

Abstract
Cardiac tropnoin I (cTnI) plays a critical role in the regulation of diastolic function, and its low expression may result in cardiac diastolic dysfunction, which is the most common form of cardiovascular disorders in older adults. In this study, cTnI expression levels were determined in mice at various ages and cardiac function was measured and compared between young adult mice (3 and 10 months) and older mice (18 months). The data indicated that the cTnI levels reached a peak high in young adult hearts (3 months), but decreased in older hearts (18 months). Furthermore, the older hearts showed a significant diastolic dysfunction observed by P–V loop and echocardiography measurements. To further define the mechanism underlying the cTnI decrease in aging hearts, we tested DNA methylation and histone acetylation modifications of cTnI gene. We found that acetylation of histone near the promoter region of cTnI gene played an important role in regulation of cTnI expression in the heart at different ages. Our study indicates that epigenetic modification caused cTnI expression decrease is one of the possible causes that result in a reduced cTnI level and diastolic dysfunction in the older hearts.
Keywords: Epigenetics, Histone acetylation, Gene expression, Troponin, Diastolic dysfunction
1. Introduction
Cardiac troponin I (cTnI) is an inhibitory subunit in troponin complex that binds to actin-tropomyosin and regulates muscle contraction and actin-activated myosin (actomyosin) ATPase activities [1]. cTnI plays a critical role in cardiac contraction and relaxation, especially the diastolic function. Our previous studies have demonstrated that deficiency or mutations of cTnI can result in an impaired relaxation in myocardial cells and diastolic dysfunction in the heart [2–9]. Diastolic dysfunction refers to an impaired relaxation and an abnormality in heart filling during diastole while left ventricular systolic function is preserved [10,11]. There is a critical need to understand the mechanisms that cause diastolic dysfunction in an aging heart and to develop target-based medications to treat the disorders.
Because of the difficulty in obtaining human cardiac tissues, the data of cTnI concentrations in the heart are very limited although the cTnI concentration in peripheral blood samples is used as an indicator for diagnosis of heart attack. At least one study has been reported that the content of cTnI in left ventricular myocardial cells decreased in older men with or without cardiac disease [12]. In our previous study, we have demonstrated that the cTnI expression is lower in fetal and new born hearts and up-regulated a week after birth and then reaches a peak high in adult hearts [2]. However, no follow-up studies have been performed to observe the cTnI levels in aging mouse hearts (18 months or older).
Several studies have shown that cTnI gene expression is controlled by the interaction of cis-elements and transcription factors [13]. Our previous studies on mouse cTnI gene have shown that 230 bp of proximal promoters are sufficient to drive cardiac-specific expression in transgenic animals [14]. Two kinds of cis-elements of this promoter, A/T rich and GATA elements have been demonstrated to play considerable roles in the regulation of cTnI gene transcription [15,16]. Mutation of each of these elements markedly reduces gene activation [14]. Cardiac core transcription factors, such as Mef2c and GATA4, can specifically bind to the elements in cardiac genes to initiate the expression. However, the mechanisms underlying the cTnI expression and the interaction between these regulatory cis-elements and transcription factors are still unclear.
Furthermore, a variety of epigenetic alterations affect all cells and tissues throughout life [17]. Epigenetic modifications involve the alteration in DNA methylation patterns, post translational modification of histones, and chromatin remodeling. DNA methylation of cardiac genes or acetylation of histone H3 near the gene promoter region can regulate the cardiac gene expression [18–21].
In this study, cTnI expression levels were determined in mice at various ages (new born, 2 weeks, 3, 10 and 18 months) and cardiac function was measured and compared between young adult mice (3 and 10 months) and older mice (18 months). The experimental data indicated that the cTnI levels were low in new born and reached a peak high in young adult hearts (3 months), but decreased in older hearts (18 months). The patterns were similar in both protein and mRNA levels. Compared to young adult hearts, the older hearts showed a significant diastolic dysfunction observed by P–V loop and echocardiography measurements. Furthermore, we found that epigenetic modifications such as acetylation of histone near the promoter region of cTnI gene played an important role in regulation of cTnI expression in the heart at different ages. Our study indicates that epigenetic modification caused cTnI expression decrease is one of the causes that result in a reduced cTnI level and diastolic dysfunction in the older hearts.
2. Materials and methods
2.1. Animals
Healthy and newborn, 2w, 3 m, and 18 m SPF class c57bl/6 mice were purchased from the Experimental Animal Center in Chongqing Medical University (Chongqing, China). All procedures on experimental animals were approved by the Animal Care and Use Committee at the Chongqing Medical University. Animal experiments were performed conform the NIH guidelines (Guide for the care and use of laboratory animals) [22]. All the mice we used in this study are female mice. And in our animal facilities, we have a strict regulation for animal breeding and a complete breeding recording. The range for animals aging over 3 months is ±1 day. The mice were killed by carbon dioxide asphyxia, and cardiac tissues were collected.
2.2. Morphological observations
Transmission Electron Microscope observation of cardiac ultra-structure was performed as well in mice at different ages. The mice were killed by carbon dioxide asphyxia, and heart samples of 3 m mouse and 18 m mouse were collected, then clipped a piece of left ventricle tissue and pruned it immediately into 1 mm [3] pieces and fixed in 2.5% glutaraldehyde for at least 2 h. Fixed heart samples were embedded after dehydration. Samples were sliced by ultra microtome (Leica, Solms, German) and stained negative using lead citrate. For EM morphological assays, 5 young adult mice (3 months) and 5 old mice (18 months) were used to take the cardiac samples for examination using a transmission electron microscope (Hitachi, Tokyo, Japan). In each group, 30 fields under EM were observed from each mouse by an expert in the EM laboratory who was blinded to the animal ages. The field with abnormal morphological changes was counted and compared finally between two age groups.
2.3. Echocardiography
Echocardiography studies were performed in our laboratory by using a Vevo 770 High-Resolution In Vivo Imaging System (VisualSonics, Toronto, ON, Canada) as described previously [6,7]. To decrease experimental bias, the echocardiography measurements were performed in the study by an examiner blinded to the genotype. All data and images were saved and analyzed with the Advanced Cardiovascular Package Software (VisualSonics, Toronto, ON, Canada) using an automated analysis or semi-automated analysis to evaluate the cardiac function.
2.4. In vivo pressure–volume loop analysis
Mouse left ventricular pressure and volume were simultaneously measured in intact experimental animals. Briefly, mice were anesthetized with 3% isoflurane and were supported by a ventilator with a maintenance dose of 2% isoflurane after tracheostomy. A 1F P–V catheter was inserted into the left ventricle through the right carotid artery. Signals of pressure and volume were continually recorded by using a P–V conductance system (MPVS Ultra, AD Instruments, INC. Houston, TX) coupled to a digital converter (PowerLab, AD Instruments). Hemodynamic parameters were measured under different preloads, which were elicited by transiently compressing the abdominal inferior vena cava. 5–8 mice were measured in each experimental group.
2.5. Western blotting
The quantities of cardiac proteins, especially troponins, were determined using Western blotting assays as described previously [2]. The immunoreactive bands were detected with a Chemiluminescence Analyzer, scanned and analyzed using Quantity One Version 4.4 software (Bio-Rad, Richmond, CA).
2.6. Total RNA extraction and quantitative real-time PCR analysis
Total RNA was extracted using a RNA Extraction kit (Bioteck, Beijing, China). Single-strand cDNA was reverse transcribed from 500 to 1000 ng RNA by using oligo dT-Adaptor primers and AMV reverse transcriptase kit (TaKaRa, Otsu, Japan). cDNA was detected using quantitative RT-PCR assay with a SYBR Green RealMasterMix kit (Tiangen, Beijing, China). The mRNA expression levels of cTnI were quantified. β-actin was used as the endogenous “housekeeping” gene to normalize the RNA sample levels. The primer sequences of cardiac-specific genes and controls were designed as follows:
The analyses of relative mRNA expression were carried out using 2−ΔΔCt method [23].
2.7. Chromatin immunopreciptitation (ChIP) assay
The heart tissues of each group were collected and prepared for ChIP assay following the manufacturer’s protocol using a ChIP assay kit (Millipore, MA, USA). Proteins and DNA were crossed link after adding formaldehyde (Sigma-Aldrich, St. Louis, USA) into heart tissue samples. The DNAs were ultrasonically cut into small fragments (200–1000 bp). Then the protein-DNA complexes were recruited and precipitated using monoclonal antibody against acetylated histone3, AcH3K9, AcH3K27, GATA4 and Mef2c (Millipore-06599, Abcam10812, Abcam4729, Santa-SC1237, Abcam-79436). The protein-DNA complexes were also precipitated using anti-RNA polymerase II antibody as positive control and using normal mouse IgG as negative control. The protein-DNA complexes were collected without antibody as input group (to show the total DNA in the samples). After removing the crossing link of proteins and DNAs, the DNAs were extracted. Specific primers were designed to determine the acetylation level of hitone H3, H3K9, K3K27, binding levels of GATA4 and Mef2c in proximal promoter regions of cTnI for quantitative Real-Time PCR (Q-PCR) analysis. The primers’ sequence was as followed, cTnI 5′-CCAACTGGAGCTTTGCACACG-3′ (forward) and 5′ –AGGACACTGAGATAAGGGGCG-3′ (reverse).
|
| |
| cTnI, | 5′-gcaggtgaagaaggaggaca-3′ (forward) |
| 5′-cgatattcttgcgccagtc-3′ (reverse). | |
| β-actin, | 5′-CACACCCGCCACCAGTTCG-3′(forward) |
| 5′-GTCCTTCTGACCCATTCCCACC-3′ (reverse). | |
|
| |
2.8. Statistical analysis
All data were expressed as mean ± SD, and statistically analyzed by one-way ANOVA. The differences were considered statistically significant when p < 0.05.
3. Results
3.1. cTnI level decreases in aging hearts
The cTnI levels in the hearts were measured using Western blotting assays at various ages. As shown in Fig. 1, the cTnI protein level increases gradually from birth to age of 2 weeks, and reaches a peak at age of 3 months. Then the cTnI level maintains stable from age of 3 months–10 months. However, a significant decrease of cTnI level is observed in mice at age of 18 months (18 m vs. 3 m, p < 0.05). The expression level of another troponin, cardiac TnT (cTnT) does not show such significant changes in the same experimental animals (Fig. 1C).
Fig. 1. cTnI and cTnT protein levels in mice at different ages.
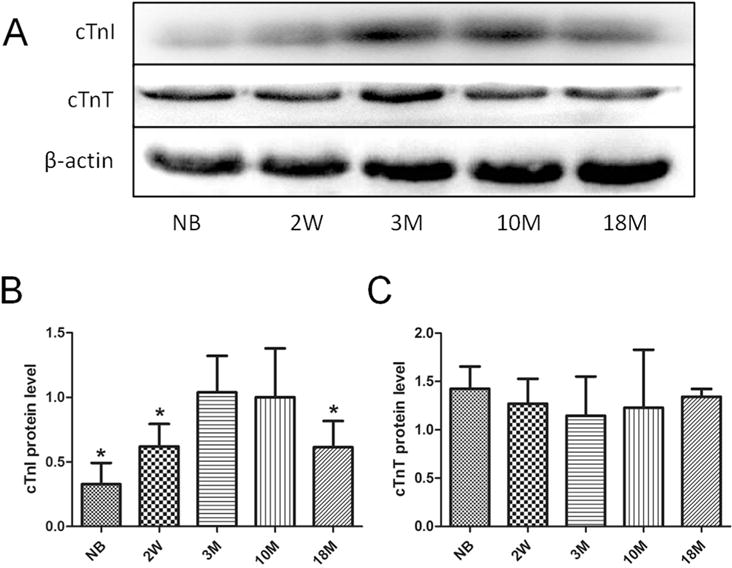
(A)Western blotting analysis using cTnI and cTnT antibodies. β-actin was used as protein loading control. Summary of Western blotting results of cTnI and cTnT are shown in Fig. 1B and C. Values are expressed as means ± SD from 4 separate experiments. The data from 3-month-old mice was set as 1 to compare with the data from other animal groups. Statistical significance was determined by ANOVA followed by Least—Significant Difference (LSD) tests. *p < 0.05 as compared with 3 m group.
We further used Q-PCR assays to measure the mRNA level of cTnI in the hearts of mice at the same time points. As shown in Fig. 2, the time-course of cTnI mRNA level is similar to that of cTnI protein expression, i.e. a significant decrease of cTnI mRNA is observed in the hearts of old mice at age of 18 months, indicating that both cTnI protein and mRNA decrease significantly in aging hearts.
Fig. 2. cTnI mRNA levels in mice at different ages.
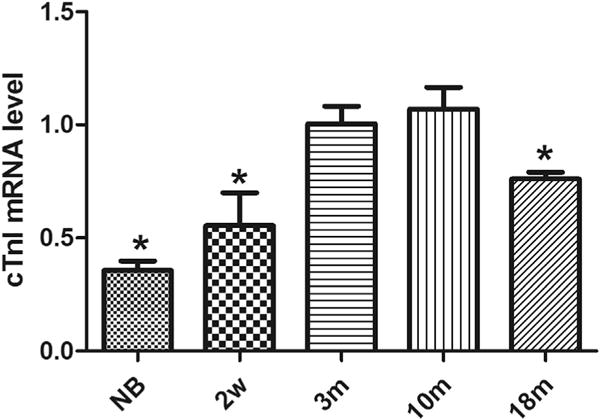
The expression pattern of cTnI mRNA was determined in the hearts of mice at various ages. The results are expressed as mean ± SD from at least three separate experiments. The data from 3-month-old mice was set as 1 to compare with the data from other animal groups. Statistical significance was determined by ANOVA followed by Least—Significant Difference (LSD) tests. *p < 0.05 as compared with 3 m group.
3.2. Cardiac morphological examination and ultra-structure evaluation of aging hearts
We compared the ultra-structures of cardiac tissues of young adult mice and old mice under a TEM. Fig. 3A showed the ultra-structures of heart tissues from left ventricle of 3-month mouse. Fig. 3B–D showed the ultra-structures of heart tissues from left ventricle of 18-month mice. The EM imaging indicated that the ultrastructure of the hearts from the mice at age of 18 months showed an enlargement of sarcoplasmic reticulum (approximately 7 per each field of 10000 × vision), a wildly thickening of Z line, broadly existence of sarcomere dissolution and occasionally hydropic degeneration (indicated by red arrows).
Fig. 3. Cardiac ultra-structures evaluation.

Fig. 3A shows the ultra-structures of heart tissue in left ventricle of 3-month mouse (10000 ×). Ultra-structures of heart tissue in left ventricle of 18-month mouse (10000 ×) are showed in Fig. 3B–D. Red arrow in Fig. 3B shows an enlargement of sarcoplasmic reticulum; red arrow in Fig. 3C displays sarcomere dissolution; and red arrows in Fig. 3D indicate hydropic degeneration (left) and thickening of Z line (right). Scale bars in A–D = 2 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Cardiac function measurements on aging mice
In vivo cardiac functions in intact mice at different ages were measured noninvasively using a high resolution mouse echocardiography. Representative echo images were illustrated as Fig. 4. As shown in Table 1, indicators of heart systolic function, such as EF value, FS value, and left ventricular isovolumic contraction time (IVCT), did not show any significant changes between young adult mice and aged mice (p > 0.05). However aged mice showed a significant prolongation of left ventricular isovolumetric relaxation time (IVRT) compared to young adult mice (15.60 ± 0.95 ms vs 12.91 ± 1.05 ms, p < 0.05). The E/A ratio measured with Doppler is significantly reduced in 18 month old mouse group (1.07 ± 0.20) compared to that in 3 month young mouse group (1.79 ± 0.14). The data from left ventricular pressure volume measurements showed a significantly reduced blood return during diastole accompanying with the reduced –dp/dt min and dp/dt max (Fig. 5A–C). Left ventricular developmental pressure (LVDP) did not show a significant change in the hearts between young adult and old mice (Fig. 5D). The Tau value was increased significantly in the hearts of old mice compared to that of young adult mice (Fig. 5E). The end diastolic pressure volume relationship (EDPVR, green line) was significantly changed in 18 month-old mice (0.74 ± 0.03 mmHg/μl) compared to that in 3 month-old mice (0.29 ± 0.01 mmHg/ml), suggesting a more restricted ventricle during the end of diastole. However, the end systolic pressure volume relationship (ESPVR, red line) did not show significant changes between 3 month-old and 18 month-old mice (Fig. S1 in Supplementary Data). All of these cardiac functional data indicate a diastolic dysfunction and a less compliant left ventricle in old mice.
Fig. 4. Representative M-mode echocardiograph images obtained from mice at ages of 3 months and 18 months.
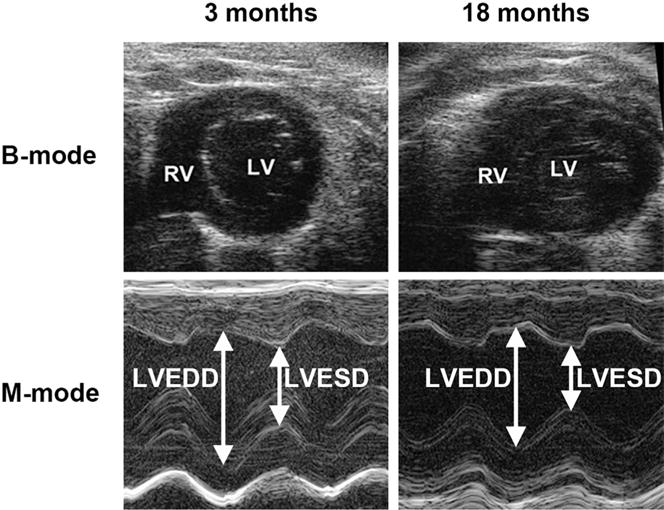
RV, right ventricle; LV, left ventricle; LVEDD, left ventricular end diastolic dimension; LVESD, left ventricular end systolic dimension.
Table 1.
Cardiac function measurements on young adult and old mice.
| Parameters | 3 months | 18 months |
|---|---|---|
| Body weight (g) | 24.04 ± 1.47 | 23.59 ± 1.32 |
| LV end diastole | ||
| IVS (mm) | 0.89 ± 0.21 | 0.92 ± 0.16 |
| LVID (mm) | 2.95 ± 0.50 | 3.05 ± 0.46 |
| LV PW (mm) | 0.77 ± 0.09 | 0.89 ± 0.30 |
| LV volume (μl) | 34.93 ± 12.25 | 36.99 ± 12.57 |
| LV end systole | ||
| IVS (mm) | 1.58 ± 0.21 | 1.77 ± 0.41 |
| LVID (mm) | 1.35 ± 0.27 | 1.25 ± 0.17 |
| LV PW (mm) | 1.32 ± 0.14 | 1.50 ± 0.36 |
| LV volume (μl) | 4.96 ± 2.27 | 3.86 ± 1.24 |
| Ejection fraction (%) | 85.99 ± 3.58 | 88.16 ± 2.36 |
| LV fraction shortening (%) | 54.09 ± 4.56 | 58.81 ± 4.19 |
| Mitral Pulse Doppler | ||
| E/A | 1.79 ± 0.14 | 1.07 ± 0.20* |
| IVRT (ms) | 12.91 ± 1.05 | 15.60 ± 0.95* |
| IVCT (ms) | 7.66 ± 1.21 | 8.27 ± 0.87 |
Values are expressed as mean ± SE for each group. LV, left ventricle; IVS, intra-ventricular septum; LVID, left ventricular internal diameter; PW, posterior wall thickness of LV; IVRT, isovolumetric relaxation time; IVCT, isovolumetric contraction time. Statistical significance was determined by ANOVA followed by post hoc Newman-Keuls (SNK) tests.
P < 0.05, compared with 3 months group.
Fig. 5. Left ventricular pressure-volume measurements in mice.
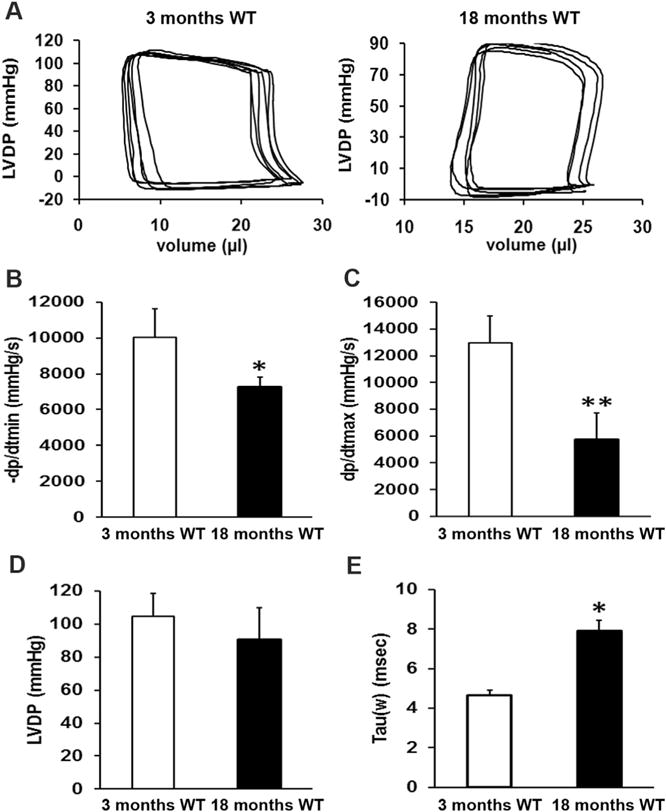
Representative pressure-volume (P–V) loops obtained from Millar catheter measurement in left ventricles of mice at ages of 3 months and 18 months, showing a significant reduced blood filling during the diastole in 18 months old mice compared to that of 3-month-old young adult mice (A). P–V loop data analyses indicate that –dp/dt min is significantly reduced in 18 month old mice compared to that of 3-month-old young adult mice (B). dp/dt max also shows a significant decrease in old mice (C). However, left ventricular developed pressure (LVDP) does not show any significant changes between old and young adult mice (D). The Tau value is significantly increased in 18 month old mice (E). These data suggest a restricted left ventricle and impaired relaxation in 18 month old hearts. The data are obtained from the measurements of 8 mice per group and the values are expressed as Means ± SD from 8 separate experiments. Statistical significance is determined using t tests. *p < 0.05, **p < 0.01.
3.4. Determination of histone acetylation in the promoter region of cTnI gene in mice at different ages
Modification of histone acetylation or deacetylation is a reversible dynamic process, which is a crucial part of epigenetic regulation in the body. To explore the potential mechanism underlying the reduced cTnI expression in aging hearts, we have determined the acetylation levels of histone 3 (AcH3), lysine 9 in H3 (AcH3K9) and lysine 27 in H3 (AcH3K27) in the promoter region of cTnI gene using ChIP-Q-PCR assays in mice at different ages. As shown in Fig. 6A, AcH3 levels near the promoter elements of cTnI are low in mice of new born or at age of 2 weeks. AcH3 levels reach the top in mice at ages of 3 months and 10 months. However, it is reduced significantly in old mice at age of 18 months (Fig. 6A). The pattern of AcH3K9 was similar to that of AcH3 between two mouse groups (Fig. 6B). However, no significant decrease was observed in AcH3K27 levels between 18 month and 3 month groups (Fig. 6C).
Fig. 6. Acetylation levels of histone3, histone3 lysine9 and histone3 lysine27 near the proximal promoter of cTnI in mice at different ages.
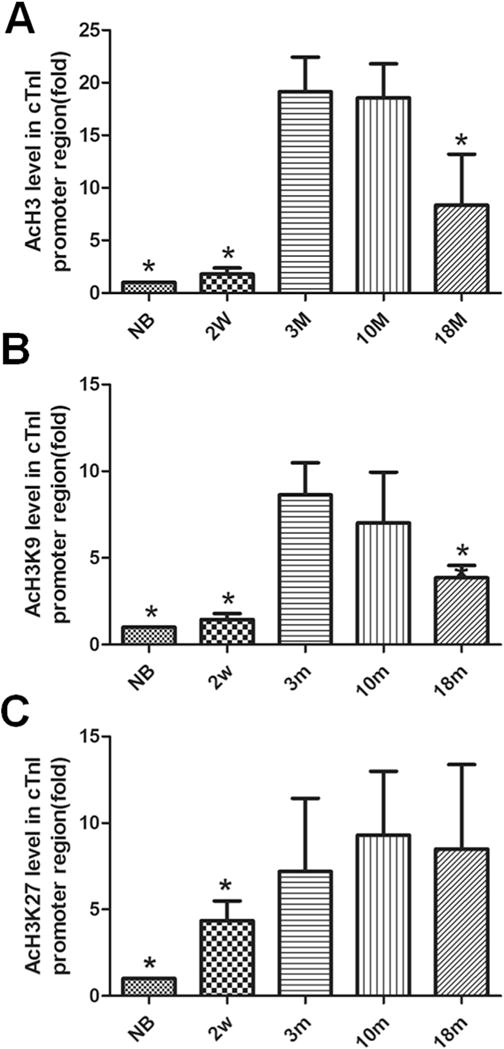
Acetylation levels of total histone 3 (AcH3) (A) and specific lysine 9 amino acid in H3 (AcH3K9)(B) and lysine 27 in H3 (AcH3K27)(C), are detected near the promoter of cTnI. The values from the input without antibody (nAb) are standardized as 1 and compared with the values of other experimental groups. Values are expressed as means ± SD from 3 separate experiments. Statistical significance was determined by ANOVA followed by Least—Significant Difference (LSD) tests. *p < 0.05as compared with 3 m group.
3.5. Determination of binding levels of transcription factors with GATA or Mef2 element of cTnI promoter in mice at different ages
ChIP assays were also applied to determine the binding levels of transcription factors with GATA4 or Mef2 element of cTnI promoter in mice at different ages. Binding of transcription factor to the GATA4 element in cTnI promoter reached top levels in mice at ages of 3 months and 10 months. But in 18 month old mouse group, the binding levels were reduced significantly just like the NB and 2w groups compared to 3 m group (p < 0.05) (Fig. 7A). Binding levels of transcription factor to Mef2 element in cTnI promoter showed a similar pattern (Fig. 7B), indicating that the binding affinity was reduced in both GATA4 and Mef2 elements in cTnI promoter in the aging hearts. These results are consistent with what we observed on histone acetylation levels in the aging mice (18 months).
Fig. 7. Binding of GATA4 and Mef2c with the proximal promoter regions of cTnI in mice at different ages.
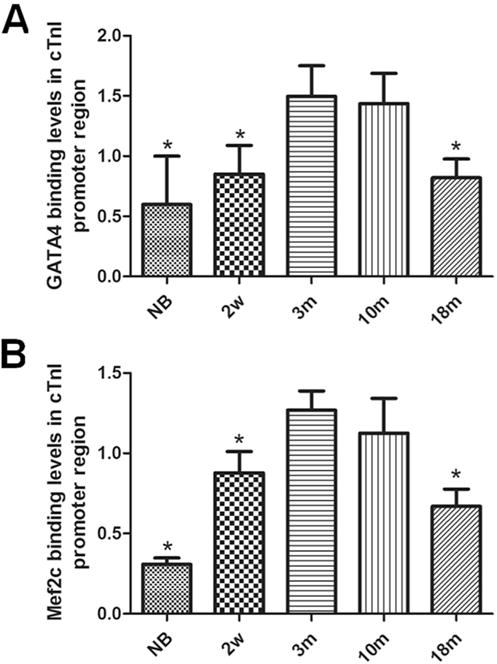
Transcription factor GATA4 binding with GATA elements of cTnI’s promoter is measured in the cardiac samples in mice at different ages (A). Transcription factor Mef2c binding with Mef2 element in the promoter of cTnI. is measured in cardiac samples in mice at different ages (B). Values are expressed as means ± SD from 3 separate experiments. Statistical significance was determined by ANOVA followed by Least—Significant Difference (LSD) tests. *p < 0.05 as compared with 3 m group.
4. Discussion
Heart failure (HF) is the leading cause of mortality in the United States [24]. In two major types of HF, HF with reduced ejection fraction (HFrEF) and HF with preserved ejection fraction (HFpEF), the latter is nearly exclusively found in older persons, particularly older women, in whom 90% of new HF cases are HFpEF [25]. The prevalence of HFpEF is raising, with morbidity, mortality, and healthcare costs now equal to HFrEF [26–29]. Outcomes following hospitalization for decompensated HFpEF are poor, with about 1/3 of patients re-hospitalized or dead within 90 days of discharge [30]. Its pathophysiology is poorly understood, and no medication trials have had positive effect on their primary end-points. A major cause of HFpEF is diastolic dysfunction which is very common in older adults. Diastolic dysfunction occurs when the ventricles cannot fill normally, and in severe conditions, it can lead to diastolic heart failure (DHF). Recently studies have revealed that cardiac diastolic dysfunction increases with advancing age, approximately 30%–40% of the population (aged 45 or older) had diastolic dysfunction, and nearly half of them were healthy individuals [31–33]. Although not every individual with diastolic dysfunction may develop to heart failure, diastolic dysfunction was still predictive of all-cause mortality, even when controlling for sex, age and EF31.
In the present study, we have measured cTnI concentration as well as cardiac function in mice at different ages including 18-month-old mice. Our data have demonstrated that cTnI concentration does reduce significantly in the hearts of aging mice accompanying with various signs of diastolic dysfunction such as the prolongation of IVRT, reduced E/A ratio detected by echocardiography and Doppler measurements In constant with the studies in elder people, diastolic dysfunction is commonly observed in aging hearts while the indexes of cardiac contraction does not show any significant changes in old mice compared to young adult mice. Although no obvious changes have been observed in cardiac tissues obtained from the old mice under histological examination (data not shown), ultra-structures of heart tissues from old mice do show some cellular changes such as enlarged sarcoplasmic reticulums, thickened Z lines and some dissolved sarcomeres and hydropic degeneration, suggesting that senile cardiac diastolic dysfunction may be associated with the alternations occurred in myocardial filament structures. These data demonstrate for the first time that loss of cTnI in aging heart might related to myocardial cellular changes and functional impairment, especially the impaired relaxation.
The deficiency of cTnI in the heart can result in diastolic dysfunction and sudden death in cTnI knockout mice [2]. Further studies have revealed that cTnI is critical in the regulation of cardiac function, especially diastolic function. Loss of cTnI or mutations on cTnI has been confirmed to be associated with impaired relaxation and diastolic heart failure [6,8]. In the present study, we have detected that the content of cTnI in the hearts from new born mice or 2 week-old mice are also low compared to 3-month-old adult mice. However, we know that ssTnI, another TnI expressed in the heart in the early life, is still expressed, which can compensate for the less expression of cTnI to maintain a normal cardiac function during the early life [2]. But, loss of cTnI in old mice is uncompensated since no expression or re-expression of ssTni occurs in aging hearts [2,3]. Decreased cTnI expression would cause a pure total TnI level decrease in the heart.
What is the potential mechanism(s) that causes the reduced cTnI expression in aging hearts? Recent studies have shown that epigenetic modifications such as DNA methylation and histone modification play important roles in the regulation of gene expression by modifying chromatin structures. In our study, we have observed interesting changes in histone acetylation levels near the promoter region of cTnI gene. When AcH3 or AcH3K9 levels are decreased in aging hearts, the cTnI expression levels and cTnI concentration in the heart of these old mice are also reduced accompanying by a diastolic dysfunction. However, no significant changes are observed on the methylation levels of cTnI gene promoters in the mice at different ages (Fig. S2 in Supplement Data). These data suggest that histone acetylation is more important as an epigenetic regulation than gene methylation on cTni gene expression regulation. It should be pointed out that histone acetylation (AcH3 or AcH3K9) data sometimes are variable with unknown reasons, which explains why some of our experimental results with a big standard deviation (Fig. 6). However, by increasing the experimental samples, a significant time course of histone acetylation in the heart can still be observed.
It is known that the transcription of a gene is dependent largely both on the activity of cis-elements in gene promoters and on the combination of transcription factors with the elements in the promoters [16]. Promote analysis has demonstrated that Mef2 and GATA4 binding sites are involved in the regulation of many cardiac genes [34–37], for instance, GATA4 binding sites are sufficient to active BNP gene in cardiac cells [13], whereas a single HF1-b/Mef2 site is crucial to active MLC2v gene in the heart [38]. Histone modifications, especially histone acetylation, can weaken the electrostatic attraction between DNA and histone, reducing chromatin compaction, and then increase the gene transcription [39]. In our study, a dynamic pattern of transcription factor binding levels with GATA4 or Mef2c elements in the proximal promoter of cTnI has been revealed. The synchronous changes in histone acetylation, transcription factor binding with GATA4 or Mef2c in cTnI promoters suggests that epigenetic regulation controls at least in part the cTnI expression in the hearts of mice at different ages.
Aging is a complex that is associated with a lot of mechanisms and factors in the heart. So far at least the following mechanisms that are considered to be associated with myocardium aging: 1) a decline of sarcoplasmic reticulum ATPase (SERCA2) protein content, 2) a decrease of cAMP levels caused by diminished beta1-receptor density, 3) a reduction in the inactivation rate of the Ca2+ current and a decrease in the magnitude of the outwardly directed K+ current, resulting in a prolonged action potential, and 4) age-related isoform switch such as myosin heavy chain in myofilament [40–43]. The data from our study indicate that reduced concentration of cTnI in the heart is one of the important mechanisms that could de associated with diastolic dysfunction in elderly population.
Supplementary Material
Acknowledgments
We appreciate Fan and Bao for his assistance with ultrathin sections of cardiac tissues and image analysis.
Founding
This study was supported by research grants from Natural Science Foundation of China (Grant Number: 81270234) and a NIH grant HL 112130-01 to XPH.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.abb.2016.05.008.
Footnotes
Conflict of interest
The authors have no conflicts of interest to declare.
References
- 1.Tobacman LS. Thin filament-mediated regulation of cardiac contraction. Annu Rev Physiol. 1996;58:447–481. doi: 10.1146/annurev.ph.58.030196.002311. http://dx.doi.org/10.1146/annurev.ph.58.030196.002311. [DOI] [PubMed] [Google Scholar]
- 2.Huang X, et al. Cardiac troponin I gene knockout: a mouse model of myocardial troponin I deficiency. Cir Res. 1999;84:1–8. doi: 10.1161/01.res.84.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Huang X, et al. Thyroid hormone regulates slow skeletal troponin I gene inactivation in cardiac troponin I null mouse hearts. J Mol Cell Cardiol. 2000;32:2221–2228. doi: 10.1006/jmcc.2000.1249. http://dx.doi.org/10.1006/jmcc.2000.1249. [DOI] [PubMed] [Google Scholar]
- 4.Du J, Zhang C, Liu J, Sidky C, Huang XP. A point mutation (R192H) in the C-terminus of human cardiac troponin I causes diastolic dysfunction in transgenic mice. Arch Biochem Biophys. 2006;456:143–150. doi: 10.1016/j.abb.2006.08.018. http://dx.doi.org/10.1016/j.abb.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 5.Du J, et al. Functional characterization of mouse fetal TnI gene promoters in myocardial cells. J Biomed Sci. 2008;15:605–613. doi: 10.1007/s11373-008-9246-y. http://dx.doi.org/10.1007/s11373-008-9246-y. [DOI] [PubMed] [Google Scholar]
- 6.Liu J, Du J, Zhang C, Walker JW, Huang X. Progressive troponin I loss impairs cardiac relaxation and causes heart failure in mice. Am J Physiol Heart Cir Physiol. 2007;293:H1273–H1281. doi: 10.1152/ajpheart.01379.2006. http://dx.doi.org/10.1152/ajpheart.01379.2006. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, et al. Correcting diastolic dysfunction by Ca2+ desensitizing troponin in a transgenic mouse model of restrictive cardiomyopathy. J Mol Cell Cardiol. 2010;49:402–411. doi: 10.1016/j.yjmcc.2010.04.017. http://dx.doi.org/10.1016/j.yjmcc.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y, et al. Dose-dependent diastolic dysfunction and early death in a mouse model with cardiac troponin mutations. J Mol Cell Cardiol. 2013;62:227–236. doi: 10.1016/j.yjmcc.2013.06.007. http://dx.doi.org/10.1016/j.yjmcc.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang L, et al. Calcium desensitizer catechin reverses diastolic dysfunction in mice with restrictive cardiomyopathy. Arch Biochem Biophys. 2015;573:69–76. doi: 10.1016/j.abb.2015.03.015. http://dx.doi.org/10.1016/j.abb.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 10.Mandinov L, Eberli FR, Seiler C, Hess OM. Diastolic heart failure. Cardiovasc Res. 2000;45:813–825. doi: 10.1016/s0008-6363(99)00399-5. [DOI] [PubMed] [Google Scholar]
- 11.Alves ML, et al. Desensitization of myofilaments to Ca2+ as a therapeutic target for hypertrophic cardiomyopathy with mutationsþ in thin filament proteins. Circ Cardiovasc Genet. 2014;7:132–143. doi: 10.1161/CIRCGENETICS.113.000324. http://dx.doi.org/10.1161/CIRCGENETICS.113.000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Welsh TM, Kukes GD, Sandweiss LM. Differences of creatine kinase MB and cardiac troponin I concentrations in normal and diseased human myocardium. Ann Clin Lab Sci. 2002;32:32–49. [PubMed] [Google Scholar]
- 13.Bhavsar PK, Dellow KA, Yacoub MH, Brand NJ, Barton PJ. Identification of cis-acting DNA elements required for expression of the human cardiac troponin I gene promoter. J Mol Cell Cardiol. 2000;32:95–108. doi: 10.1006/jmcc.1999.1058. http://dx.doi.org/10.1006/jmcc.1999.1058. [DOI] [PubMed] [Google Scholar]
- 14.Di Lisi R, et al. Combinatorial cis-acting elements control tissue-specific activation of the cardiac troponin I gene in vitro and in vivo. J Biol Chem. 1998;273:25371–25380. doi: 10.1074/jbc.273.39.25371. [DOI] [PubMed] [Google Scholar]
- 15.Di Lisi R, Picard A, Ausoni S, Schiaffino S. GATA elements control repression of cardiac troponin I promoter activity in skeletal muscle cells. BMC Mol Biol. 2007;8:78. doi: 10.1186/1471-2199-8-78. http://dx.doi.org/10.1186/1471-2199-8-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stefanovic S, et al. GATA-dependent regulatory switches establish atrioventricular canal specificity during heart development. Nat Commun. 2014;5:3680. doi: 10.1038/ncomms4680. http://dx.doi.org/10.1038/ncomms4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. http://dx.doi.org/10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gronbaek K, Hother C, Jones PA. Epigenetic changes in cancer, APMIS Acta Pathologica. Microbiol Immunol Scand. 2007;115:1039–1059. doi: 10.1111/j.1600-0463.2007.apm_636.xml.x. http://dx.doi.org/10.1111/j.1600-0463.2007.apm_636.xml.x. [DOI] [PubMed] [Google Scholar]
- 19.Glass JL, et al. CG dinucleotide clustering is a species-specific property of the genome. Nucleic Acids Res. 2007;35:6798–6807. doi: 10.1093/nar/gkm489. http://dx.doi.org/10.1093/nar/gkm489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Backs J, Olson EN. Control of cardiac growth by histone acetylation/deacetylation. Cir Res. 2006;98:15–24. doi: 10.1161/01.RES.0000197782.21444.8f. http://dx.doi.org/10.1161/01.RES.0000197782.21444.8f. [DOI] [PubMed] [Google Scholar]
- 21.Jenuwein T, Allis CD. Translating the histone code. Sci (New York, N Y) 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 22.National Research Council (US) Guide for the Care and Use of Laboratory Animals. eighth. National Academies Press (US); Washington (DC): 2011. Committee for the Update of the Guide for the Care and Use of Laboratory Animals. [Google Scholar]
- 23.Livaka Kenneth J, Schmittgen Thomas D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 ΔDDCT method. Methods Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Braunwald E. The management of heart failure: the past, the present, and the future. Circ Heart Fail. 2008;1:58–62. doi: 10.1161/CIRCHEARTFAILURE.107.752162. http://dx.doi.org/10.1161/CIRCHEARTFAILURE.107.752162. [DOI] [PubMed] [Google Scholar]
- 25.Gottdiener JS, et al. Predictors of congestive heart failure in the elderly: the Cardiovascular Health Study. J Am Coll Cardiol. 2000;35:1628–1637. doi: 10.1016/s0735-1097(00)00582-9. [DOI] [PubMed] [Google Scholar]
- 26.Bhatia RS, et al. Outcome of heart failure with preserved ejection fraction in a population-based study. N Engl J Med. 2006;355:260–269. doi: 10.1056/NEJMoa051530. http://dx.doi.org/10.1056/NEJMoa051530. [DOI] [PubMed] [Google Scholar]
- 27.Liao L, et al. Costs for heart failure with normal vs reduced ejection fraction. Arch Intern Med. 2006;166:112–118. doi: 10.1001/archinte.166.1.112. http://dx.doi.org/10.1001/archinte.166.1.112. [DOI] [PubMed] [Google Scholar]
- 28.Owan TE, et al. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355:251–259. doi: 10.1056/NEJMoa052256. http://dx.doi.org/10.1056/NEJMoa052256. [DOI] [PubMed] [Google Scholar]
- 29.Steinberg BA, et al. Trends in patients hospitalized with heart failure and preserved left ventricular ejection fraction: prevalence, therapies, and outcomes. Circulation. 2012;126:65–75. doi: 10.1161/CIRCULATIONAHA.111.080770. http://dx.doi.org/10.1161/CIRCULATIONAHA.111.080770. [DOI] [PubMed] [Google Scholar]
- 30.Fonarow GC, et al. Association between performance measures and clinical outcomes for patients hospitalized with heart failure. JAMA. 2007;297:61–70. doi: 10.1001/jama.297.1.61. http://dx.doi.org/10.1001/jama.297.1.61. [DOI] [PubMed] [Google Scholar]
- 31.Redfield MM, et al. Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA. 2003;289:194–202. doi: 10.1001/jama.289.2.194. [DOI] [PubMed] [Google Scholar]
- 32.Kane GC, et al. Progression of left ventricular diastolic dysfunction and risk of heart failure. JAMA. 2011;306:856–863. doi: 10.1001/jama.2011.1201. http://dx.doi.org/10.1001/jama.2011.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roger VL, et al. Trends in heart failure incidence and survival in a community-based population. JAMA. 2004;292:344–350. doi: 10.1001/jama.292.3.344. http://dx.doi.org/10.1001/jama.292.3.344. [DOI] [PubMed] [Google Scholar]
- 34.Pikkarainen S, Tokola H, Kerkela R, Ruskoaho H. GATA transcription factors in the developing and adult heart. Cardiovasc Res. 2004;63:196–207. doi: 10.1016/j.cardiores.2004.03.025. http://dx.doi.org/10.1016/j.cardiores.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 35.Kuisk IR, Li H, Tran D, Capetanaki Y. A single MEF2 site governs desmin transcription in both heart and skeletal muscle during mouse embryogenesis. Dev Biol. 1996;174:1–13. doi: 10.1006/dbio.1996.0046. http://dx.doi.org/10.1006/dbio.1996.0046. [DOI] [PubMed] [Google Scholar]
- 36.van Oort RJ, et al. MEF2 activates a genetic program promoting chamber dilation and contractile dysfunction in calcineurin-induced heart failure. Circulation. 2006;114:298–308. doi: 10.1161/CIRCULATIONAHA.105.608968. http://dx.doi.org/10.1161/CIRCULATIONAHA.105.608968. [DOI] [PubMed] [Google Scholar]
- 37.He A, et al. Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat Commun. 2014;5:4907. doi: 10.1038/ncomms5907. http://dx.doi.org/10.1038/ncomms5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ross RS, Navankasattusas S, Harvey RP, Chien KR. An HF-1a/HF-1b/MEF-2 combinatorial element confers cardiac ventricular specificity and established an anterior-posterior gradient of expression. Development. 1996;122:1799–1809. doi: 10.1242/dev.122.6.1799. [DOI] [PubMed] [Google Scholar]
- 39.Pan B, et al. Alcohol consumption during gestation causes histone3 lysine9 hyperacetylation and an alternation of expression of heart development-related genes in mice. Alcohol Clin Exp Res. 2014;38:2396–2402. doi: 10.1111/acer.12518. http://dx.doi.org/10.1111/acer.12518. [DOI] [PubMed] [Google Scholar]
- 40.Schmidt U, et al. Restoration of diastolic function in senescent rat hearts through adenoviral gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase. Circulation. 2000;101:790–796. doi: 10.1161/01.cir.101.7.790. [DOI] [PubMed] [Google Scholar]
- 41.Weisfeldt M. Aging, changes in the cardiovascular system, and responses to stress. Am J Hypertens. 1998;11:41S–45S. doi: 10.1016/s0895-7061(98)00009-0. [DOI] [PubMed] [Google Scholar]
- 42.Yin FC, Weisfeldt ML, Milnor WR. Role of aortic input impedance in the decreased cardiovascular response to exercise with aging in dogs. J Clin InvestigInvestation. 1981;68:28–38. doi: 10.1172/JCI110245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin FC, Spurgeon HA, Rakusan K, Weisfeldt ML, Lakatta EG. Use of tibial length to quantify cardiac hypertrophy: application in the aging rat. Am J Physiol Genomics. 1982;243:H941–H947. doi: 10.1152/ajpheart.1982.243.6.H941. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.