3 expression on both transcriptional and translational levels.
Conclusion
Moderate hypothermia protects neurons from hypoxia-induced cell death. The expression of the cold-shock protein RBM3 is induced by moderate hypothermia and could be one possible mediator of hypothermia-induced neuroprotection.
Keywords: RBM3, cold-inducible RNA-binding protein, hypothermia, neuroprotection, hypoxia
Introduction
Perinatal asphyxia is the third most frequent cause of death during the neonatal period and a major cause of long-term neurodevelopmental disability in Western industrialized nations.1 It is a medical condition under which the neonatal brain suffers from oxygen deprivation, which leads to hypoxic neuronal damage.2 The only evidence-based therapy to prevent neuronal damage due to perinatal asphyxia is to lower the infant’s body temperature to 33°C–34°C for 72 hours, as observed in several multicenter randomized controlled trials, to improve survival rates and neurological outcome in term infants.3–5 Moderate therapeutic hypothermia (32.0°C–33.9°C) is also recommended for other clinical situations, leading to hypoxic–ischemic brain injury, such as out-of-hospital cardiac arrest.6,7 Yet, many patients with hypoxic–ischemic brain injury survive with a poor neurological outcome or die. Therefore, it is important to further understand the molecular mechanisms of hypothermia-induced neuroprotection and to identify key targets of hypothermia in order to improve current therapy as well as to develop new intervention strategies.
Although the understanding of hypothermia-induced neuroprotection remains incomplete, many experimental studies have shown that hypothermia is able to mitigate several injury processes in the hypoxic–ischemic brain. Hypothermia reduces cerebral metabolism by ~5%–10% per degree cooling and, therefore, decreases oxygen demand in neurons.8,9 The accumulation of excitatory amino acids such as glutamate released during hypoxic–ischemic brain injury is diminished by hypothermia,10,11 as well as decreased the production of free radicals and reactive oxygen species (ROS).12 Furthermore, hypothermia reduces cell death following hypoxia and attenuates inflammatory responses through the inhibition of proinflammatory cytokine expression.13,14
Whereas lowering the body temperature inhibits global protein synthesis, a small group of RNA-binding proteins is induced by mild-to-moderate hypothermia. One of these “cold-shock proteins” is the RNA-binding motif 3 (RBM3). First described by Derry et al, RBM3 consists of 157 amino acids, has a molecular weight of 17 kDa, and has an arginine–glycine-rich domain, called the RNA recognition motif (RRM), to bind RNA.15,16 RBM3 is ubiquitously expressed in the body, but in a temperature-dependent manner. Exposure to moderate hypothermia (32°C) for 24 hours has been observed to increase RBM3 mRNA expression, whereas exposure to elevated temperatures (39°C) resulted in decreased expression.17 RBM3 expression was also observed to be enhanced in hippocampal murine slices in response to mild hypothermia (33.5°C) on both the transcriptional and translational levels.18 Furthermore, RBM3 seems to be participating in the oxygen stress response as it has been observed to be upregulated upon exposure to mild or severe hypoxia (8% or 1% O2, respectively) in non-neuronal cells by an HIF-1-independent mechanism.19 RBM3 is also highly expressed in the brain of young rats and mice as well as in areas with high translational rates in cerebral tissue of adult rats.20
The molecular and cellular function of RBM3 is thought to be multifarious and remains poorly understood. RBM3 is able to bind to the 60s subunit of ribosomes and increases translation.21,22 Overexpression of RBM3 reduces apoptosis, whereas RBM3 knockout decreases cell viability and inhibits cell proliferation.23,24 Peretti et al reported that RBM3 is involved in synapse regeneration in a mouse model of neurodegenerative disease.25 Cold-shock proteins such as RBM3 could be possible mediators of hypothermia-induced neuroprotection. Therefore, an in vitro model with human SK-N-SH neurons was established to investigate the effects of hypoxia and hypothermia on neuronal cell survival and to examine the regulation of RBM3 gene expression.
Methods
Cell culture
SK-N-SH human neuroblastoma cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were routinely cultured in Dubecco’s Modified Eagle’s Medium (DMEM) with 10% heat-inactivated fetal calf serum (FCS), 1 mM sodium pyruvate, and 1% penicillin/streptomycin at 37°C and 5% CO2 in a fully humidified incubator. Culture medium was changed every 2 days and the cells were passed upon reaching ~80% confluency. Cell culture flasks, culture dishes, and 96-well plates were coated with 0.2% gelatin in phosphate-buffered saline (PBS), and the cells were allowed to settle for 24 hours before the experimental start.
Hypoxia/reoxygenation
Hypoxia was induced in the SK-N-SH neurons by using a three-gas-regulated incubator (Binder, Tuttlingen, Germany) with 0.2% O2 (hypoxia), or 8% O2 (subatmospheric oxygen tension), 5% CO2 and N2 balance at 37°C for 24 hours, followed by reoxygenation at atmospheric normoxic conditions (95% air and 5% CO2) for up to an additional 72 hours. Control groups were maintained at atmospheric normoxia (21% O2) for the duration of the experiment (Figure 1). SKN-SH neurons were cultured in DMEM with 10% FCS and 1% penicillin/streptomycin.
Figure 1.
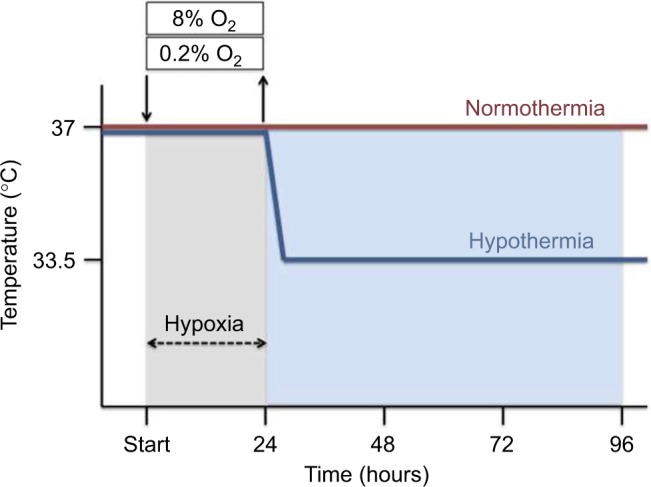
Time temperature protocol. SK-N-SH cells were exposed to either 0.2% or 8% O2 for 24 hours followed by normothermia (37°C) or hypothermia (33.5°C) for 24, 48 or 72 hours (total 48, 72 and 96 hours).
Moderate therapeutic hypothermia
Post-hypoxia moderate therapeutic hypothermia was subsequently applied by cooling the neurons to 33.5°C for 24, 48, or 72 hours in a CO2 incubator with 95% air, 5% CO2, and 95% humidity (Hereaus, Hanau, Germany), in accordance with the current guidelines for treatment of perinatal asphyxia. Normothermic experimental groups were maintained at 37°C (Figure 1).
Immunofluorescent microscopy
To examine cell morphology, SK-N-SH cells were stained for neuron-specific beta III-Tubulin (Covance, Princeton, NJ, USA) and 4′,6-diamidin-2-phenylindol (DAPI; Pierce, Rockford, IL, USA) nuclear counterstain according to an established protocol. Briefly, cells were fixed with 4% paraformaldehyde for 20 minutes, permeabilized with 0.3% Triton X-100 in PBS, and blocked with 10% goat serum for nonspecific binding. The primary antibody (anti-beta III-Tubulin, 1:2000) was diluted in 0.5% bovine serum albumin in PBS, and cells were incubated overnight at 4°C with gentle agitation. Cells were then washed and incubated with the secondary antibody (Texas Red; Thermo Fisher Scientific, Waltham, MA, USA) for 1 hour at room temperature and protected from light with gentle agitation. Subsequently, cells were washed and then incubated with DAPI (1:2000) for 10 minutes at room temperature and protected from light with gentle agitation. After a final washing with PBS, cover slips were mounted onto glass slides and examined by fluorescent microscopy.
Cell death
For quantification of cell death, the enzymatic activity of lactate dehydrogenase (LDH) released from the cytosol of damaged cells was determined by using a colorimetric Cytotoxicity Detection Kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instructions. Extinctions were measured at 570 nm in a microtiter plate reader (Multiscan Ascent; Thermo Fisher Scientific). Optical density values were normalized to the normoxic and normothermic control for each corresponding time point. In addition, release of neuron-specific enolase (NSE) from damaged cells was measured by ELISA (Roche Diagnostics).
ATP concentration
Intracellular ATP levels were quantified by using the CellTiterGlo®Luminescent Cell Viability Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Luminescence intensity was measured with an integrity time of 500 ms.
Quantitative real time polymerase chain reaction analysis
SK-N-SH cells were seeded in 22 cm2 cell culture dishes at a density of 2×105 cells per dish 24 hours before the experimental start. Total cellular RNA extraction was performed by using the peqGOLD RNAPure™ kit (PEQLAB Biotechnology, Erlangen, Germany) according to the manufacturer’s instructions. RNA concentration was analyzed employing a NanoDrop spectrophotometer (Thermo Scientific, Karlsruhe, Germany). Two thousand nanograms of total RNA was reversely transcribed into cDNA by using Moloney murine leukemia virus reverse transcriptase and random primer (Promega, Madison, WI, USA) in a reaction mixture including DNAse I treatment. The products of RBM3 and GAPDH as housekeeping gene were quantified in real time by using the following dye-labeled fluorogenic reporter oligonucleotide TaqMan™ probes (Thermo Scientific Fisher): RBM3 Hs00943160_g1 and GAPDH Hs03929097_g1.
Protein extraction and Western blot analysis
SK-N-SH cells were seeded in 60 cm2 cell culture dishes at cell densities between 1×106 and 4×106 cells per dish differing for each time point. Cells were allowed to settle 24 hours before experiments. After experiments, SK-N-SH cells were mechanically scratched off the dish surface and lysated by adding a protein lyses buffer ([20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM PMSF, and 1:100 of protease inhibitor cocktail]; Sigma-Aldrich, Munich, Germany). Protein concentration was analyzed employing the BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA). Equivalent amounts of protein were loaded on a 12% gel for SDS-PAGE. Immunoblotting to a nitrocellulose membrane (Biostep GmbH, Jahnsdorf, Germany) was performed by using the tank blotting transfer method (Bio-Rad Laboratories GmbH, Munich, Germany). For immunodetection, anti-RBM3 (1:1000, Cat#14363-1-AP; Proteintech, Chicago, IL, USA) and anti-beta-actin (1:10,000, Cat#A5441; Sigma-Aldrich) were used as primary antibodies followed by incubation with anti-rabbit IgG-HRP (1:10,000, Cat#711-035-152, Jackson, ImmunoResearch, West Grove, PA, USA) as secondary antibody. Proteins were visualized by using SuperSignal West Dura Chemiluminescent Substrate according to the manufacturer’s instructions (Thermo Fisher Scientific, Bonn, Germany). Densitometry analysis was performed with Image Lab (Bio-Rad Laboratories GmbH, Munich, Germany).
Statistical analysis
Data were graphed and analyzed with Prism GraphPad 6 (GraphPad Software Inc., Lajolla, CA, USA). All experiments were performed at minimum three times, and results are presented as mean ± SD. Variability between groups was compared by one-way analysis of variance (ANOVA) followed by Bonferroni’s post-test for correction of multiple comparisons. Statistical comparison of two non-paired samples was performed by using the Student’s t-test. For all analyses, p<0.05 was considered statistically significant.
Results
LDH
Cell death or cytotoxicity is typically quantified by cellular membrane damage, which results in the release of intracellular proteins such as the LDH into the cultured medium. In the authors’ in vitro model, exposure of the SK-N-SH neurons to hypoxia (0.2% O2 for 24 hours) did not result in a significant increase of LDH release compared to the atmospheric normoxia control directly after exposure (Figure 2A). However, 24 hours after the hypoxic event, LDH release was significantly higher compared to groups maintained at 21% O2 for the same duration (48 hours after experimental start). Application of moderate hypothermia (33.5°C) resulted in decreased LDH release in comparison to neurons maintained at normothermic conditions after hypoxia, reaching a significant level at 48 and 72 hours of hypothermic treatment (eg, 72 and 96 hours after experimental start). SK-N-SH neurons that were only exposed to hypothermia for 24, 48, or 72 hours at atmospheric normoxia (21% O2) at all times showed the lowest LDH releases.
Figure 2.

LDH release in SK-N-SH cells exposed to (A) 0.2% or (B) 8% O2 for 24 hours followed by normothermia/hypothermia for 24, 48, or 72 hours (total 48, 72 and 96 hours, n=3–4, *p<0.05).
Abbreviation: LDH, lactate dehydrogenase.
Exposure to subatmospheric oxygen tension (8% O2 for 24 hours) had no observable impact on SK-N-SH neuron viability. The LDH release did not differ from that in neurons under atmospheric normoxia (Figure 2B). However, exposure to moderate hypothermia for 24, 48, or 72 hours significantly decreased LDH release in SK-N-SH neurons that were exposed to 8% O2 as compared to normothermic controls at 37°C. The amount of LDH release was comparable with that of SK-N-SH treated with moderate hypothermia at 21% O2.
NSE
NSE is an established biomarker for quantification of neurological damage. In accordance with the LDH data, hypoxia (0.2% O2 for 24 hours) followed by 72 hours of atmospheric normoxia resulted in a significant increase in NSE release in the SK-N-SH cells, indicating cellular injury (Figure 3). Moderate hypothermia (33.5°C) for 72 hours significantly reduced NSE concentrations and, therefore, attenuated the neurological damage due to hypoxia. Furthermore, NSE concentration in cooled post-hypoxic SK-N-SH was comparable to that in the undamaged control group kept at 21% O2 and normothermia (37°C) at all observed time points. The release of NSE was significantly lower in SK-N-SH neurons that were only exposed to moderate hypothermia for 72 hours compared to undamaged SK-N-SH at 37°C.
Figure 3.

(A) NSE release in SK-N-SH cells exposed to hypoxia (0.2% O2 for 24 hours) followed by normothermia/hypothermia for 72 hours compared to SK-N-SH at atmospheric normoxia and hypothermia/normothermia for 72 hours (n=3, *p<0.05). (B) Intracellular ATP concentration in SK-N-SH cells exposed to hypoxia (0.2% O2 for 24 hours) followed by normothermia/hypothermia compared to SK-N-SH at atmospheric normoxia and hypothermia/normothermia (n=4, *p<0.05).
Abbreviation: NSE, neuron-specific enolase.
ATP
Intracellular ATP concentration was quantified in SK-N-SH cells exposed to hypoxia and then treated with moderate hypothermia (Figure 3B). Hypoxia (0.2% O2 for 24 hours) significantly decreased ATP levels. Twenty-four hours after reoxygenation, ATP concentration significantly increased and was almost as high as in the control at 21% O2. Moderate hypothermia for 24, 48, or 72 hours led to slightly increased intracellular ATP levels in SK-N-SH cells already exposed to hypoxia, but the differences were not significant. Neurons that were exposed to hypothermia at atmospheric normoxia (21% O2) had similar ATP levels as those maintained at 37°C.
Cell morphology is maintained under hypothermic conditions
In addition to decreased viable cell counts, exposure to hypoxia (0.2% O2 for 24 hours) followed by 72 hours at atmospheric normoxia also led to morphological changes, as observed in swollen and enlarged somata in the SK-N-SH neurons (Figure 4). Treatment with moderate hypothermia (33.5°C) for 72 hours attenuated the morphological changes induced by hypoxia. The cooled cells returned to their physiological appearance, and no sign of cell swelling was observed.
Figure 4.

Cell morphology after 24 hours of atmospheric normoxia, subatmospheric oxygen tension, or hypoxia (21%, 8%, or 0.2% O2) followed by 72 hours of normothermia (37°C) or hypothermia (33.5°C). SK-N-SH cells are stained with anti-beta III-Tubulin (red) and DAPI (blue).
Abbreviation: DAPI, 4′,6-diamidin-2-phenylindol.
Subatmospheric oxygen tension (8% O2 for 24 hours followed by 72 hours normoxia) resulted in only small morphological changes (Figure 4). SK-N-SH neuron cell somata were only slightly enlarged, but physiological shape was preserved, and no signs of cell swelling were observed. No observable differences were seen in the morphological appearance of SK-N-SH cells that were cooled after exposure to subatmospheric oxygen tension as compared to those maintained at atmospheric normoxia (21% O2).
RBM3 mRNA expression (reverse transcriptase-quantitative polymerase chain reaction [RT-qPCR])
Moderate hypothermia for 48 hours resulted in significantly increased levels of RBM3 transcripts (mRNA) in human SK-N-SH cells (Figure 5A). The level of RBM3 mRNA was threefold higher compared to SK-N-SH cells at normothermia. No impact on the regulation of RBM3 transcription by hypoxia or subatmospheric oxygen tension (0.2% or 8% O2 for 24 hours) was observed (Figure 5B). Moderate hypothermia increased RBM3 mRNA levels in SK-N-SH cells that had undergone hypoxia (0.2% O2 for 24 hours) beforehand, with significant differences 72 and 96 hours after the experimental start (Figure 6). Likewise, moderate hypothermia resulted in a significant upregulation of RBM3 mRNA in SK-N-SH cells exposed to 8% O2 for 24 hours before reaching a significant level 48 and 72 hours after the experimental start (Figure 6). The temperature-regulated induction of RBM3 mRNA in post-hypoxic SK-N-SH cells was similar to that in SK-N-SH cells exposed to atmospheric conditions, at all times. Therefore, no additional or attenuating effect of hypoxia or subatmospheric oxygen tension on the regulation due to moderate hypothermia on RBM3 transcription was observed.
Figure 5.

Expression of RBM3 mRNA in SK-N-SH after (A) 48 hours of moderate hypothermia compared to a normothermic control group and after (B) 24 hours of 0.2% O2 or 8% O2 oxygen tension compared to an atmospheric normoxic control group at 21% O2 (n=3–6, *p<0.05).
Abbreviations: RBM3, RNA-binding motif 3; m, messenger.
Figure 6.

Expression of RBM3 mRNA in SK-N-SH exposed to 0.2% O2 (A) or 8% O2 (B) for 24 hours followed by normothermia/hypothermia for 24, 48, or 72 hours (total 48, 72 and 96 hours, n=3, *p<0.05).
Abbreviation: RBM3, RNA-binding motif 3.
RBM3 protein expression (Western blot analysis)
In accordance with the presented RT-qPCR data, RBM3 protein levels were significantly increased in SK-N-SH neurons in response to moderate hypothermia (33.5°C) for 48 hours (Figure 7A). Neither reduction to 0.2% nor to 8% O2 for 24 hours had an observable impact on RBM3 expression (Figure 7B). Moderate hypothermia (33.5°C) for 24, 48, or 72 hours significantly increased RBM3 protein concentration in SK-N-SH cells that were either exposed to hypoxia or subatmospheric oxygen tension for 24 hours (Figure 8). The overexpression of RBM3 protein in response to moderate hypothermia was similar in SK-N-SH cells already exposed to hypoxia or kept at 21% O2. No additional effects on RBM3 expression could be observed. The results on RBM3 protein expression correlate with the findings in the regulation of RBM3 transcripts analyzed via RT-qPCR.
Figure 7.

Western blots and densitometric quantification of RBM3 protein versus beta-actin in SK-N-SH after (A) 48 hours of normothermia (37°C) versus hypothermia (33.5°C) and after (B) 24 hours of 0.2% or 8% O2 tension compared to 24 hours of atmospheric normoxia (n=4–8, *p<0.05).
Abbreviation: RBM3, RNA-binding motif 3.
Figure 8.

Western blots and densitometric quantification of RBM3 protein versus beta-actin in SK-N-SH cells after (A) 24 hours of hypoxia (0.2% O2) or (B) 24 hours of subatmospheric oxygen tension (8% O2) followed by 24, 48, or 72 hours of normothermia/hypothermia (total 48, 72 and 96 hours, n=4, *p<0.05).
Abbreviation: RBM3, RNA-binding motif 3.
Discussion
Cell death
Reperfusion or reoxygenation injury is an acknowledged mechanism of cellular death characterized by excessive formation of ROS, leading to an inflammatory response and consequently cell death.26 In the present study, the authors demonstrated that reoxygenation after exposure of SK-N-SH neurons to hypoxia (0.2% O2) for 24 hours results in significantly increased cell death. This effect was not immediately observed at the end of the hypoxic phase, but rather 24 hours after reoxygenation, thus, indicating reoxygenation after hypoxia as the crucial effector of cell death in the authors’ cell culture model. Induced cell death was still significantly observable 72 hours after hypoxic insult, as quantified by increased NSE release into the cultured medium. Treatment with moderate hypothermia for 48 and 72 hours effectively attenuated cell death in the post-hypoxic SK-N-SH neurons as observed in significantly reduced LDH and NSE releases, thus further supporting clinical indications of therapeutic hypothermia as a potent neuroprotective strategy against hypoxic–ischemic brain injury. The observations are also supportive of previous in vitro findings where a beneficial effect of mild-to-moderate hypothermia on neuronal cell survival has been demonstrated.27–29
Exposure to subatmospheric oxygen tension (8% O2) for 24 hours, however, did not lead to observable increased cell death in the authors’ neuronal cell culture model. The amount of cell death was comparable to neurons maintained at atmospheric normoxia (21% O2). Although ambient oxygen (21% O2) is commonly used as a normoxic control in the majority of in vitro models, it is higher than in vivo O2 levels (1%–11%) in the mammalian brain. Previous studies have even demonstrated enhanced survival of neuronal cells under subatmospheric oxygen tension.30 The findings are comparable to those of Parmar et al who also observed no significant impact of subatmospheric oxygen tension on the number of viable neurons, nor on cell morphology in rat cortices exposed to 8% O2 for 3 hours.31 Therefore, it can be concluded that a moderate decrease in oxygen from ambient level has to be considered as a physiological cell environment for neurons and does not result in increased cell death.
Nonetheless, treatment of SK-N-SH neurons exposed to 8% O2 for 24 hours with moderate hypothermia still resulted in reduced LDH releases and, therefore, attenuated cell death. This cytoprotective effect of hypothermia is also observed in SK-N-SH cells that were only cooled to 33.5°C under atmospheric conditions, and it is suspected that this effect is not due to less cell proliferation in the hypothermic group, as no significant differences in BrdU cell proliferation assay were observed (data not shown). Therefore, it is assumed that hypothermia has a beneficial impact on SK-N-SH cell survival under physiological conditions.
The authors investigated if the observable cell death in the culture model is due to apoptosis via activation of caspase 3 and did not observe any significant differences in the normothermia versus hypothermia group (data not shown). Therefore, cell death in the authors’ model is most likely due to necrosis or via a caspase-independent pathway, as has been shown in other studies examining neuronal cell death due to asphyxia.32
Intracellular ATP levels are significantly reduced immediately after exposure to hypoxia, but are restored in 24 hours after oxygen deprivation. Even though slightly higher ATP concentrations are observed in post-hypoxic SK-N-SH neurons that were cooled compared to those at 37°C, the difference was not significant, however this cooling may contribute to keeping the neurons viable but does not seem to be the most significant mechanism for neuroprotection induced by moderate hypothermia.
Cell morphology
Cell volume and intracellular ionic concentrations are strictly regulated to maintain a physiological cell function.33 Due to hypoxia, several changes in cellular pathways occur, leading to ionic imbalances that result in an intracellular accumulation of calcium and sodium. Consequently, normal homeostasis of affected cells is disrupted and fluid streams into the intracellular space result in cell swelling, referred to as osmotic stress.34 During reoxygenation, this process intensifies the excessive production of ROS. While glial cells are able to regulate their cell volume and ionic balance, neurons are not.35,36 Thus, osmotic stress leads to neuronal cell death, eventually. These morphological changes could be observed in the authors’ cell culture model with human SK-N-SH neurons stained with anti-beta III-Tubulin/DAPI after being exposed to hypoxia (0.2% O2 for 24 hours) followed by 72 hours of reoxygenation. Moderate hypothermia (33.5°C for 72 hours) was able to diminish the swelling of cell somata, thus protecting SK-N-SH from hypoxic injury. Subatmospheric oxygen tensions (8% O2 for 24 hours) did not lead to cell swelling and, therefore, seem to have no impact on ionic balances in human SK-N-SH.
Regulation of RBM3
There is a growing interest in stress responsive genes such as RBM3 which could possibly mediate hypothermia-induced neuroprotection, hence the authors strive to identify key targets of hypothermia to better understand its beneficial effects on cell survival. In the authors’ cell culture model, an upregulation of RBM3 transcripts and proteins in response to moderate hypothermia (33.5°C for 24, 48, or 72 hours) was demonstrated. This overexpression was observed in SK-N-SH cells that were exposed to hypoxia or 8% O2 concentration for 24 hours before induction of hypothermia, as well as in the cells that were kept under atmospheric normoxia at all times. There was no additional effect of hypoxia or 8% O2 concentration on the upregulation of RBM3 due to moderate hypothermia, and the amount of the increase was similar in all groups (21%, 8%, or 0.2% O2). The findings are in correlation with previously published data, where the temperature-related regulation of RBM3 was examined. Danno et al17 observed an increase in RBM3 expression after moderate hypothermia (32°C) in several non-neuronal human cell lines. The authors demonstrated in a previous study an upregulation of RBM3 transcripts and protein in murine hippocampal brain slices and in murine hippocampal neurons in response to moderate hypothermia.18 This study is, according to the authors’ knowledge, the first one to examine the effects of both hypothermia and hypoxia (in different ranges) on the regulation of RBM3 in neuronal cells. Wellmann et al demonstrated a hypoxia-induced upregulation of RBM3 transcripts and proteins in non-neuronal cells in an HIF-1α-independent manner.19 In contrast, no change in RBM3 expression in the neuronal SK-N-SH cell line in response to hypoxia or subatmospheric oxygen tension (0.2% or 8%) for 24 hours was observed, neither on the transcriptional nor on the translational level. The RBM3 regulation in response to reduced oxygen concentrations in other neuronal cells (murine hippocampal HT-22, murine primary neurons, data not shown) was also examined and the authors could not find any regulation, neither on transcriptional nor on the translational level. Trollmann et al exposed pregnant mice to hypoxia (6% O2 for 6 hours) at late pregnancy and analyzed the gene expression of the fetal brains and the placenta via microarray analysis.37 They also did not see an upregulation of RBM3, but saw a suppression of RBM3 expression in response to hypoxia. In conclusion, the oxygen-regulated expression of RBM3 seems to distinguish between different cell types and possibly also depends on other stress factors that occur simultaneously such as additional glucose deprivation during ischemia.
Through which specific molecular mechanism is RBM3 protecting neurons from cell death remains an unanswered question. Prior studies implicate that RBM3 is involved in protein translation, as it is able to bind ribosome subunit 60S and increases the formation of polysomes.21,22 RBM3 might act as a chaperone to ensure basal protein translation under hypothermic conditions. Moreover, RBM3 seems to have a regulatory function in the expression of microRNAs, although its exact function remains unclear.38–40 It has been demonstrated that RBM3 is able to mitigate cell death due to neurodegenerative diseases.25,41 Interestingly, in that context, a recent study identified RTN3, a protein that is involved in synapse function, as a target of RBM3.42 Taken together, further research is needed to address the possible targets and molecular functions of RBM3.
Conclusion
The presented cell culture model, by using human SK-N-SH neurons, reflects several important clinical aspects of therapeutic hypothermia and is suitable for investigating the induced pathways and gene expression kinetics. The beneficial effects of moderate hypothermia after hypoxic brain injury with regard to cell survival and morphology were demonstrated, which support the findings of previous studies. Moreover, a temperature-related regulation of the cold-shock protein RBM3 correlating with the protective effects of hypothermia was shown. The findings suggest that RBM3 is a promising key target of hypothermia and, therefore, a possible mediator of hypothermia-induced neuroprotection. Further research is needed to elucidate cellular functions and molecular pathways of RBM3 and its role as a cytoprotective effector.
Acknowledgments
Constanze Pfitzer is a participant in the BIH Charité Junior Clinician Scientist Program funded by the Charité – Universitaetsmedizin Berlin and the Berlin Institute of Health. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Lawn JE, Cousens S, Zupan J. 4 million neonatal deaths: when? where? why? Lancet. 2005;365(9462):891–900. doi: 10.1016/S0140-6736(05)71048-5. [DOI] [PubMed] [Google Scholar]
- 2.Johnston MV, Fatemi A, Wilson MA, Northington F. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol. 2011;10(4):372–382. doi: 10.1016/S1474-4422(11)70016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacobs SE, Berg M, Hunt R, Tarnow-Mordi WO, Inder TE, Davis PG. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev. 2013;1:CD003311. doi: 10.1002/14651858.CD003311.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simbruner G, Mittal RA, Rohlmann F, Muche R, neo.nEURO.network Trial Participants Systemic hypothermia after neonatal encephalopathy: outcomes of neo.nEURO.network RCT. Pediatrics. 2010;126(4):e771–e778. doi: 10.1542/peds.2009-2441. [DOI] [PubMed] [Google Scholar]
- 5.Shankaran S, Laptook AR, McDonald SA, et al. Whole-body hypothermia for neonates with hypoxic–ischemic encephalopathy. N Engl J Med. 2005;353(15):1574–1584. doi: 10.1056/NEJMcps050929. [DOI] [PubMed] [Google Scholar]
- 6.Bernard SA, Gray TW, Buist MD, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346(8):557–563. doi: 10.1056/NEJMoa003289. [DOI] [PubMed] [Google Scholar]
- 7.Peberdy MA, Callaway CW, Neumar RW, et al. Part 9: post-cardiac arrest care: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122(18 Suppl 3):S768–S786. doi: 10.1161/CIRCULATIONAHA.110.971002. [DOI] [PubMed] [Google Scholar]
- 8.Laptook AR, Corbett RJ, Sterett R, Garcia D, Tollefsbol G . Quantitative relationship between brain temperature and energy utilization rate measured in vivo using 31P and 1H magnetic resonance spectroscopy. Pediatr Res. 1995;38(6):919–925. doi: 10.1203/00006450-199512000-00015. [DOI] [PubMed] [Google Scholar]
- 9.Hagerdal M, Harp J, Nilsson L, Siesjö BK. The effect of induced hypothermia upon oxygen consumption in the rat brain. J Neurochem. 1975;24(2):311–316. doi: 10.1111/j.1471-4159.1975.tb11881.x. [DOI] [PubMed] [Google Scholar]
- 10.Thoresen M, Penrice J, Lorek A, et al. Mild hypothermia after severe transient hypoxia-ischemia ameliorates delayed cerebral energy failure in the newborn piglet. Pediatr Res. 1995;37(5):667–670. doi: 10.1203/00006450-199505000-00019. [DOI] [PubMed] [Google Scholar]
- 11.Bruno VM, Goldberg MP, Dugan LL, Giffard RG, Choi DW. Neuroprotective effect of hypothermia in cortical cultures exposed to oxygen-glucose deprivation or excitatory amino acids. J Neurochem. 1994;63(4):1398–1406. doi: 10.1046/j.1471-4159.1994.63041398.x. [DOI] [PubMed] [Google Scholar]
- 12.Globus MY, Busto R, Lin B, Schnippering H, Ginsberg MD. Detection of free radical activity during transient global ischemia and recirculation: effects of intraischemic brain temperature modulation. J Neurochem. 1995;65(3):1250–1256. doi: 10.1046/j.1471-4159.1995.65031250.x. [DOI] [PubMed] [Google Scholar]
- 13.Xiong M, Yang Y, Chen GQ, Zhou WH. Post-ischemic hypothermia for 24h in P7 rats rescues hippocampal neuron: association with decreased astrocyte activation and inflammatory cytokine expression. Brain Res Bull. 2009;79(6):351–357. doi: 10.1016/j.brainresbull.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 14.Diestel A, Troeller S, Billecke N, Sauer IM, Berger F, Schmitt KR. Mechanisms of hypothermia-induced cell protection mediated by microglial cells in vitro. Eur J Neurosci. 2010;31(5):779–787. doi: 10.1111/j.1460-9568.2010.07128.x. [DOI] [PubMed] [Google Scholar]
- 15.Derry JM, Kerns JA, Francke U. RBM3, a novel human gene in Xp11.23 with a putative RNA-binding domain. Hum Mol Genet. 1995;4(12):2307–2311. doi: 10.1093/hmg/4.12.2307. [DOI] [PubMed] [Google Scholar]
- 16.Sutherland LC, Rintala-Maki ND, White RD, Morin CD. RNA binding motif (RBM) proteins: a novel family of apoptosis modulators? J Cell Biochem. 2005;94(1):5–24. doi: 10.1002/jcb.20204. [DOI] [PubMed] [Google Scholar]
- 17.Danno S, Nishiyama H, Higashitsuji H, et al. Increased transcript level of RBM3, a member of the glycine-rich RNA-binding protein family, in human cells in response to cold stress. Biochem Biophys Res Commun. 1997;236(3):804–807. doi: 10.1006/bbrc.1997.7059. [DOI] [PubMed] [Google Scholar]
- 18.Tong G, Endersfelder S, Rosenthal LM, et al. Effects of moderate and deep hypothermia on RNA-binding proteins RBM3 and CIRP expressions in murine hippocampal brain slices. Brain Res. 2013;1504:74–84. doi: 10.1016/j.brainres.2013.01.041. [DOI] [PubMed] [Google Scholar]
- 19.Wellmann S, Bührer C, Moderegger E, et al. Oxygen-regulated expression of the RNA-binding proteins RBM3 and CIRP by a HIF-1-independent mechanism. J Cell Sci. 2004;117(Pt 9):1785–1794. doi: 10.1242/jcs.01026. [DOI] [PubMed] [Google Scholar]
- 20.Pilotte J, Cunningham BA, Edelman GM, Vanderklish PW. Developmentally regulated expression of the cold-inducible RNA-binding motif protein 3 in euthermic rat brain. Brain Res. 2009;1258:12–24. doi: 10.1016/j.brainres.2008.12.050. [DOI] [PubMed] [Google Scholar]
- 21.Dresios J, Aschrafi A, Owens GC, Vanderklish PW, Edelman GM, Mauro VP. Cold stress-induced protein Rbm3 binds 60S ribosomal subunits, alters microRNA levels, and enhances global protein synthesis. Proc Natl Acad Sci U S A. 2005;102(6):1865–1870. doi: 10.1073/pnas.0409764102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smart F, Aschrafi A, Atkins A, et al. Two isoforms of the cold-inducible mRNA-binding protein RBM3 localize to dendrites and promote translation. J Neurochem. 2007;101(5):1367–1379. doi: 10.1111/j.1471-4159.2007.04521.x. [DOI] [PubMed] [Google Scholar]
- 23.Chip S, Zelmer A, Ogunshola OO, et al. The RNA-binding protein RBM3 is involved in hypothermia induced neuroprotection. Neurobiol Dis. 2011;43(2):388–396. doi: 10.1016/j.nbd.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 24.Wellmann S, Truss M, Bruder E, et al. The RNA-binding protein RBM3 is required for cell proliferation and protects against serum deprivation-induced cell death. Pediatr Res. 2010;67(1):35–41. doi: 10.1203/PDR.0b013e3181c13326. [DOI] [PubMed] [Google Scholar]
- 25.Peretti D, Bastide A, Radford H, et al. RBM3 mediates structural plasticity and protective effects of cooling in neurodegeneration. Nature. 2015;518(7538):236–239. doi: 10.1038/nature14142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maxwell SR, Lip GY. Reperfusion injury: a review of the pathophysiology, clinical manifestations and therapeutic options. Int J Cardiol. 1997;58(2):95–117. doi: 10.1016/s0167-5273(96)02854-9. [DOI] [PubMed] [Google Scholar]
- 27.Khaspekov LG, Lyzhin AA, Victorov IV, Dupin AM, Erin AN. Hypoxic and posthypoxic neuronal injury in hippocampal cell culture: attenuation by lipophylic antioxidant U-18 and superoxide dismutase. Int J Neurosci. 1995;82(1–2):33–45. doi: 10.3109/00207459508994288. [DOI] [PubMed] [Google Scholar]
- 28.Hua Y, Hisano K, Morimoto Y. Effect of mild and moderate hypothermia on hypoxic injury in nearly pure neuronal culture. J Anesth. 2010;24(5):726–732. doi: 10.1007/s00540-010-0999-x. [DOI] [PubMed] [Google Scholar]
- 29.Bossenmeyer-Pourie C, Koziel V, Daval JL. Effects of hypothermia on hypoxia-induced apoptosis in cultured neurons from developing rat forebrain: comparison with preconditioning. Pediatr Res. 2000;47(3):385–391. doi: 10.1203/00006450-200003000-00017. [DOI] [PubMed] [Google Scholar]
- 30.Kaplan FS, Brighton CT, Boytim MJ, et al. Enhanced survival of rat neonatal cerebral cortical neurons at subatmospheric oxygen tensions in vitro. Brain Res. 1986;384(1):199–203. doi: 10.1016/0006-8993(86)91240-0. [DOI] [PubMed] [Google Scholar]
- 31.Parmar J, Jones NM. Hypoxic preconditioning can reduce injury-induced inflammatory processes in the neonatal rat brain. Int J Dev Neurosci. 2015;43:35–42. doi: 10.1016/j.ijdevneu.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 32.Takada SH, dos Santos Haemmerle CA, Motta-Teixeira LC, et al. Neonatal anoxia in rats: hippocampal cellular and subcellular changes related to cell death and spatial memory. Neuroscience. 2015;284:247–259. doi: 10.1016/j.neuroscience.2014.08.054. [DOI] [PubMed] [Google Scholar]
- 33.Pedersen SF, O’Donnell ME, Anderson SE, Cala PM. Physiology and pathophysiology of Na+/H+ exchange and Na+ -K+ -2Cl-cotransport in the heart, brain, and blood. Am J Physiol Regul Integr Comp Physiol. 2006;291(1):R1–R25. doi: 10.1152/ajpregu.00782.2005. [DOI] [PubMed] [Google Scholar]
- 34.Song M, Yu SP. Ionic regulation of cell volume changes and cell death after ischemic stroke. Transl Stroke Res. 2014;5(1):17–27. doi: 10.1007/s12975-013-0314-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McManus ML, Strange K. Acute volume regulation of brain cells in response to hypertonic challenge. Anesthesiology. 1993;78(6):1132–1137. doi: 10.1097/00000542-199306000-00017. [DOI] [PubMed] [Google Scholar]
- 36.Andrew RD, Lobinowich ME, Osehobo EP. Evidence against volume regulation by cortical brain cells during acute osmotic stress. Exp Neurol. 1997;143(2):300–312. doi: 10.1006/exnr.1996.6375. [DOI] [PubMed] [Google Scholar]
- 37.Trollmann R, Rehrauer H, Schneider C, et al. Late-gestational systemic hypoxia leads to a similar early gene response in mouse placenta and developing brain. Am J Physiol Regul Integr Comp Physiol. 2010;299(6):R1489–R1499. doi: 10.1152/ajpregu.00697.2009. [DOI] [PubMed] [Google Scholar]
- 38.Zhu X, Buhrer C, Wellmann S. Cold-inducible proteins CIRP and RBM3, a unique couple with activities far beyond the cold. Cell Mol Life Sci. 2016;73(20):3839–3859. doi: 10.1007/s00018-016-2253-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pilotte J, Dupont-Versteegden EE, Vanderklish PW. Widespread regulation of miRNA biogenesis at the Dicer step by the cold-inducible RNA-binding protein, RBM3. PLoS One. 2011;6(12):e28446. doi: 10.1371/journal.pone.0028446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong JJ, Au AY, Gao D, et al. RBM3 regulates temperature sensitive miR-142-5p and miR-143 (thermomiRs), which target immune genes and control fever. Nucleic Acids Res. 2016;44(6):2888–2897. doi: 10.1093/nar/gkw041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kita H, Carmichael J, Swartz J, et al. Modulation of polyglutamine-induced cell death by genes identified by expression profiling. Hum Mol Genet. 2002;11(19):2279–2287. doi: 10.1093/hmg/11.19.2279. [DOI] [PubMed] [Google Scholar]
- 42.Bastide A, Peretti D, Knight JR, et al. RTN3 is a novel cold-induced protein and mediates neuroprotective effects of RBM3. Curr Biol. 2017;27(5):638–650. doi: 10.1016/j.cub.2017.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]