

Abstract
Obesity greatly increases the risk for cardiovascular, metabolic, and renal diseases and is one of the most significant and preventable causes of increased blood pressure (BP) in patients with essential hypertension. This review high-lights recent advances in our understanding of central nervous system (CNS) signaling pathways that contribute to the etiology and pathogenesis of obesity-induced hypertension. We discuss the role of excess adiposity and activation of the brain leptin-melanocortin system in causing increased sympathetic activity in obesity. In addition, we highlight other potential brain mechanisms by which increased weight gain modulates metabolic and cardiovascular functions. Unraveling the CNS mechanisms responsible for increased sympathetic activation and hypertension and how circulating hormones activate brain signaling pathways to control BP offer potentially important therapeutic targets for obesity and hypertension.
Keywords: Blood pressure, Leptin, Melanocortins, Sympathetic nervous system, Metabolism
Introduction
There have been substantial increases in the prevalence of obesity during the last 20–30 years in the USA and worldwide. More than one-third (34.9 % or 78.6 million) of US adults are considered to be obese with body mass index (BMI) of 30 or greater. Obesity rates are higher in middle age (40–59 years, 39.5 %) than in younger adults (aged 20–39, 30.3 %) or adults aged 60 or above (35.4 %). Even more alarming, however, is the fact that obesity has more than doubled in children and quadrupled in adolescents in the past 30 years. Recent estimates indicate that approximately 17 % of adolescents are obese, and even higher rates of obesity are observed in African-American, Native American, and Hispanic children [1–3]. The annual medical costs associated with obesity are over $190.2 billion in the USA (Centers for Disease Control and Prevention).
Being overweight or obese greatly increases the risk for several major diseases including hypertension, coronary heart disease, stroke, type 2 diabetes, cancer, and chronic kidney disease [4–6]. For example, risk estimates from population studies suggest that weight gain may contribute as much as 85 % of the risk for diabetes and 65 to 78 % of the risk for essential hypertension [7]. Excess weight gain shifts the frequency distribution of blood pressure (BP) towards higher levels. Therefore, obese subjects not classified as being hypertensive usually have lower BP when they reduce body weight [4]. There is a nearly linear relationship between BMI and BP in population studies, and excess weight gain predicts future development of hypertension [4–6]. In addition, weight loss helps prevent development of hypertension and reduces BP in most hypertensive individuals [8, 9].
SNS Activation in Obesity-Induced Hypertension
Excess weight gain, when followed by increased visceral adiposity, is associated with increased sympathetic nervous system (SNS) activity which has been shown to contribute to development of hypertension in obese experimental animals as well as humans [4, 10]. Increases in SNS activity in diet-induced obesity develop early after exposure to high-fat diets in experimental animals [11, 12] and weight gain in humans is associated with increased SNS activity [4, 10]. However, increases in SNS activity in obesity are modest and occur only in certain organs and tissues instead of generalized whole-body sympathetic activation. For instance, in obese subjects, SNS activities in the kidney and skeletal muscle are elevated while cardiac sympathetic activity is minimally increased, or even reduced, most likely due to baroreflex inhibition [13, 14].
Increased renal SNS activity in obese subjects contributes to sodium retention, increased renin release, impaired renal-pressure natriuresis, and elevated BP [10]. However, SNS activation in obese subjects is generally not great enough to directly cause peripheral vasoconstriction in most tissues [15]. In fact, blood flow in the kidneys and many other tissues as well as cardiac output are often increased in obesity, although the ability to vasodilate further during stresses such as exercise may be impaired due to endothelial dysfunction and increased vascular stiffness [16]. Also, increased SNS activity in obesity appears to vary according to ethnicity and body fat distribution with visceral obesity eliciting greater SNS activation than subcutaneous obesity [17].
Brain Centers Involved in Obesity-Induced SNS Activation
The rostral ventral lateral medulla (RVLM) is a key brain center for controlling SNS activity [18–20]. This center is modulated by inputs from several other regions of the central nervous system (CNS) including the paraventricular nucleus of the hypothalamus (PVN) and the spinal sympathetic intermediolateral nucleus (IML). Neurons in the PVN that project to the RVLM display an autorhythmicity which closely correlates with sympathetic discharge rate [21, 22]. In addition, the PVN receives extensive neuronal inputs from several other regions of the brain, including the arcuate nucleus (ARC), subfornical (SO), and median preoptic nuclei, lateral hypothalamus, limbic nuclei, lateral parabrachial nucleus, nucleus tractus solitarius (NTS), dorsal motor nucleus of the vagus (DMV), and other areas. Beside the PVN, the dorsomedial hypothalamus (DMH), which contains connections with sympathetic and parasympathetic systems, is influenced by peripheral afferents via the NTS, the parabrachial nucleus, and sympathetic IML [23]. The DMH also interconnects to the lateral hypothalamus and the circumventricular organs.
Although the brain areas involved in obesity-induced hypertension have not been fully elucidated, hypothalamic (ARC, PVN, and DMH) and hindbrain regions (NTS/DMV, RVLM) as well as the IML appear to play a crucial role in mediating increases in SNS activity and BP [24–27]. The ventromedial hypothalamus (VMH) plays a major role in regulating food intake, and VMH lesions cause severe obesity that have been reported by some, but not all, investigators to be accompanied by hypertension and elevated plasma norepinephrine [28, 29]. These observations, if correct, suggest that either the VMH may not be critical for SNS activation and hypertension associated with obesity. Alternatively, VMH neurons may even exert an inhibitory influence on SNS activity and BP since some studies suggest that destruction of these neurons may cause SNS activation and hypertension [30].
Several populations of preautonomic neurons located in the caudal hindbrain are critical in mediating the effect of cytokines/adipokines, which are increased in obesity, on sympathetic outflow [31,32]. Although the hypothalamus and hindbrain areas are importantly involved in cardiovascular control in obesity, additional studies are needed to identify specific brain circuits that mediate increases in SNS activity and BP in obesity.
Mechanisms of SNS Activation in Obesity
Multiple mechanisms have been proposed to increase SNS activity in obesity, including brain oxidative stress, inflammation, impaired baroreflex sensitivity, angiotensin II (Ang II), hyperinsulinemia, sleep apnea and hypoxia, hypoghrelinemia, hypoadiponectemia, and hyperleptinemia (Fig. 1).
Fig. 1.
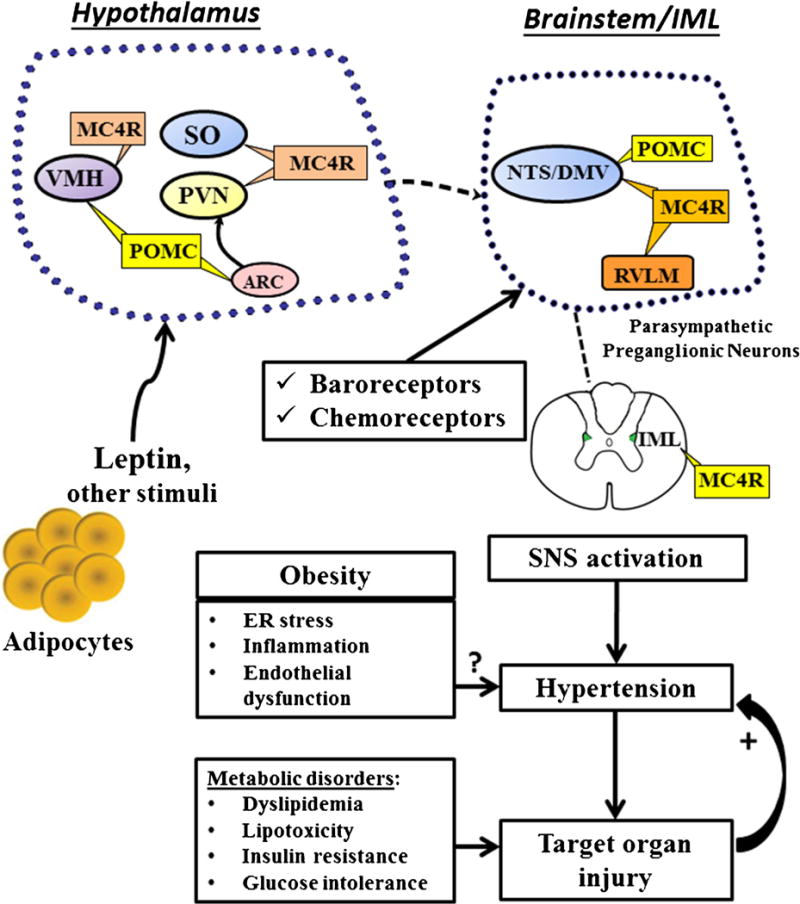
The CNS leptin-melanocortin system and hypothalamic and brainstem centers that may contribute to the sympathetic nervous system (SNS) activation and hypertension in obesity. POMC (proopiomelanocortin), MC4R (melanocortin-4 receptor), SO (subfornical organ), ARC (arcuate nucleus), PVN (paraventricular nucleus of the hypothalamus), VMH (ventromedial hypothalamus), NTS (nucleus, nucleus tractus solitaries), DMV (dorsal motor nucleus of the vagus), RVLM rostral ventral lateral medulla, IML (intermediolateral nucleus)
Nagae et al. [33] reported that oxidative stress via NAD(P)H oxidase in the brain, mainly in the hypothalamus, contributes to increased SNS activation in obesity-induced hypertension in rats fed a high-fat diet. Low-grade CNS inflammation has also been suggested to be involved in the pathogenesis of SNS activation and hypertension associated with obesity [34]. Previous studies also suggest that an imbalance of nitric oxide (NO) and reactive oxygen species (ROS) in the autonomic nuclei in the brain may mediate obesity-induced SNS activation and hypertension, contributing to the inflammatory process and progression of hypertension [35, 36]. However, there are no studies, to our knowledge, showing that anti-inflammatory drugs reduce SNS activity and BP in obese subjects.
Obesity is associated with impaired glucose tolerance, insulin resistance, and increased plasma insulin. Acute hyperinsulinemia has been reported to cause SNS activation and sodium retention and has been suggested to link obesity with increased BP [37, 38]. However, multiple studies have shown that chronic hyperinsulinemia causes peripheral vasodilation, but does not elevate BP in dogs or in humans, although some studies suggest that high levels of insulin may modestly increase BP in rodents [16]. Administration of an insulin antagonist ICV caused a similar small reduction in BP (~3–4 mmHg) in lean as well as obese rabbits fed a high-fat diet, but no change in renal SNS activity [12]. Thus, most of the available evidence suggests that hyperinsulinemia plays a minor role, if any, in stimulating SNS activity and raising BP in obesity.
Ghrelin and adiponectin deficiency have also been suggested to influence BP regulation in obesity. Obesity is associated with decreases in plasma adiponectin, which are inversely related to insulin resistance. Further, adiponectin knockout mice have severe insulin resistance and atherogenesis [38, 39]. However, loss of function adiponectin gene mutations do not cause hypertension, although BP may become more salt-sensitive [40]. Ghrelin levels are also reduced in obesity and weight loss increases ghrelin levels [41]. However, there have been no studies, to our knowledge, that have found an important role for ghrelin or adiponectin deficiency in mediating increased SNS activity and hypertension in obesity.
The role of the renin-angiotensin-aldosterone system (RAAS) in obesity has been reviewed previously [42, 43••]. Obese subjects often have modest increases in plasma renin activity, plasma angiotensinogen, Ang II, and aldosterone [38, 43••]. Although Ang II and mineralocorticoid activation contributes importantly to obesity-induced hypertension [16, 44], there is no compelling evidence that increases in Ang II or aldosterone mediate SNS activation in obesity. Instead, increased SNS activity is an important factor in stimulating renin release and activating the RAAS in obese subjects [16, 43••].
Baroreflex Dysfunction in Obesity
Although the arterial baroreceptors clearly provide powerful moment-to-moment control of BP, their role in long-term BP regulation and in obesity hypertension is unclear. Previous studies demonstrated that baroreflex control of SNS activity is impaired in obese subjects, in parallel with metabolic abnormalities such as hyperglycemia, dyslipidemia, hyperleptinemia, hyperinsulinemia, and elevated BP [4, 45]. However, the role of these multiple metabolic abnormalities in causing impaired baroreflex regulation is unclear. In addition, impaired arterial baroreceptor function could be, at least partly, secondary to elevated BP in obesity hypertension.
Chronic electrical stimulation of the carotid sinus nerves reduces sympathetic activity and BP in obese dogs [46], consistent with the hypothesis that strong activation of arterial baroreceptors can have significant long-term effects on BP regulation. This finding, however, does not necessarily indicate that impaired baroreflexes actually cause obesity hypertension. However, increased lability of BP and periodic large increases in BP that occur with baroreceptor dysfunction may eventually cause target organ injury, especially kidney injury that could contribute to worsening of hypertension. Currently, the importance of baroreflex dysfunction in mediating obesity-induced SNS activation and hypertension is unclear.
Obstructive Sleep Apnea and Intermittent Hypoxia in Obesity
Obesity is a major risk factor for obstructive sleep apnea (OSA), and chronic intermittent hypoxemia (CIH) caused by OSA has been suggested to cause resistant hypertension though SNS activation [47, 48]. Renal denervation not only reduces BP in obese subjects with resistant hypertension, but also has been suggested to attenuate OSA in these patients [49].
Even in the absence of OSA, obesity may tend to cause hypoxemia since obese subjects have increased metabolic rate and decreased cardiac/blood flow reserve [4, 16]; this mismatch between metabolic rate and blood flow is especially evident during exercise. Thus, it is possible that chronic hypoxemia may cause chemoreflex activation and subsequent sympathoexcitation in obesity. Support for this hypothesis comes from recent studies showing that carotid sinus denervation, which eliminates baroreceptor and chemoreceptor input to the CNS, reduces BP and ventilation rate in obese dogs [50].
Although the precise CNS mechanism by which hypoxemia increases sympathetic activation is unclear, previous studies suggested that in CIH altered PVN activity leads to impairment of sympathetic and parasympathetic tone [24]. However, further experiments are needed to unravel the CNS areas involved in CIH-induced increase in SNS activation and hypertension and the importance of this mechanism in obesity-induced hypertension.
Role of Leptin in Mediating SNS Activation in Obesity
Although multiple factors may contribute to increased SNS activity in obesity, leptin has emerged as a key contributor (Fig. 1). Leptin, a peptide hormone secreted by adipocytes in direct proportion to adiposity, crosses the blood brain barrier to activate its receptors in various regions of the CNS, especially in the hypothalamus and brainstem. Leptin has powerful effects to decrease appetite and to increase energy expenditure by increasing SNS activity [51, 52, 53••]. The major brain areas of leptin’s actions are ARC, ventromedial, and DMH of hypothalamus, NTS, and SO [54••], which are all important areas involved in SNS regulation. Leptin is perhaps the body’s most powerful hormonal regulator of energy balance; leptin deficiency or loss of function mutations of the leptin receptor (LR) lead to early-onset, morbid obesity in humans and experimental animals [55, 56].
Evidence consistent with a role for leptin in contributing to increased SNS activity and development of obesity hypertension comes from studies in rodents and humans showing that acute infusions of leptin increase renal and muscle SNS activity [57, 58]. Also, chronic infusion of leptin in lean normotensive rodents, at rates that raise plasma levels to those observed in severe obesity, causes gradual elevation in BP that can be completely abolished by intravenous administration of α and β adrenergic receptor blockers [59, 60]. Leptin-induced elevation in BP in lean animals occurs in parallel with marked reductions in appetite and significant weight loss which would normally decrease BP [43••, 61]. Therefore, in the absence of these metabolic actions to promote weight loss, leptin’s effects on SNS activity and BP may be exacerbated. Leptin’s effects on BP are also exacerbated when nitric oxide synthesis is impaired [62] as may occur in obese subjects with endothelial dysfunction.
Infusion of leptin antagonists reduced renal SNS activity and BP in obese rabbits fed a high-fat diet [11, 12], suggesting an important role of leptin in obesity-induced SNS activation and increased BP. The importance of leptin in linking obesity with hypertension is further supported by the observation that mice with leptin deficiency (ob/ob) have severe obesity and many characteristics of the metabolic syndrome, including insulin resistance, hyperinsulinemia, hyperglycemia, and dyslipidemia, but maintain lower BP and SNS activity compared to control mice [63]. In addition, humans with leptin deficiency, although exhibiting morbid obesity and many characteristics of the metabolic syndrome [55], are not hypertensive and do not have increased SNS activity [64]. Therefore, clinical and experimental data are consistent with the hypothesis that leptin may act as an important link between obesity, increased SNS activity and elevated BP.
Selective Leptin Resistance in Obesity: Neuronal-Specific Activation of LRs and Intracellular Signaling Events
Although plasma leptin is markedly increased in obesity, leptin’s ability to suppress appetite is attenuated whereas its effects on SNS activity appear to be sustained, suggesting that obesity is associated with resistance to the anorexic but not the SNS effects of leptin [54••]. In fact, leptin infusion causes normal or enhanced renal SNS activation and BP responses in obese animals fed a high-fat diet compared to lean controls [54••, 65]. The mechanisms by which obesity leads to this “selective” leptin resistance have not been fully elucidated but clues have recently emerged.
LRs are widely distributed in the brain and deletion of LR signaling in specific neuronal populations has produced only modest obesity, failing to recapitulate the severe obesity observed when LRs are deleted in the entire brain. Vong and colleagues [66] showed that deleting LRs in GABAergic neurons recapitulates most of the obese phenotype observed in leptin deficiency; however, GABAergic neurons are widely distributed and it is still unclear which neuronal types or brain sites are most important in mediating the effects of leptin on body weight homeostasis.
Neurons in the hypothalamus and brainstem clearly play a key role in mediating the actions of leptin on SNS activity and BP regulation. For instance, deletion of LRs in the ARC significantly reduces the acute effects of leptin to increase renal SNS activity and attenuates the rise in BP induced by a high-fat feeding diet [67]. In addition, we demonstrated that selective deletion of LRs in proopiomelanocortin (POMC) neurons, located in the ARC and in the hindbrain, completely abolished the chronic effects of leptin to raise BP whereas leptin’s ability to reduce appetite and to promote weight loss remained intact [68]. These findings are consistent with the possibility that POMC neurons may be an important component contributor to selective leptin resistance in obesity.
The VMH and DMH as well as extra-hypothalamic centers may also contribute to the acute effects of leptin on SNS activity. For example, mice with deletion of LRs only in SO neurons had normal brown adipose tissue sympathetic nerve activity (BATSNA) responses to acute leptin injection but increases in renal sympathetic activity (RSNA) were abolished [69].
Hindbrain regions also appear to mediate the acute effects of leptin on SNS activity. Microinjections of leptin into NTS increased renal RSNA and acutely raised BP, while BATSNA remained unaffected [70]. Together, these findings suggest that several brain regions contribute to leptin’s effects on SNS and that these neurons may be differentially regulated.
After binding to its brain receptors, leptin increases Janus Tyrosine Kinase 2 (JAK2) activity and activates 3 main signaling pathways: (1) latent signal transducers and activators of transcription 3 (STAT3) which regulates transcription of leptin target genes, (2) Src homology protein 2 (SHP2) which activates mitogen-activated protein kinase (MAPK), and (3) insulin receptor substrate 2 (IRS2) which activates phosphatidylinositol 3-kinase (PI3K). CNS deletion of each of these signaling pathways results in varying degrees of obesity.
Neuron-specific deletion of STAT3 mimics the obesity and the hyperphagia found in leptin-deficient animals [71]. However, deletion of STAT3 in the entire CNS may have other effects on food intake besides preventing leptin-mediated anorexia. Deletion of SHP2 in forebrain neurons also causes early-onset obesity associated with hyperphagia and impaired glucose regulation, although obesity and hyperphagia are not as pronounced as with STAT3 deletion. SHP2 deletion in forebrain neurons also attenuated leptin’s ability to reduce food intake and raise BP [72]. We also found that SHP2 signaling in POMC neurons contributes to the chronic BP and glucose-lowering effects of leptin but plays only a modest role in body weight regulation [73••], suggesting that SHP2 in POMC neurons may also contribute to the effects of leptin on SNS activity and BP. IRS2-PI3K signaling may also mediate leptin’s effect on SNS activity and BP. Pharmacological blockade of PI3K abolished the acute effects of leptin on RSNA [74]. Recently, we found that IRS2 signaling in the entire brain, and particularly in POMC neurons, is essential for the chronic effects of leptin on BP but not for leptin’s actions on appetite and glucose regulation [75], suggesting that while IRS2 contributes only modestly to body weight regulation it has a major role in the effects of leptin on SNS activity and BP.
Role of CNS Proopiomelanocortin Pathway in SNS Activation
Mice with melanocortin 4 receptor (MC4R) deficiency are hyperphagic and obese and exhibit most of the characteristics of metabolic syndrome that are observed in leptin deficiency [76]. Mutations in POMC or MC4R genes also lead to severe early-onset obesity and pronounced hyperphagia in humans as well as in rodents [76, 77].
Besides its effects on food intake and body weight regulation, MC4R may also link obesity and hyperleptinemia with increased SNS activity and hypertension. MC4R-deficient mice are obese but normotensive when compared to lean controls, and they are resistant to the pressor effects of chronic leptin administration [76]. MC4R mutations in humans are associated with reduced 24-h norepinephrine spillover, reduced diastolic and systolic BPs, and reduced prevalence of hypertension compared to obese individuals with normal MC4R function [78, 79]. In addition, pharmacological activation of MC4R in humans elevates BP [80].
We previously demonstrated that even in non-obese models of hypertension such as spontaneously hypertensive rats or hypertension induced by the nitric oxide synthase inhibitor (L-NAME), the CNS melanocortin system contributes to the maintenance of adrenergic tone and BP [81–83]. In addition, MC4R blockade caused greater BP reduction in obese compared to lean Zucker rats [84], suggesting a key role for MC4R in the regulation of SNS activity and BP even in obese models that lack normal leptin actions. Thus, in humans as well as in rodents, chronic MC4R activation raises BP and the presence of a functional POMC-MC4R pathway appears to be necessary for hyperleptinemia and other obesity-related factors to increase SNS activity and BP.
Despite evidence that leptin-MC4R pathway is important for weight gain to be associated with increased SNS activity and hypertension, the increase in BP measured during chronic administration of leptin or MC4R agonists is modest. One potential explanation is that the hypertensive effects of leptin and MC4R agonists have been conducted in lean animals and that obesity is associated with other factors, such as impaired endothelial and renal NO formation, that potentiate the pressor actions of the leptin-MC4R pathway.
Even though the powerful effects of MC4R agonists on body weight and glucose homeostasis make them potential anti-obesity agents, the side effects of SNS activation, increased BP, and increased heart rate have been major limitations. Therefore, development of MC4R agonists capable of triggering the beneficial anorexic and antidiabetic effects of MC4R activation without eliciting detrimental effects on cardiovascular function is of great interest.
Although the physiological and behavior factors that regulate body weight homeostasis are still not well understood, complex interactions of adipokines, gastrointestinal hormones, and CNS pathways that control food intake and energy expenditure are clearly involved. The most powerful pathways that regulate appetite and other metabolic functions, such as the leptin-CNS melanocortin system, also influence SNS activity and BP regulation. Unfortunately, in obesity, many of the beneficial metabolic actions of these systems are greatly attenuated whereas the harmful effects of SNS activation and increased BP are preserved. Understanding the mechanisms involved in the adaptations to obesity may provide opportunities to design better therapies for obesity and its associated metabolic and cardiovascular disorders.
Conclusions
Obesity is a major contributor to hypertension and cardiometabolic diseases worldwide. While obesity-induced increases in BP are multifactorial, SNS activation contributes to renal dysfunction and hypertension in obese subjects. The mechanisms linking obesity with SNS activation and hypertension are not fully understood, but leptin and activation of the CNS melanocortin pathway may play important roles. Abnormal function of the leptin-MC4R axis in obesity may lead to impaired control of appetite and other metabolic actions while effects on SNS activity and BP are maintained or enhanced. The neural circuits and molecular pathways by which the leptin-MC4R controls RSNA and BP independently of its effects on food intake and other metabolic functions are still unclear. Better understanding of the molecular pathways and neuronal-specific actions of LR and MC4R in controlling appetite, metabolic functions, and SNS activity is critical for the development of anti-obesity drugs without deleterious cardiovascular effects.
Acknowledgments
We thank Sydney P. Moak, Jackson Browning, and Calvin Torrey for technical assistance with our experimental studies cited in this review.
Sources of Funding This research was supported by National Heart, Lung and Blood Institute Grant (PO1HL-51971), the National Institute of General Medical Sciences (P20GM104357), and by an American Heart Association Scientist Development Grant to Jussara M. do Carmo.
Footnotes
This article is part of the Topical Collection on Secondary Hypertension: Nervous System Mechanisms
Conflict of Interest Dr. do Carmo reports grants from National Institutes of Health, American Heart Association, and Palatin Technologies. Dr. Hall reports grants from National Institutes of Health and Palatin Technologies. Drs. da Silva, Wang, Fang, Aberdein, and de Lara Rodriguez report no conflicts of interest.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
•• Of major importance
- 1.Anderson SE, Whitaker RC. Prevalence of obesity among US pre-school children in different racial and ethnic groups. Arch Pediatr Adolesc Med. 2009;163(4):344–8. doi: 10.1001/archpediatrics.2009.18. [DOI] [PubMed] [Google Scholar]
- 2.Bauer KW, Marcus MD, El ghormli L, Ogden CL, Foster GD. Cardio-metabolic risk screening among adolescents: understanding the utility of body mass index, waist circumference and waist to height ratio. Pediatr Obes. 2015;10(5):329–37. doi: 10.1111/ijpo.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ogden CL, Carroll MD, Flegal KM. Prevalence of obesity in the United States. JAMA. 2014;312(2):189–90. doi: 10.1001/jama.2014.6228. [DOI] [PubMed] [Google Scholar]
- 4.Hall JE, Crook ED, Jones DW, Wofford MR, Dubbert PM. Mechanisms of obesity-associated cardiovascular and renal disease. Am J Med Sci. 2002;324(3):127–37. doi: 10.1097/00000441-200209000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Jones DW, Kim JS, Andrew ME, Kim SJ, Hong YP. Body mass index and blood pressure in Korean men and women: the Korean National Blood Pressure Survey. J Hypertens. 1994;12(12):1433–7. doi: 10.1097/00004872-199412000-00018. [DOI] [PubMed] [Google Scholar]
- 6.Wilson PW, D’Agostino RB, Sullivan L, Parise H, Kannel WB. Overweight and obesity as determinants of cardiovascular risk: the Framingham experience. Arch Intern Med. 2002;162(16):1867–72. doi: 10.1001/archinte.162.16.1867. [DOI] [PubMed] [Google Scholar]
- 7.Garrison RJ, Kannel WB, Stokes J, 3rd, Castelli WP. Incidence and precursors of hypertension in young adults: the Framingham Offspring Study. Prev Med. 1987;16(2):235–51. doi: 10.1016/0091-7435(87)90087-9. [DOI] [PubMed] [Google Scholar]
- 8.Obarzanek E, Kimm SY, Barton BA, Van Horn LL, Kwiterovich PO, Jr, Simons-Morton DG, et al. Long-term safety and efficacy of a cholesterol-lowering diet in children with elevated low-density lipoprotein cholesterol: seven-year results of the Dietary Intervention Study in Children (DISC) Pediatrics. 2001;107(2):256–64. doi: 10.1542/peds.107.2.256. [DOI] [PubMed] [Google Scholar]
- 9.Stevens VJ, Obarzanek E, Cook NR, Lee IM, Appel LJ, Smith West D, et al. Long-term weight loss and changes in blood pressure: results of the Trials of Hypertension Prevention, phase II. Ann Intern Med. 2001;134(1):1–11. doi: 10.7326/0003-4819-134-1-200101020-00007. [DOI] [PubMed] [Google Scholar]
- 10.Hall ME, do Carmo JM, da Silva AA, Juncos LA, Wang Z, Hall JE. Obesity, hypertension, and chronic kidney disease. Int J Nephrol Renovasc Dis. 2014;7:75–88. doi: 10.2147/IJNRD.S39739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Head GA, Lim K, Barzel B, Burke SL, Davern PJ. Central nervous system dysfunction in obesity-induced hypertension. Curr Hypertens Rep. 2014;16(9):466. doi: 10.1007/s11906-014-0466-4. [DOI] [PubMed] [Google Scholar]
- 12.Lim K, Burke SL, Head GA. Obesity-related hypertension and the role of insulin and leptin in high-fat-fed rabbits. Hypertension. 2013;61(3):628–34. doi: 10.1161/HYPERTENSIONAHA.111.00705. [DOI] [PubMed] [Google Scholar]
- 13.Esler M. The 2009 Carl Ludwig Lecture: Pathophysiology of the human sympathetic nervous system in cardiovascular diseases: the transition from mechanisms to medical management. J Appl Physiol. 2010;108(2):227–37. doi: 10.1152/japplphysiol.00832.2009. [DOI] [PubMed] [Google Scholar]
- 14.Straznicky NE, Grima MT, Eikelis N, Nestel PJ, Dawood T, Schlaich MP, et al. The effects of weight loss versus weight loss maintenance on sympathetic nervous system activity and metabolic syndrome components. J Clin Endocrinol Metab. 2011;96(3):E503–8. doi: 10.1210/jc.2010-2204. [DOI] [PubMed] [Google Scholar]
- 15.Hall JE. The kidney, hypertension, and obesity. Hypertension. 2003;41(3 Pt 2):625–33. doi: 10.1161/01.HYP.0000052314.95497.78. [DOI] [PubMed] [Google Scholar]
- 16.Hall JE, Granger JP, do Carmo JM, da Silva AA, Dubinion J, George E, et al. Hypertension: physiology and pathophysiology. Compr Physiol. 2012;2(4):2393–142. doi: 10.1002/cphy.c110058. [DOI] [PubMed] [Google Scholar]
- 17.Davy KP, Hall JE. Obesity and hypertension: two epidemics or one? Am J Physiol Regul Integr Comp Physiol. 2004;286(5):R803–13. doi: 10.1152/ajpregu.00707.2003. [DOI] [PubMed] [Google Scholar]
- 18.McAllen RM. Action and specificity of ventral medullary vasopressor neurones in the cat. Neuroscience. 1986;18(1):51–9. doi: 10.1016/0306-4522(86)90178-8. [DOI] [PubMed] [Google Scholar]
- 19.McAllen RM. Identification and properties of sub-retrofacial bulbospinal neurones: a descending cardiovascular pathway in the cat. J Auton Nerv Syst. 1986;17(2):151–64. doi: 10.1016/0165-1838(86)90090-1. [DOI] [PubMed] [Google Scholar]
- 20.Sugiyama Y, Suzuki T, Yates BJ. Role of the rostral ventrolateral medulla (RVLM) in the patterning of vestibular system influences on sympathetic nervous system outflow to the upper and lower body. Exp Brain Res. 2011;210(3–4):515–27. doi: 10.1007/s00221-011-2550-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen QH, Toney GM. In vivo discharge properties of hypothalamic paraventricular nucleus neurons with axonal projections to the rostral ventrolateral medulla. J Neurophysiol. 2010;103(1):4–15. doi: 10.1152/jn.00094.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dampney RA, Horiuchi J, Tagawa T, Fontes MA, Potts PD, Polson JW. Medullary and supramedullary mechanisms regulating sympathetic vasomotor tone. Acta Physiol Scand. 2003;177(3):209–18. doi: 10.1046/j.1365-201X.2003.01070.x. [DOI] [PubMed] [Google Scholar]
- 23.ter Horst GJ, Luiten PG. The projections of the dorsomedial hypothalamic nucleus in the rat. Brain Res Bull. 1986;16(2):231–48. doi: 10.1016/0361-9230(86)90038-9. [DOI] [PubMed] [Google Scholar]
- 24.Carmichael CY, Wainford RD. Hypothalamic signaling mechanisms in hypertension. Curr Hypertens Rep. 2015;17(5):39. doi: 10.1007/s11906-015-0550-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirooka Y, Kishi T, Ito K, Sunagawa K. Potential clinical application of recently discovered brain mechanisms involved in hypertension. Hypertension. 2013;62(6):995–1002. doi: 10.1161/HYPERTENSIONAHA.113.00801. [DOI] [PubMed] [Google Scholar]
- 26.Kishi T, Hirooka Y, Ogawa K, Konno S, Sunagawa K. Calorie restriction inhibits sympathetic nerve activity via anti-oxidant effect in the rostral ventrolateral medulla of obesity-induced hypertensive rats. Clin Exp Hypertens. 2011;33(4):240–5. doi: 10.3109/10641963.2011.583969. [DOI] [PubMed] [Google Scholar]
- 27.Seravalle G, Grassi G. Sympathetic nervous system, hypertension, obesity and metabolic syndrome. High Blood Press Cardiovasc Prev. 2016 doi: 10.1007/s40292-016-0137-4. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 28.Gauthier P, Reis DJ, Nathan MA. Arterial hypertension elicited either by lesions or by electrical stimulations of the rostral hypothalamus in the rat. Brain Res. 1981;211(1):91–105. doi: 10.1016/0006-8993(81)90069-x. [DOI] [PubMed] [Google Scholar]
- 29.Valensi P, Doare L, Perret G, Germack R, Paries J, Mesangeau D. Cardiovascular vagosympathetic activity in rats with ventromedial hypothalamic obesity. Obes Res. 2003;11(1):54–64. doi: 10.1038/oby.2003.10. [DOI] [PubMed] [Google Scholar]
- 30.Reisin E, Wilson JR, Frohlich ED. Hypertension and obesity in rats with ventromedial-hypothalamic lesions and low salt intake. J Hypertens. 1987;5(2):173–8. doi: 10.1097/00004872-198704000-00007. [DOI] [PubMed] [Google Scholar]
- 31.Grill HJ, Hayes MR. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab. 2012;16(3):296–309. doi: 10.1016/j.cmet.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rinaman L. Hindbrain noradrenergic A2 neurons: diverse roles in autonomic, endocrine, cognitive, and behavioral functions. Am J Physiol Regul Integr Comp Physiol. 2011;300(2):R222–35. doi: 10.1152/ajpregu.00556.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagae A, Fujita M, Kawarazaki H, Matsui H, Ando K, Fujita T. Sympathoexcitation by oxidative stress in the brain mediates arterial pressure elevation in obesity-induced hypertension. Circulation. 2009;119(7):978–86. doi: 10.1161/CIRCULATIONAHA.108.824730. [DOI] [PubMed] [Google Scholar]
- 34.Winklewski PJ, Radkowski M, Wszedybyl-Winklewska M, Demkow U. Brain Inflammation and hypertension: the chicken or the egg? J Neuroinflammation. 2015;12:85. doi: 10.1186/s12974-015-0306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campese VM, Ku E, Park J. Sympathetic renal innervation and resistant hypertension. Int J Hypertens. 2011;2011:814354. doi: 10.4061/2011/814354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campese VM, Ye S, Zhong H, Yanamadala V, Ye Z, Chiu J. Reactive oxygen species stimulate central and peripheral sympathetic nervous system activity. Am J Physiol Heart Circ Physiol. 2004;287(2):H695–703. doi: 10.1152/ajpheart.00619.2003. [DOI] [PubMed] [Google Scholar]
- 37.Stocker SD, Gordon KW. Glutamate receptors in the hypothalamic paraventricular nucleus contribute to insulin-induced sympathoexcitation. J Neurophysiol. 2015;113(5):1302–9. doi: 10.1152/jn.00764.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kotsis V, Stabouli S, Papakatsika S, Rizos Z, Parati G. Mechanisms of obesity-induced hypertension. Hypertens Res. 2010;33(5):386–93. doi: 10.1038/hr.2010.9. [DOI] [PubMed] [Google Scholar]
- 39.Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. 2001;86(5):1930–5. doi: 10.1210/jcem.86.5.7463. [DOI] [PubMed] [Google Scholar]
- 40.Ohashi K, Kihara S, Ouchi N, Kumada M, Fujita K, Hiuge A, et al. Adiponectin replenishment ameliorates obesity-related hypertension. Hypertension. 2006;47(6):1108–16. doi: 10.1161/01.HYP.0000222368.43759.a1. [DOI] [PubMed] [Google Scholar]
- 41.Kawczynska-Drozdz A, Olszanecki R, Jawien J, Brzozowski T, Pawlik WW, Korbut R, et al. Ghrelin inhibits vascular superoxide production in spontaneously hypertensive rats. Am J Hypertens. 2006;19(7):764–7. doi: 10.1016/j.amjhyper.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 42.DeMarco VG, Aroor AR, Sowers JR. The pathophysiology of hypertension in patients with obesity. Nat Rev Endocrinol. 2014;10(6):364–76. doi: 10.1038/nrendo.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43••.Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res. 2015;116(6):991–1006. doi: 10.1161/CIRCRESAHA.116.305697. Provides a comprehensive review of the neurohumoral mechanisms in obesity-induced hypertension. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Paula RB, da Silva AA, Hall JE. Aldosterone antagonism attenuates obesity-induced hypertension and glomerular hyperfiltration. Hypertension. 2004;43(1):41–7. doi: 10.1161/01.HYP.0000105624.68174.00. [DOI] [PubMed] [Google Scholar]
- 45.Van Vliet BN, Hall JE, Mizelle HL, Montani JP, Smith MJ., Jr Reduced parasympathetic control of heart rate in obese dogs. Am J Physiol. 1995;269(2 Pt 2):H629–37. doi: 10.1152/ajpheart.1995.269.2.H629. [DOI] [PubMed] [Google Scholar]
- 46.Lohmeier TE, Warren S, Cunningham JT. Sustained activation of the central baroreceptor pathway in obesity hypertension. Hypertension. 2003;42(1):96–102. doi: 10.1161/01.HYP.0000076092.10923.FD. [DOI] [PubMed] [Google Scholar]
- 47.Dewan NA, Nieto FJ, Somers VK. Intermittent hypoxemia and OSA: implications for comorbidities. Chest. 2015;147(1):266–74. doi: 10.1378/chest.14-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mansukhani MP, Kara T, Caples SM, Somers VK. Chemoreflexes, sleep apnea, and sympathetic dysregulation. Curr Hypertens Rep. 2014;16(9):476. doi: 10.1007/s11906-014-0476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Witkowski A, Prejbisz A, Florczak E, Kadziela J, Sliwinski P, Bielen P, et al. Effects of renal sympathetic denervation on blood pressure, sleep apnea course, and glycemic control in patients with resistant hypertension and sleep apnea. Hypertension. 2011;58(4):559–65. doi: 10.1161/HYPERTENSIONAHA.111.173799. [DOI] [PubMed] [Google Scholar]
- 50.Lohmeier TE, Iliescu R, Tudorancea I, Cazan R, Cates AW, Georgakopoulos D, et al. Chronic interactions between carotid baroreceptors and chemoreceptors in obesity hypertension. Hypertension. 2016 doi: 10.1161/HYPERTENSIONAHA.116.07232. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Collins S, Kuhn CM, Petro AE, Swick AG, Chrunyk BA, Surwit RS. Role of leptin in fat regulation. Nature. 1996;380(6576):677. doi: 10.1038/380677a0. [DOI] [PubMed] [Google Scholar]
- 52.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404(6778):661–71. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 53••.Simonds SE, Pryor JT, Ravussin E, Greenway FL, Dileone R, Allen AM, et al. Leptin mediates the increase in blood pressure associated with obesity. Cell. 2014;159(6):1404–16. doi: 10.1016/j.cell.2014.10.058. This study shows that the effects of leptin on blood pressure are mediated by neurons in the dorsomedial hypothalamus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54••.Mark AL. Selective leptin resistance revisited. Am J Physiol Regul Integr Comp Physiol. 2013;305(6):R566–81. doi: 10.1152/ajpregu.00180.2013. Provides a comprehensive review on the sympathetic and cardiovascular actions of leptin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341(12):879–84. doi: 10.1056/NEJM199909163411204. [DOI] [PubMed] [Google Scholar]
- 56.Harris RB. Leptin-much more than a satiety signal. Annu Rev Nutr. 2000;20:45–75. doi: 10.1146/annurev.nutr.20.1.45. [DOI] [PubMed] [Google Scholar]
- 57.Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI. Receptor-mediated regional sympathetic nerve activation by leptin. J Clin Invest. 1997;100(2):270–8. doi: 10.1172/JCI119532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Machleidt F, Simon P, Krapalis AF, Hallschmid M, Lehnert H, Sayk F. Experimental hyperleptinemia acutely increases vasoconstrictory sympathetic nerve activity in healthy humans. J Clin Endocrinol Metab. 2013;98(3):E491–6. doi: 10.1210/jc.2012-3009. [DOI] [PubMed] [Google Scholar]
- 59.Carlyle M, Jones OB, Kuo JJ, Hall JE. Chronic cardiovascular and renal actions of leptin: role of adrenergic activity. Hypertension. 2002;39(2 Pt 2):496–501. doi: 10.1161/hy0202.104398. [DOI] [PubMed] [Google Scholar]
- 60.Shek EW, Brands MW, Hall JE. Chronic leptin infusion increases arterial pressure. Hypertension. 1998;31(1 Pt 2):409–14. doi: 10.1161/01.hyp.31.1.409. [DOI] [PubMed] [Google Scholar]
- 61.Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, et al. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem. 2010;285(23):17271–6. doi: 10.1074/jbc.R110.113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuo JJ, Jones OB, Hall JE. Inhibition of NO synthesis enhances chronic cardiovascular and renal actions of leptin. Hypertension. 2001;37(2 Pt 2):670–6. doi: 10.1161/01.hyp.37.2.670. [DOI] [PubMed] [Google Scholar]
- 63.Mark AL, Shaffer RA, Correia ML, Morgan DA, Sigmund CD, Haynes WG. Contrasting blood pressure effects of obesity in leptin-deficient ob/ob mice with agouti yellow obese mice. J Hypertens. 1999;17(12 Pt 2):1949–53. doi: 10.1097/00004872-199917121-00026. [DOI] [PubMed] [Google Scholar]
- 64.Ozata M, Ozdemir IC, Licinio J. Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J Clin Endocrinol Metab. 1999;84(10):3686–95. doi: 10.1210/jcem.84.10.5999. [DOI] [PubMed] [Google Scholar]
- 65.Dubinion JH, da Silva AA, Hall JE. Chronic blood pressure and appetite responses to central leptin infusion in rats fed a high fat diet. J Hypertens. 2011;29(4):758–62. doi: 10.1097/HJH.0b013e328344280b. [DOI] [PubMed] [Google Scholar]
- 66.Vong L, Ye C, Yang Z, Choi B, Chua S, Jr, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71(1):142–54. doi: 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harlan SM, Morgan DA, Agassandian K, Guo DF, Cassell MD, Sigmund CD, et al. Ablation of the leptin receptor in the hypothalamic arcuate nucleus abrogates leptin-induced sympathetic activation. Circ Res. 2011;108(7):808–12. doi: 10.1161/CIRCRESAHA.111.240226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.do Carmo JM, da Silva AA, Cai Z, Lin S, Dubinion JH, Hall JE. Control of blood pressure, appetite, and glucose by leptin in mice lacking leptin receptors in proopiomelanocortin neurons. Hypertension. 2011;57(5):918–26. doi: 10.1161/HYPERTENSIONAHA.110.161349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Young CN, Morgan DA, Butler SD, Mark AL, Davisson RL. The brain subfornical organ mediates leptin-induced increases in renal sympathetic activity but not its metabolic effects. Hypertension. 2013;61(3):737–14. doi: 10.1161/HYPERTENSIONAHA.111.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mark AL, Agassandian K, Morgan DA, Liu X, Cassell MD, Rahmouni K. Leptin signaling in the nucleus tractus solitarii increases sympathetic nerve activity to the kidney. Hypertension. 2009;53(2):375–80. doi: 10.1161/HYPERTENSIONAHA.108.124255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, et al. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci USA. 2004;101(13):4661–6. doi: 10.1073/pnas.0303992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.do Carmo JM, da Silva AA, Sessums PO, Ebaady SH, Pace BR, Rushing JS, et al. Role of Shp2 in forebrain neurons in regulating metabolic and cardiovascular functions and responses to leptin. Int J Obes (Lond) 2014;38(6):775–83. doi: 10.1038/ijo.2013.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73••.do Carmo JM, da Silva AA, Ebaady SE, Sessums PO, Abraham RS, Elmquist JK, et al. Shp2 signaling in POMC neurons is important for leptin’s actions on blood pressure, energy balance, and glucose regulation. Am J Physiol Regul Integr Comp Physiol. 2014;307(12):R1438–47. doi: 10.1152/ajpregu.00131.2014. This study shows that Shp2 signaling in POMC neurons contributes to the long-term blood pressure and antidiabetic effects of leptin but has only a modest role in body weight regulation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rahmouni K, Morgan DA, Morgan GM, Liu X, Sigmund CD, Mark AL, et al. Hypothalamic PI3K and MAPK differentially mediate regional sympathetic activation to insulin. J Clin Invest. 2004;114(5):652–8. doi: 10.1172/JCI21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.do Carmo JM, da Silva AA, Wang Z, Freeman NJ, Alsheik AJ, Adi A, et al. Regulation of blood pressure, appetite, and glucose by leptin after inactivation of insulin receptor substrate 2 signaling in the entire brain or in proopiomelanocortin neurons. Hypertension. 2016;67(2):378–86. doi: 10.1161/HYPERTENSIONAHA.115.06153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tallam LS, da Silva AA, Hall JE. Melanocortin-4 receptor mediates chronic cardiovascular and metabolic actions of leptin. Hypertension. 2006;48(1):58–64. doi: 10.1161/01.HYP.0000227966.36744.d9. [DOI] [PubMed] [Google Scholar]
- 77.Aslan IR, Ranadive SA, Valle I, Kollipara S, Noble JA, Vaisse C. The melanocortin system and insulin resistance in humans: insights from a patient with complete POMC deficiency and type 1 diabetes mellitus. Int J Obes (Lond) 2014;38(1):148–51. doi: 10.1038/ijo.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Farooqi IS. Genetic aspects of severe childhood obesity. Pediatr Endocrinol Rev. 2006;3(Suppl 4):528–36. [PubMed] [Google Scholar]
- 79.Greenfield JR. Melanocortin signalling and the regulation of blood pressure in human obesity. J Neuroendocrinol. 2011;23(2):186–93. doi: 10.1111/j.1365-2826.2010.02088.x. [DOI] [PubMed] [Google Scholar]
- 80.Greenfield JR, Miller JW, Keogh JM, Henning E, Satterwhite JH, Cameron GS, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360(1):44–52. doi: 10.1056/NEJMoa0803085. [DOI] [PubMed] [Google Scholar]
- 81.da Silva AA, do Carmo JM, Dubinion JH, Bassi M, Mokhtarpouriani K, Hamza SM, et al. Chronic central nervous system MC3/4R blockade attenuates hypertension induced by nitric oxide synthase inhibition but not by angiotensin II infusion. Hypertension. 2015;65(1):171–7. doi: 10.1161/HYPERTENSIONAHA.114.03999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.da Silva AA, do Carmo JM, Wang Z, Hall JE. The brain melanocortin system, sympathetic control, and obesity hypertension. Physiology(Bethesda) 2014;29(3):196–202. doi: 10.1152/physiol.00061.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.da Silva AA, do Carmo JM, Kanyicska B, Dubinion J, Brandon E, Hall JE. Endogenous melanocortin system activity contributes to the elevated arterial pressure in spontaneously hypertensive rats. Hypertension. 2008;51(4):884–90. doi: 10.1161/HYPERTENSIONAHA.107.100636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.do Carmo JM, da Silva AA, Rushing JS, Hall JE. Activation of the central melanocortin system contributes to the increased arterial pressure in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol. 2012;302(5):R561–7. doi: 10.1152/ajpregu.00392.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]