

Abstract
The extracellular aggregation of amyloid β (Aβ) peptides and the intracellular hyperphosphorylation of tau at specific epitopes are pathological hallmarks of neurodegenerative diseases such as Alzheimer's disease (AD). Cdk5 phosphorylates tau at AD-specific phospho-epitopes when it associates with p25. p25 is a truncated activator, which is produced from the physiological Cdk5 activator p35 upon exposure to Aβ peptides. We show that neuronal infections with Cdk5 inhibitory peptide (CIP) selectively inhibit p25/Cdk5 activity and suppress the aberrant tau phosphorylation in cortical neurons. Furthermore, Aβ1−42-induced apoptosis of these cortical neurons was also reduced by coinfection with CIP. Of particular importance is our finding that CIP did not inhibit endogenous or transfected p35/Cdk5 activity, nor did it inhibit the other cyclin-dependent kinases such as Cdc2, Cdk2, Cdk4 and Cdk6. These results, therefore, provide a strategy to address, and possibly ameliorate, the pathology of neurodegenerative diseases that may be a consequence of aberrant p25 activation of Cdk5, without affecting ‘normal' Cdk5 activity.
Keywords: Alzheimer's disease, Cdk5 inhibitory peptide (CIP), hyperphosphorylation, p25, tau
Introduction
Cdk5 is a member of the Cdk family of serine/threonine kinases, most of which are key regulators of the cell cycle. Cdk5 activity is regulated through association with its neuron-specific activators, p35 and p39 (Dhavan and Tsai, 2001). Recent evidence suggests that aberrant Cdk5 activity induced by the conversion of p35 to p25 plays a role in the pathogenesis of neurodegenerative diseases such as Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS) and Niemann's Pick type-C disease (NPD) (Julien and Mushynski, 1998; Lee et al, 1999; Ahlijanian et al, 2000; Nguyen et al, 2001; Bu et al, 2002; Lau et al, 2002; Cruz et al, 2003). The calpain-directed proteolysis of p35 releases an N-terminal myristoylated membrane tether and the dissociated Cdk5/p25 complexes are induced to phosphorylate a number of cytosolic proteins (Kusakawa et al, 2000; Lee et al, 2000). Transgenic mice, overexpressing human p25, show cytoskeletal disruptions and hyperphosphorylation of tau in certain areas of the brain, resembling AD pathology (Ahlijanian et al, 2000; Cruz et al, 2003). Inhibitors of Cdk5 and calpain applied to rat hippocampal neurons stabilized p35 and markedly reduced p25 neurotoxicity induced by the amyloid β (Aβ) peptide Aβ1−42, suggesting that p25 is a downstream effector of Aβ peptides. Additionally, the stabilization of its activator p35 also decreases Aβ-mediated toxicity (Alvarez et al, 1999; Lee et al, 2000; Town et al, 2002; Li et al, 2003b). Abnormal hyperphosphorylation of tau after Aβ1−42 treatment destabilizes microtubules, contributing to neurite degeneration and the formation of paired helical filaments (PHFs) containing neurofibrillary tangles (NFTs), one of the principal lesions of AD (Busciglio et al, 1995; Michaelis et al, 2002; Hardy, 2003).
We have reported that a Cdk5 inhibitory peptide (CIP), a 125-residue peptide consisting of p35154−279 with a much higher affinity for Cdk5 than p25, effectively and specifically inhibits the activity of Cdk5 in vitro (Amin et al, 2002). It also inhibits the phosphorylation of tau induced by Cdk5/p25 expression in transfected HEK293 cells without affecting their endogenous Cdc2 kinase activity (Zheng et al, 2002). This study shows that CIP inhibited p25/Cdk5- and Aβ1−42-induced Cdk5 hyperactivation in cortical neurons. We show that in these paradigms CIP was able to inhibit neuronal apoptosis in Aβ1−42-treated neurons. This raises the intriguing possibility that such an agent might be therapeutic for AD and other neurodegenerative diseases, which exhibit abnormal phosphorylation of neuronal cytoskeletal proteins by p25/Cdk5.
Results
Expression of Cdk5, p35, p25 and CIP in neurons
Initially, the lentiviral-based infection system was used to introduce the appropriate genes into primary rat cortical neurons and overexpress Cdk5, p25, p35 and CIP. This was carried out to determine whether CIP could inhibit Cdk5 even when its activity was significantly increased. Typically, neurons were infected to approximately 80–90% efficiency in single infections with double and triple coinfections showing approximately 60–70% efficiency (data not shown). This has been, by far, the most efficient method of gene delivery in neurons. We utilized Western blotting to examine the expression of Cdk5, p35 and p25 genes in cortical neurons (Figure 1A). E18 neurons were infected at day 7 in culture (7-DIC) and lysates were harvested after 72 h to ensure proper integration and infection rates. The infections were extremely efficient at introducing the genes into neurons and the neurons produced high levels of proteins. In Figure 1A, Western blots show the presence of Cdk5 using C-8 antibody. Since endogenous Cdk5 is expressed at high levels in these neurons, saturation of the Cdk5 signals is easily achieved. Higher signals were obtained with infected Cdk5 (Figure 1A, lanes 2–7) when compared to empty vector (EV) (Figure 1A, lane 1). Endogenous p35 (Figure 1A, lanes 1–3) was expressed at much lower levels when compared to endogenous Cdk5 (Figure 1A, lane 1), while infected p35 and p25 showed dramatic increases in the amounts of expressed protein (Figure 1A, lanes 2–7). It is noteworthy that endogenous p25 was not detected in EV-infected neurons (lane 1). Moreover, it was not detected in neurons infected with p35 (Figure 1A, lanes 4, 5 and 7).
Figure 1.
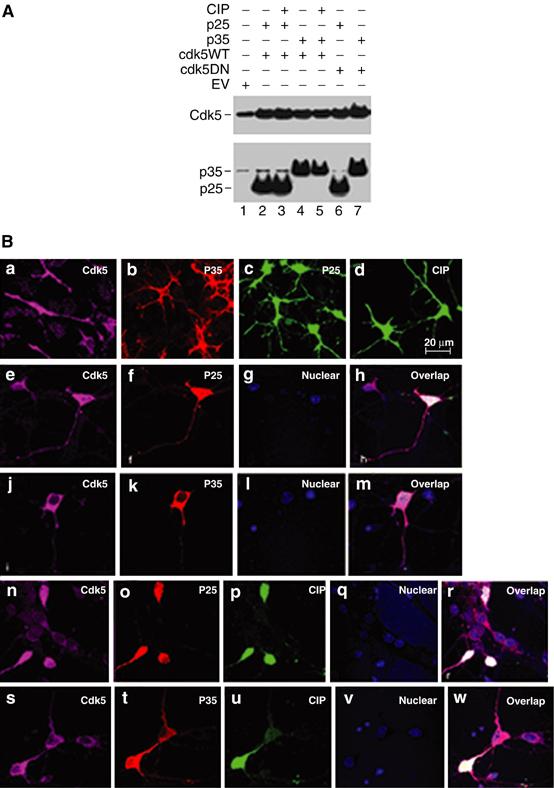
Cdk5, p35, p25 and CIP expression in coinfected primary cortical neurons. E18 rat embryonic cortical neurons were infected with the following expression constructs: empty LV-GFP vector (EV), p25 and wild-type Cdk5 (Cdk5WT) with or without CIP, p35 and wild-type Cdk5 with or without CIP, p25 and dominant-negative Cdk5 (Cdk5DN) and p35 and Cdk5DN. (A) After 3 days of infection, neuronal lysates were resolved on 4–20% SDS–PAGE gradient gels and Western blotting was employed to detect endogenous and transfected Cdk5 (C-8; top panel), p35 and p25 (C-19; bottom panel). Lane 1: EV only; lanes 2 and 3: p25+Cdk5WT with and without CIP respectively; lanes 4 and 5: p35+Cdk5WT with and without CIP respectively; lane 6: p25+Cdk5DN; lane 7: p35+Cdk5DN. (B) Confocal images illustrate the cortical neurons single infections with Cdk5, p35, p25 and CIP (panels a–d, respectively), double infection of Cdk5 and p25 (panels e–h), double infection of Cdk5 and p35 (panels j–m), triple infections of Cdk5, p25 and CIP (panels n–r) and Cdk5, p35 and CIP (panels s–w). The scale bar represents 20 μm.
We used immunocytochemical analyses to confirm the results obtained by Western blotting (Figure 1B). Infected Cdk5 was expressed throughout the neurons, in the perikarya and nuclei, and as previously reported p25 showed a similar pattern. CIP was found in both compartments colocalizing with both Cdk5 and p25 (Figure 1B, panels n–r). However, p35 was found primarily in the perikarya and not in nuclei of infected neurons (Figure 1B, panels b and k). p35 was immunodetected using the C-19 antibody, while p25 and CIP were identified by using their GFP tags. p25 and CIP were distinguished by immunostaining using the C-19 antibody, which detects p25 and not CIP.
CIP specifically inhibits p25/Cdk5 activity without affecting endogenous p35/Cdk5 activity of neurons
Given the high levels of expression of all the introduced genes, we now asked whether CIP inhibited Cdk5 kinase activity. We examined Cdk5 activity in immunoprecipitates from infected neurons using in vitro kinase assays with histone H1 as the substrate. 7-DIC neurons were infected with EV, p25/Cdk5, p25/dominant-negative Cdk5, p25/Cdk5/CIP, p25 only and p25/CIP and harvested after 72 h. Cdk5 was immmunoprecipitated from equal amounts of lysates and assayed for kinase activity using both C-8 polyclonal and J-3 monoclonal antibodies. The results presented in Figure 2A show that neurons infected with p25/Cdk5 exhibited robust phosphorylation of histone H1 in C-8 and J-3 immunoprecipitates (Figure 2A, lane 3), while noninfected (NI) and EV-infected neurons showed basal levels of phosphorylation (Figure 2A, lanes 1 and 2). Coinfection of p25 and dominant-negative Cdk5 and, more importantly, coinfection of p25, Cdk5 and CIP diminished the histone H1 phosphorylation by approximately two times compared to p25/Cdk5 (Figure 2A, compare lane 3 with lanes 4 and 5). Neurons infected with p25 only showed an elevated level of histone H1 phosphorylation compared to NI and EV-infected neurons (Figure 2A, compare lane 6 with lanes 1 and 2), suggesting activation of endogenous Cdk5. The coinfection of p25/CIP decreased this phosphorylation (Figure 2A, lane 7). The presence of Cdk5 in the C-8 immunoprecipitates was confirmed by immunodetection using J-3 antibody, while the presence of Cdk5 in the J-3 immunoprecipitates was confirmed by immunodetection with C-8 antibody. The presence of p25 and p35 in the J-3 immunoprecipitates was confirmed using the C-19 antibody. From these data, it was clearly evident that infected CIP did not mask the ability to immunoprecipitate Cdk5 from neurons; each immunoprecipitate contained a p25/Cdk5 complex (lanes 3–7) with the exception of NI and EV neurons, which contained endogenous p35/Cdk5 complexes.
Figure 2.
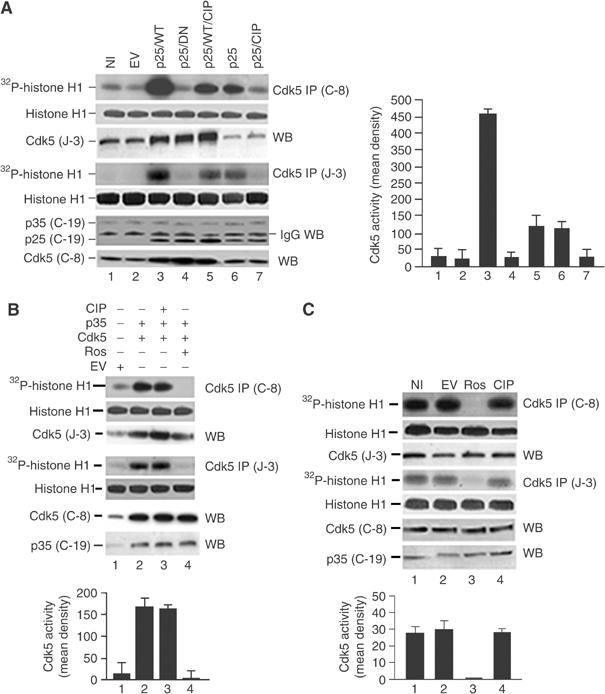
CIP specifically inhibited p25/Cdk5 but not p35/Cdk5 activity in coinfected primary cortical neurons. Infections of primary cortical neurons were carried out and Cdk5 was immunoprecipitated using C-8 and J-3 antibodies from equal amounts of lysates. Immunoprecipitates were then subjected to in vitro histone H1 kinase assays and Western blot for immunodetection. All bar graphs represent mean optical density measurements of phospho-histone (32P-histone H1) of both Cdk5 activities from C-8 and J-3 IPs measured from autoradiographs (first and fourth panels in (A–C)). Data are expressed as mean±s.e.m. of four separate experiments. Coomassie-stained gels show equal amounts of histone present in all assays (histone H1; second and fifth panels in (A–C)). (A) NI (lane 1), EV (lane 2), p25+wild-type Cdk5 (p25/WT; lane 3), p25+dominant-negative Cdk5 (p25/DN; lane 4), p25+WT+CIP (p25/Cdk5/CIP; lane 5), p25 only (lane 6) and p25+CIP (p25/CIP; lane 7). Cdk5 was immunodetected using polyclonal J-3 antibody (third panel) in the C-8 IP. In the J-3 IP, p25 and p35 were immunodetected using polyclonal C-19 antibody (sixth panel); Cdk5 was detected using polyclonal C-8 antibody (seventh panel). (B) The CIP effect on infected p35/Cdk5 activity was investigated using in vitro kinase assays after infections of 7-DIC cortical neurons. EV, lane 1; p35/Cdk5, lane 2; p35/Cdk5/CIP, lane 3; p35/Cdk5+roscovitine, lane 4. Cdk5 IP and Western blot immunodetection were performed using the same antibodies as in (A). (C) CIP effect on endogenous Cdk5 activity was investigated using in vitro kinase assays in neurons. NI (lane 1), EV infected (lane 2), NI+roscovitine (lane 3) and CIP infected (lane 4). Cdk5 IP and Western blot immunodetection were performed using the same antibodies as in (A).
A parallel series of experiments was carried out with those shown in Figure 2A to address the question whether CIP inhibits p35/Cdk5 activity under similar conditions. Cortical neurons were coinfected with p35/Cdk5 and p35/Cdk5/CIP and similar immunoprecipitations (IPs) to those seen in Figure 2A were performed. In both C-8 and J-3 IPs, we found that CIP did not inhibit p35/Cdk5 activity (Figure 2B, lanes 2 and 3) while roscovitine completely inhibited p35/Cdk5 activity (Figure 2B, lane 4). Again the presence of CIP did not prevent the pull-down of a p35/Cdk5 complex.
An important question we wanted to address was whether CIP inhibited endogenous p35/Cdk5 activity. To determine CIP effect on endogenous Cdk5 activity, the same experiments were carried out with NI, EV-infected, CIP-infected or roscovitine-treated neurons as a control. IPs with both C-8 and J-3 antibodies illustrated in Figure 2C show that CIP did not inhibit endogenous Cdk5 activity (Figure 2C, compare lane 4 with lanes 1 and 2), while roscovitine completely inhibited endogenous Cdk5 activity (Figure 2C, compare lane 3 with lanes 1, 2 and 4).
CIP inhibits p25/Cdk5-mediated phosphorylation of tau
To determine whether CIP, when coinfected with Cdk5/p25, inhibits endogenous tau hyperphosphorylation, cortical neurons were infected with EV, p25/Cdk5 or p25/Cdk5/CIP for 3 days, lysed and prepared for Western blotting. As shown in Figure 3A, endogenous levels of tau phosphorylation at residue 199/202 were at basal levels in NI and EV lysates (Figure 3A, lanes 1 and 2). p25/Cdk5-infected neurons exhibited robust phosphorylation of tau at the above epitopes and coinfected CIP was able to reduce this phosphorylation two-fold (Figure 3A, lanes 3 and 4 and bar graph). We used the Cdk5 inhibitor roscovitine to show that in its presence tau phosphorylation was significantly inhibited (Figure 3A, lane 5). In p25-infected neurons, tau phosphorylation was elevated compared to NI and EV-infected neurons, showing stimulation of endogenous Cdk5 activity. This phosphorylation was inhibited when CIP was coinfected with p25 (Figure 3A, lanes 6 and 7). CIP exhibited a two-fold inhibition of tau phosphorylation when phospho-tau 199/202 was examined. Additionally, when we used two other phospho-tau epitope antibodies (phospho-serine 404 and AT-8) to confirm this observation, we found a two-fold inhibition of phosphorylation between p25/Cdk5 and p25/Cdk5/CIP when phospho-tau 404 and AT-8 antibodies were quantified (Figure 3A, see bar graph).
Figure 3.
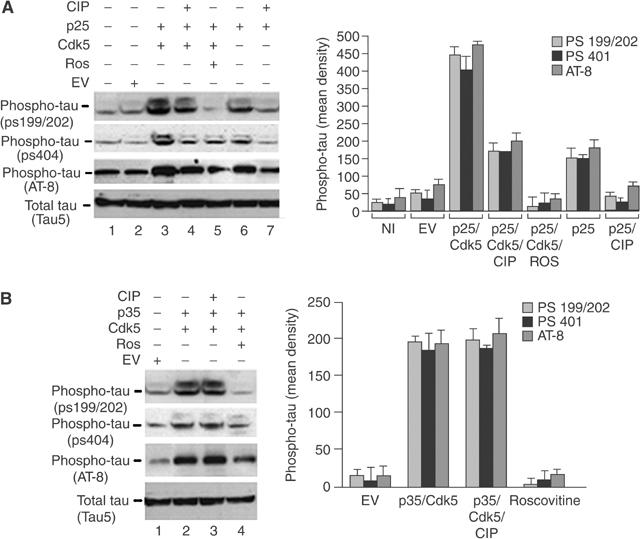
CIP specifically inhibits tau hyperphosphorylation induced by p25/Cdk5 but not tau phosphorylation induced by p35/Cdk5. (A) Cortical neurons were infected with the following expression constructs: EV (lane 2), p25+Cdk5 (lane 3), p25+Cdk5+CIP (lane 4), p25+Cdk5+roscovitine treatment (lane 5), p25 only (lane 6) and p25+CIP (lane 7). The top three panels show phospho-tau with three different phospho-tau antibodies (anti-pS199/202, anti-pS404 and AT8), while the bottom panel shows total tau (Tau5). (B) Cortical neurons were infected with the following expression constructs: EV (lane 1), p35+Cdk5 (lane 2), p35+Cdk5+CIP (lane 3) and p35+Cdk5 treated with roscovitine (lane 4). The top three panels show Western blots for phospho-tau using the same phospho-tau antibodies mentioned above and the bottom panel shows total tau (Tau5). The bar graphs show the mean optical density of phospho-tau (ps199/202, ps404 and AT-8). The results are expressed as mean±s.e.m. of four independent experiments.
Since CIP was highly efficient at inhibiting p25/Cdk5 activity, it was important to determine the specificity of CIP inhibition. We investigated the phosphorylation of endogenous tau in p35/Cdk5-and p35/Cdk5/CIP-infected neuronal lysates (Figure 3B). We found that EV infection did not result in large increases in tau phosphorylation at the same epitopes investigated in Figure 3A (Figure 3B, lane 1). Infection with p35/Cdk5 caused an increase in tau phosphorylation, which was unaffected when CIP was coinfected with p35/Cdk5 (Figure 3B, lanes 2 and 3). Roscovitine reduced tau phosphorylation to barely detectable levels (Figure 3B, lane 4), an effect expected of this potent Cdk5 inhibitor. These data on endogenous tau phosphorylation are consistent with the specificity of CIP inhibition of p25/Cdk5 activity without affecting p35/Cdk5 activity (Figure 3B, lanes 2 and 3). The use of all three phospho-tau antibodies revealed that phosphorylation of tau was unchanged in p35/Cdk5- and p35/Cdk5/CIP-infected neurons (Figure 3B, see bar graph).
CIP inhibits Aβ1−42-mediated endogenous p25/Cdk5 hyperactivity and tau hyperphosphorylation
To test whether CIP can inhibit the neurotoxic effects of Aβ1−42, 7-DIC cortical neurons infected with EV or with CIP were treated with Aβ1−42 for 6 h, lysed and prepared for Cdk5 IP using both C-8 and J-3 antibodies. The resulting IPs were used in kinase assays as before. From the data shown in Figure 4A, it is evident that Aβ1−42 treatment caused an approximately 10-fold increase in phosphorylation of histone H1 by endogenous Cdk5 (Figure 4A, lane 2 and bar graph). In the presence of CIP, Cdk5 activity exhibited a four-fold reduction in Aβ1−42-treated neurons (Figure 4A, lane 3). To detect Cdk5 in the C-8 immunoprecipitates, we used the J-3 antibody (Figure 4A, third panel). The kinase assays using the J-3 antibody immunoprecipitate showed identical results as the C-8 immunoprecipitates (Figure 4A, first and fourth panels). To confirm that J-3 IPs did indeed pull down a p25/Cdk5 complex responsible for Aβ1−42 tau hyperphosphorylation, we detected p25 and p35 using the polyclonal C-19 antibody (Figure 4A, sixth panel) and Cdk5 using C-8 (Figure 4A, seventh panel). Some degree of crossreactivity with the light chain of IgG was evident, but we clearly detected p25 and p35 species. In both IPs, CIP did not mask the ability of the antibodies to pull down Cdk5.
Figure 4.
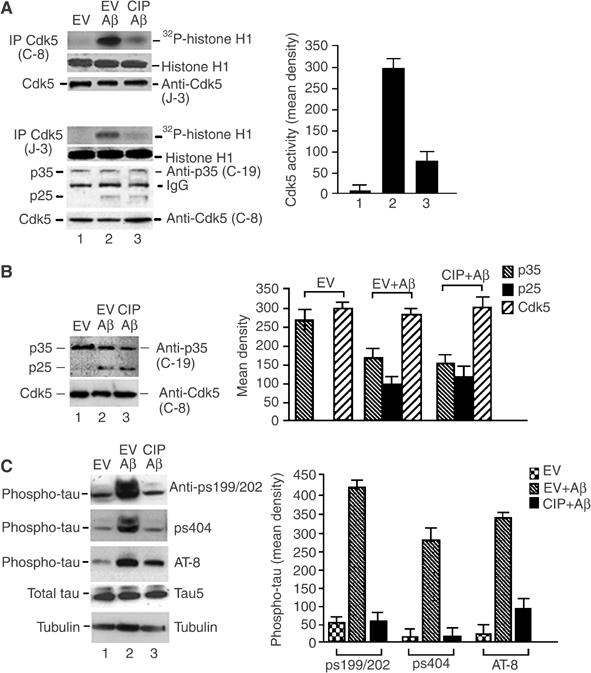
CIP inhibited Aβ1−42-induced endogenous p25/Cdk5 hyperactivity and tau hyperphosphorylation in cortical neurons. (A) 7-DIC cortical neurons were infected with EV or CIP and treated with 10 μM Aβ1−42 for 6 h. Cdk5 was immunoprecipitated from equal amounts of lysates with polyclonal C-8 and J-3 antibodies, and subjected to histone H1 kinase assays. The bar graph represents average mean optical density±s.e.m. of phospho-histone levels in four separate experiments. Coomassie panels show equal amounts of histone present in the assay. To determine Cdk5, p25 and p35 levels in the Cdk5 immunoprecipitates, Cdk5 was immunodetected using polyclonal J-3 antibody in the C-8 IP. In the J-3 IP, p25 and p35 were immunodetected using polyclonal C-19 antibody; Cdk5 was detected using rabbit polyclonal C-8. (B) Western blots were performed to detect the presence of p35, p25 and Cdk5 using lysates from (A). The bar graph shows mean density±s.e.m. measurements of each of the three proteins of four independent experiments. (C) Lysates from (A) were resolved by SDS–PAGE. Lane 1: EV; lane 2: EV+Aβ1−42; lane 3: CIP+Aβ1−42. The top three panels show phospho-tau using different phospho-tau antibodies (anti-pS199/202, anti-pS404 and AT8), the fourth panel shows total tau (Tau5) and the bottom panel shows tubulin. The bottom two panels show equal amounts of total tau (Tau5) and tubulin in the lysates. The bar graph shows mean optical densities of phospho-tau ps199/202, ps404 and AT-8 of four separate experiments.
Western blot assays of whole-cell lysates from similarly infected cortical neurons treated with Aβ1−42 revealed production of p25 with a concomitant reduction in p35 expression (Figure 4B, top panel, lanes 2 and 3 and bar graph). Significantly, in these lysates, the increased production of p25 correlated with an increased level (8- to 10-fold) of endogenous tau phosphorylation at residues 199/202 and 404 and those detected by the AT-8 tau antibody (Figure 4C, third panel, lane 2 and bar graph). CIP, under these conditions, exhibited a striking inhibition of the Aβ1−42-mediated tau hyperphosphorylation, restoring it to basal levels (Figure 4C, lane 3). Total levels of tau were shown using the tau-5 antibody where the major band resolves at approximately 55 kDa (Figure 4C, fourth panel). Equal loading was also confirmed by immunodetection of tubulin in the lysates (Figure 4C, bottom panel).
CIP inhibits neuronal apoptosis mediated by p25/Cdk5 and Aβ1−42
AD is characterized by signature lesions such as intracellular tau NFTs and extracellular amyloid plaques containing the Aβ1−42 peptide, and these processes lead to neuronal apoptosis (Patrick et al, 1999; Hardy, 2003). Accordingly, we set out to determine the effect of CIP on neuronal apoptosis induced by the overexpression of p25/Cdk5 or by treatment with Aβ1−42. The data in Figure 5 show that p25/Cdk5- and Aβ1−42-mediated activation of p25/Cdk5 may contribute to this neuronal apoptosis. In this instance, as an assay of apoptosis, we used the expression of cleaved caspase-3, a downstream marker of apoptosis (Nicholson et al, 1995). The coinfection of p25/Cdk5 tripled the levels of cleaved caspase-3 compared to EV-infected neurons (Figure 5A, lanes 1 and 2 and bar graph). On the other hand, coinfection of p25/Cdk5/CIP reduced cleaved caspase-3 back to basal levels (Figure 5A, lane 3). The coinfection of p25/dominant-negative Cdk5, p35/Cdk5 and p35/Cdk5/CIP all displayed basal levels of cleaved caspase-3 (Figure 5A, lanes 4–6) as did the NI and EV lysates (Figure 5A, lanes 1 and 7).
Figure 5.
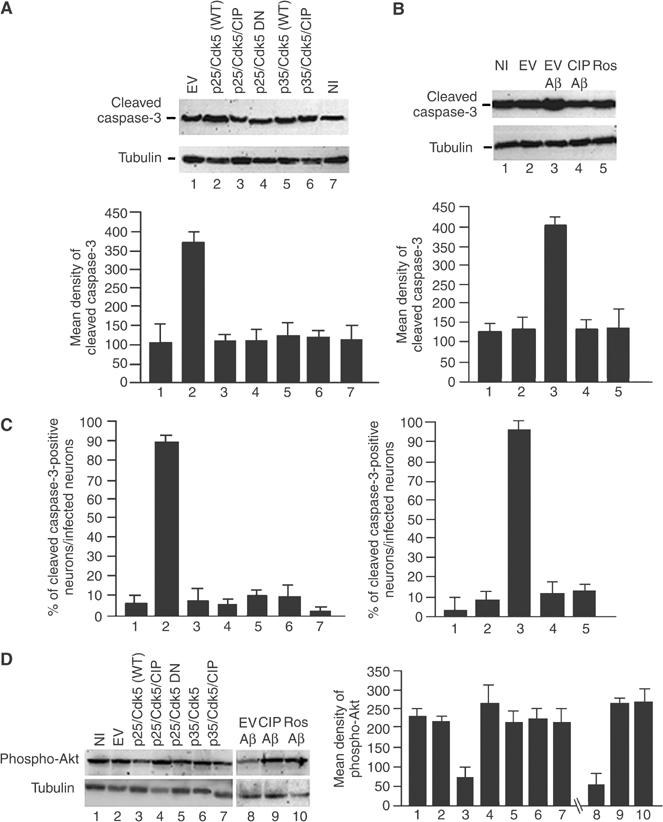
CIP reduced neuronal apoptosis induced by p25/Cdk5 overexpression as well as Aβ1−42 treatment of cortical neurons. (A) Levels of cleaved caspase-3, a well-established marker of apoptosis, were investigated after E18 rat cortical neurons were infected with the following constructs: lane 1, EV; lane 2, p25+wild-type Cdk5 (p25/WT); lane 3, p25+wild-type Cdk5+CIP (p25/WT/CIP); lane 4, p25+dominant-negative Cdk5 (p25/DN); lane 5, p35+wild-type Cdk5 (p35/WT); lane 6, p35+wild-type Cdk5+CIP (p35/WT/CIP); lane 7, NI control. The bar graph shows measurements of cleaved caspase-3 expressed as mean density±s.e.m. of four separate experiments. (B) Cortical neurons were infected with EV (lane 2), EV and treated with Aβ1−42 (lane 3), infected with CIP and treated with Aβ1−42 (lane 4), NI, treated with roscovitine and then 1 h later with Aβ1−42 (lane 5) and NI control (lane 1). All Aβ1−42 treatments were carried out for 6 h. Cell lysates were resolved by SDS–PAGE and cleaved caspase-3 (top panel) and tubulin (bottom panel) were immunodetected. The bar graph shows mean density measurements of cleaved caspase-3 expressed as ±s.e.m. of four separate experiments. (C) To confirm the Western blot data, we performed immunocytochemical assays of the infections as in (A, B) using cleaved caspase-3 as a marker. The bar graphs show cell counts of cleaved caspase-3-positive neurons with the coinfections and Aβ1−42 treatment with or without CIP in cortical neurons. (D) As another measure of apoptosis/survival afforded by CIP, we used phospho-Akt, a well-established marker of survival. Lysates from (A, B) were resolved by SDS–PAGE and immunodetected using anti-phospho-Akt antibody. The bar graph shows mean optical density±s.e.m. of four separate experiments of phospho-Akt.
To determine whether CIP can inhibit neuronal apoptosis induced by Aβ1−42, cortical neurons infected with or without CIP were treated with Aβ1−42 for 6 h, lysed and prepared for Western blots, using cleaved caspase-3 antibody as an assay for apoptosis. As seen in Figure 5B, infection of EV and treatment with Aβ1−42 caused an increase in cleaved caspase-3 levels (Figure 5B, lane 3). This was prevented in neurons treated with Aβ1−42 but infected with CIP (Figure 5B, lane 4). In these neurons, cleaved caspase-3 levels were reduced to control EV levels. Cells treated with roscovitine exhibited reduced levels of cleaved caspase-3 comparable to that seen with CIP infection, confirming that the effect was mediated by activated Cdk5 (Figure 5B, lane 5). NI and EV-infected neurons exhibited similar, basal levels of cleaved caspase-3 (Figure 5B, lanes 1 and 2). To confirm the Western blotting data with cleaved caspase-3, we performed cleaved caspase-3 immunocytochemical cell counting assays in coinfected and Aβ1−42-treated neurons. In Figure 5C, we confirm the Western blotting data and show that approximately 90% of p25/Cdk5-infected neurons exhibited cleaved caspase-3 immunoreactivity (Figure 5C, left graph, lane 2). The coinfection of p25/Cdk5/CIP into neurons reduced this number to less than 10% (Figure 5C, left graph, lane 3). The treatment of neurons with Aβ1−42 caused approximately 90% of neurons to express cleaved caspase-3 (Figure 5C, right graph, lane 3), while the coinfection of CIP and treatment with Aβ1−42 reduced this number to approximately 10% (Figure 5C, right graph, lane 4).
The levels of cleaved caspase-3 expression in NI and EV-infected neurons were comparable to those with CIP infections shown in Figure 5A and B and, likewise, the immunocytochemical assay of cleaved caspase-3-positive neurons exhibited similar levels when CIP was present.
As an additional confirmation of the anti-apoptotic effects of CIP, we utilized a phospho-Akt antibody as a measure of survival afforded by CIP (Datta et al, 1999). The coinfection of p25/Cdk5 caused a reduction in phospho-Akt levels compared to EV and NI neurons (Figure 5D, compare lane 3 with lanes 1 and 2). The coinfection of p25/Cdk5/CIP increased phospho-Akt levels compared to p25/Cdk5-infected neurons (Figure 5D, compare lanes 3 and 4). Likewise, the treatment of neurons with Aβ1−42 caused a reduction in phospho-Akt levels compared to CIP-infected Aβ1−42-treated neurons (Figure 5D, lanes 8 and 9). The treatment of neurons with Aβ1−42 and roscovitine showed levels of phospho-Akt comparable to those with CIP-infected Aβ1−42-treated neurons (Figure 5D, lanes 9 and 10).
As an additional measure of the effect of CIP on Aβ1−42-induced apoptosis, a TUNEL assay was compared to an immunocytochemical analysis of cleaved caspase-3 expression in neurons. 3-DIC primary cortical neurons were transfected with CIP-GFP using Lipofectamine 2000. After 24 h, they were treated with Aβ1−42 for 6 h, fixed and subjected to TUNEL staining, while CIP was visualized by GFP. CIP clearly inhibited Aβ1−42-mediated neuronal apoptosis; the only neurons that were TUNEL negative were the CIP-transfected neurons (Figure 6A–D). The quantitative data are shown in the accompanying bar graph. We confirmed the TUNEL data by repeating the experiment using cleaved caspase-3 and found that the CIP-transfected neurons were cleaved caspase-3 negative and approximately 85% of nontransfected neurons were positive (Figure 6E–H). DAPI staining of the nuclei showed that the CIP-transfected neurons exhibited a normal looking nucleus while nontransfected neurons surrounding it exhibited fragmented nuclei and positive TUNEL staining. The presence of ‘normal' looking nuclei that were not transfected with CIP and were TUNEL negative can be attributed to these neurons progressing but not having reached the apoptotic stage determined by TUNEL and cleaved caspase-3 staining. When the treatment of neurons with Aβ1−42 was increased to 16 h, all nontransfected neurons were TUNEL and cleaved caspase-3 positive (data not shown). In these studies, 3-DIC cortical neurons were used due to their higher efficiency when transfected by Lipofectamine 2000 compared to 7-DIC neurons. This provided us with the means to perform reliable immunocytochemical counts.
Figure 6.
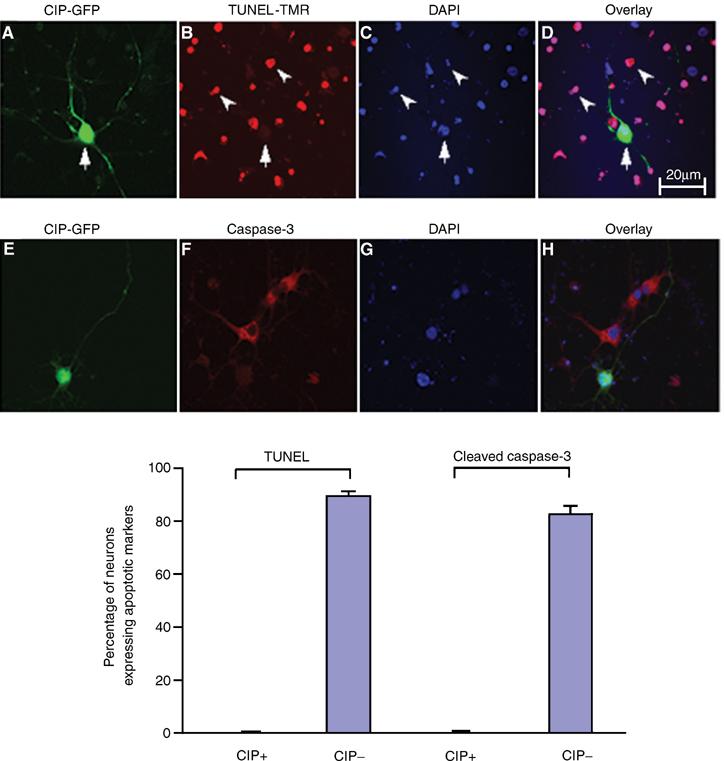
CIP reduces neuronal apoptosis induced by Aβ1−42 treatment in cortical neurons as measured by TUNEL assay and ICC assay of cleaved caspase-3. p35 Promoter-CIP-hrIRES-GFP(CIP-GFP) and p35 Promoter-empty vector hrIRES-GFP (GFP) were transfected into 3-DIC cortical neurons and after 24 h neurons were treated with 10 μM Aβ1−42 for 6 h. Neurons were fixed and stained in separate experiments for TUNEL and caspase-3 (cleaved) to show apoptotic neurons. In the first row (A–D), neurons were stained for TUNEL. (A) CIP-GFP; (B) TUNEL-TMR; (C) nuclear staining with DAPI; (D) overlay of CIP and TUNEL. In the second row (E–H), neurons are stained for cleaved caspase-3 expression. (E) CIP-GFP; (F) cleaved caspase-3; (G) nuclear staining with DAPI; (H) overlay of CIP and cleaved caspase-3. Cell counts were performed as follows: 10 independent fields were analyzed with a total of 500 neurons where TUNEL and cleaved caspase-3 could be counted. DAPI staining gave the total number of neurons and CIP was identified by GFP. The bar graph shows the quantity of neuronal apoptosis expressed as mean±s.e.m. from four separate transfections.
CIP does not affect mitotic Cdk activity and inhibits Cdk5 hyperactivity in a dose-dependent manner
To address the effect of CIP on the mitotic kinases, we studied the effect of Aβ1−42 treatment of cortical neurons on Cdk5 and Cdc2, Cdk2, Cdk4 and Cdk6 activities. We immunoprecipitated Cdk5 and Cdc2 using C-8 and PSTAIRE antibodies, respectively, and performed histone H1 kinase assays as described previously (Vincent et al, 1997). To test whether CIP inhibits Cdc2, we used GST-CIP in the kinase assays and found that the robust increase in Cdk5 activity was inhibited by GST-CIP (Figure 7A, lanes 1–3). However, the increase in Cdc2-mediated histone phosphorylation, although not as robust as seen with Cdk5, was not inhibited by GST-CIP (Figure 7A, lanes 4–6). We also infected cortical neurons with CIP, treated these neurons with Aβ1−42 for 6 h and found that CIP did not inhibit the increase in Cdc2-mediated histone phosphorylation (Figure 7A, lanes 7 and 8). To confirm the specificity of the Cdc2 antibody, we immunodetected Cdk2, 4 and 6 in the PSTAIRE immunoprecipitates and found that the PSTAIRE antibody did not pull down the other Cdks (see Supplementary figure). To determine if CIP can inhibit the other mitotic Cdks, we immunoprecipitated Cdk2, 4 and 6 using their specific antibodies, from untreated, Aβ1−42-treated and Aβ1−42-treated CIP-infected neurons and performed histone H1 kinase assays. It was found that CIP did not inhibit Cdk2, 4 or 6 activities (Figure 7B). Activities of Cdk2 and Cdk4 were higher than that of Cdk6 in these neurons and, interestingly, we did not observe any significant increases in their activities upon Aβ1−42 treatment compared to those of Cdk5 and Cdc2.
Figure 7.
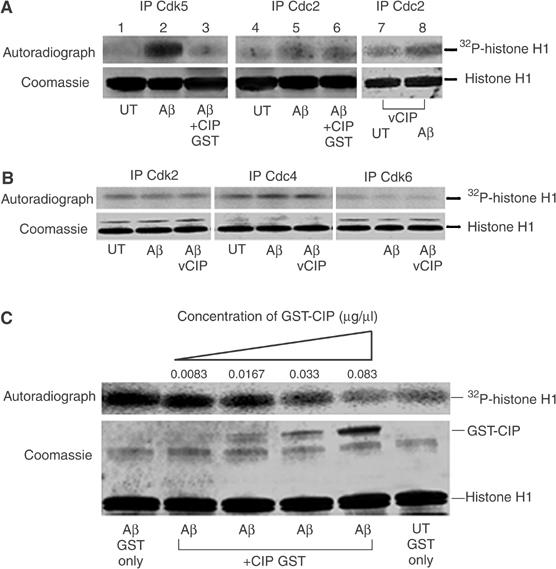
CIP did not affect endogenous neuronal Cdc2 activity nor the activity of other cyclin-dependent kinases, but inhibited Cdk5 hyperactivity in a dose-dependent manner. (A) 7-DIC cortical neurons were treated with 10 μM Aβ1−42 or PBS (UT) for 6 h and subjected to in vitro kinase assays using 5 μg GST-CIP to investigate its inhibitory properties. Cdk5 was immunoprecipitated using C-8 antibody and Cdc2 was immunoprecipitated using the PSTAIRE antibody. In lanes 7 and 8, we infected CIP prior to treatment with Aβ1−42 for 6 h, immunoprecipitated Cdc2 and performed histone H1 kinase assays. The top panel shows the autoradiograph, while the bottom panel shows Coomassie-stained histone H1 bands. (B) 7-DIC cortical neurons were infected with the CIP expression construct using the lentiviral system (lane 3 only) and neurons were treated with 10 μM Aβ1−42 or PBS (UT) for 6 h. Endogenous Cdk2, Cdk4 and Cdk6 were immunoprecipitated from equal amounts of lysates and immunoprecipitates were then subjected to in vitro histone H1 kinase assays. The top panel shows the autoradiograph while the bottom panel shows Coomassie-stained histone H1 bands. (C) CIP affects p25/Cdk5 activity in a dose-dependent manner. Cdk5 was immunoprecipitated using C-8 antibody from equal amounts of lysates of cortical neurons treated or untreated with 10 μM Aβ1−42 for 6 h. Immunoprecipitates were then subjected to in vitro histone H1 kinase assays with the addition of increasing amounts of GST-CIP in a reaction volume of 60 μl. The top panel is the autoradiograph and the bottom panel shows the corresponding Coomassie-stained histone H1 bands. GST-CIP titration experiments were reproducible in four separate assays.
To determine an effective concentration of CIP required to inhibit Cdk5 activity induced by Aβ1−42 treatment, in vitro histone H1 kinase assays using varying amounts of GST-CIP (Figure 7C) were carried out. We found that 0.083 μg/μl of GST-CIP was sufficient to inhibit Aβ1−42-mediated Cdk5 hyperactivity. Since CIP is approximately one-third the size of the full fusion protein, the effective CIP amount required for inhibition of aberrant Cdk5 hyperactivation by Aβ1−42 is approximately 0.028 μg/μl of CIP protein. The IC50 obtained from this experiment for effective CIP inhibition is 0.165 μM, which is very close to our previously reported IC50 value of 0.125 μM (Zheng et al, 2002). The slight difference in values may be attributed to different experimental systems used: previously, recombinant p25/Cdk5 was used and in the present experiments endogenous and infected p25/Cdk5 were used. This gives us tangible information regarding the amount of CIP required for inhibiting Aβ1−42-mediated p25/Cdk5 hyperactivity.
Discussion
The p35/Cdk5 and p39/Cdk5 complexes are required for the proper development and function of the nervous system because they target a wide variety of substrates involved in neuronal migration, cellular adhesion, synaptic activity, dopamine signaling and ‘crosstalk' with other kinase and signal transduction pathways (Bibb et al, 1999; Ohshima et al, 1999; Kwon et al, 2000; Rosales et al, 2000; Li et al, 2001, 2003a; Sharma et al, 2002; Kesavapany et al, 2004). Several laboratories, including ours, have observed that in neurons exposed to experimental stress or neurodegenerative disorders such as AD, Cdk5 activity is deregulated. Under neurotoxic conditions that increase intracellular Ca2+ concentrations, p35 is abnormally cleaved to p25, a more stable molecule that binds and hyperactivates Cdk5 (Patrick et al, 1999; Lee et al, 2000). It has been proposed that this deregulation of Cdk5 leads to abnormal phosphorylation of neuronal cytoskeletal proteins such as tau and neurofilaments, which may result in the formation of NFTs and neuronal apoptosis (Patrick et al, 1999; Ahlijanian et al, 2000; Lee et al, 2000; Cruz et al, 2003; Noble et al, 2003).
Initially, this hypothesis was based on the observation that the ratio of p25 to p35 in post-mortem AD brain tissue was 20- to 40-fold higher compared to normal brains and Cdk5 activity was significantly elevated (Patrick et al, 1999). Although these observations have been disputed (Yoo and Lubec, 2001; Kerokoski et al, 2002; Tandon et al, 2003), overwhelming experimental evidence, using transgenic mice, have shown that p25/Cdk5 is indeed a causative factor in neuronal apoptosis and degeneration (Ahlijanian et al, 2000; Cruz et al, 2003; Noble et al, 2003; Saura et al, 2004). If deregulation of Cdk5 neuronal activity, through its association with p25, leads to neuronal degeneration and cell death, then a specific inhibitor of the p25/Cdk5 complex might serve as an effective means of preventing the abnormal hyperphosphorylation of neuronal cytoskeletal proteins such as tau and the development of neuronal pathologies of AD and other neurodegenerative disorders.
Our previous studies identified a central fragment of p35 (residues 154–279), called CIP, that can effectively inhibit p25/Cdk5 activity in vitro and also in transfected HEK293 cells (Amin et al, 2002; Zheng et al, 2002). An essential issue was to determine if CIP exhibits a similar specific inhibitory role in primary neurons and in situations in which Cdk5 activity is deregulated.
In a number of different experimental situations involving rat cortical neurons, we have clearly shown that CIP specifically inhibited the abnormally activated p25/Cdk5 complex without affecting the normally acting p35/Cdk5 complex. This is true in neurons coinfected with CIP and the respective complexes, but more significantly in neurons containing endogenous Cdk5 that has been activated by Aβ1−42. Previous reports have shown that the toxic peptides derived from the amyloid precursor protein (APP), particularly Aβ1−42, cause the production of p25, which then activates Cdk5 to cause tau hyperphosphorylation (Lee et al, 2000; Hashiguchi et al, 2002). We have confirmed the effect of Aβ1−42 on neurons showing that p35 is converted in part to p25 with the consequent stimulation of p25/Cdk5 hyperphosphorylation of tau. Our results show that CIP specifically inhibited this endogenous p25/Cdk5-induced tau hyperphosphorylation. Most noteworthy, however, is that cortical neurons coinfected with p35/Cdk5 and p35/Cdk5/CIP exhibited similar activation of Cdk5 histone phosphorylation and endogenous tau phosphorylation, but coinfected CIP had no inhibitory effect on Cdk5 activity in these neurons.
CIP can also play a role in neuronal survival by inhibiting elements of the apoptotic pathways. Infection of p25/Cdk5 in cortical neurons leads to an increased level of cleaved caspase-3, a marker of apoptosis. Coinfection of CIP in these cells prevented the overexpression of cleaved caspase-3. Additionally, Aβ1−42 treatment of cortical neurons also induced neuronal apoptosis marked by an increase of cleaved caspase-3 levels. Infected CIP prevented this increase presumably by inhibiting p25/Cdk5 activity, since roscovitine, a Cdk5 inhibitor, also prevented cleaved caspase-3 overexpression. CIP inhibition of Aβ-induced apoptosis was more dramatically seen in TUNEL assays where virtually all cells transfected with CIP survive the Aβ treatment while 90–95% of uninfected cells die.
Alternative scenarios for neuronal apoptosis have been proposed that do not necessarily stem from the deregulation of Cdk5 activity. Neuronal cell death in neurodegenerative disorders and in many experimentally induced stress conditions has been attributed to an attempt by post-mitotic neurons to enter the cell cycle (Vincent et al, 1996, 1997, 2003; Busser et al, 1998; Pei et al, 2002; Love, 2003). We believe that the data presented here satisfactorily demonstrated that CIP specifically inhibited p25/Cdk5 without affecting Cdc2 activity since CIP had no effect on the modestly increased Cdc2 activity observed in Aβ1−42-treated neurons. Furthermore, we have also explored the effect of CIP on other cyclin-dependent kinases involved in the cell cycle: Cdk2, Cdk4, and Cdk6. Here, too, CIP had no inhibitory effect, even on the modest elevations of activity seen after Aβ treatment. These results are consistent with our previous observations that transfected CIP in cycling HEK cells had no effect on the Cdc2 activity and cell proliferation (Zheng et al, 2002). Our data, however, do not eliminate the possibility that some neuronal cell death occurs as a result of aberrant recruitment of post-mitotic neurons into the cell cycle.
How can we account for this specificity since both p35 and p25 share identical central residues corresponding to the CIP peptide? The reasons for this specificity are not fully understood, but a hypothesis to explain the specificity may be that the N-terminal p35 domain, which is absent in p25, could obstruct CIP access to the Cdk5 active site. In vitro binding experiments with Cdk5, p25 or p35 in the presence and absence of CIP, coupled to kinetic analysis and crystallography, might provide a clue as to the mechanisms underlying CIP specificity.
To be therapeutically useful, CIP has to only target p25/Cdk5 but not p35/Cdk5 and also avoid toxic and nonspecific effects seen with the Cdk5 inhibitors roscovitine and oloumoucine. In our hands, CIP has already overcome a number of hurdles: CIP is specific for p25/Cdk5 and not p35/Cdk5 or other cyclin-dependent kinases, and we have shown that it specifically targets the aberrant Cdk5 activity induced by Aβ1−42, a factor inducing tau hyperphosphorylation and cell death. Using CIP to combat the deleterious effects of p25/Cdk5 in the AD and p25 transgenic mouse models would be of great interest. This would provide a possible therapeutic route for intervention to prevent or reduce the effects of the neurodegenerative diseases where aberrant p25/Cdk5 hyperactivity or Aβ1−42- induced pathology is involved.
Materials and methods
Plasmids and constructs with lentiviral vectors and virus infection
The lentiviral vector packaging system has previously been described (Dull et al, 1998). Vector DNA was constructed from the third-generation self-inactivating vector DNA LV-lac (Pfeifer et al, 2001) by replacing the LacZ gene between Xba1 and BamH1 with a polylinker Asc1-Swa1-Age1-Nhe1-Pme1-PspOM1-BsiW1 to create LV-CMV-Link. The CIP, Cdk5, p25 and p35 coding regions were cloned after PCR into LV-CMV-Link between Asc1 and BsiW1. The p35 promoter was as described previously (Tanaka et al, 2001) and was inserted 5′ of the CIP sequence. The procedure used for the production of viruses has been described previously (Dull et al, 1998).
Primary cultures of cortical neurons and treatment
Primary cultures of rat cortical neurons were prepared from E18 rat fetuses as described previously (Kesavapany et al, 2004). After 7 days, neurons were infected using the lentiviral vector packaging system, independently or coinfected with the appropriate constructs. After infection for 3 days, neurons were treated with roscovitine (15 μM) (BIOMOL) or aged Aβ1−42 (incubated at 37°C for 7 days before being used) (10 μM) (Sigma) for 6 h. The cells were fixed for immunohistochemistry analyses or lysed with lysis buffer for IP and Western blot analyses.
Transient transfection of 3-DIC cortical neurons
Cortical neuronal cultures were prepared and plated as described earlier. Neurons were transfected with EV-p35P-GFP or CIP-p35P-GFP using Lipofectamine 2000 following the manufacturer's instructions.
Antibodies
Anti-Cdk5 (J-3, C-8), anti-p35 (C-19), anti-Cdc2 and anti-tau phospho-serine 404 antibodies were obtained from Santa Cruz Biotechnology Inc. Anti-PS199/202 and TAU-5 mAb antibodies were obtained from Biosource International Inc. and used at 1:1000 and 1:500 dilutions, respectively. AT-8 antibody was purchased from Innogenetics and used at 1:500. Anti-cleaved caspase-3 (Asp175) and anti-phospho-Akt (Ser473) antibodies were obtained from Cell Signaling Technologies and anti-tubulin antibody from Sigma was used at 1:2000 dilution. Anti-PSTAIRE antibody was obtained from Upstate Cell Signaling Solutions and 4 μg was used to immunoprecipitate Cdc2.
Western blot analysis
Cells lysates and Western blotting analysis were carried out as previously described (Kesavapany et al, 2004).
Immunoprecipitation and Cdc2/Cdk5 kinase assays in vitro
IPs and kinase assays were performed as described previously (Veeranna et al, 1998; Kesavapany et al, 2004).
Recombinant expression of GST-CIP
Expression and purification of the GST and GST-CIP fusion protein were performed essentially according to the manufacturer's instructions (AP Biotech). Briefly, CIP was generated by PCR inserted into the pGEX recombinant plasmid and expressed and purified as described previously (Amin et al, 2002). Prior to the kinase assays, GST-CIP was incubated with the Cdks/Cdk5 immunoprecipitate for 1 h at 30°C before addition of [γ-32P]ATP.
Immunofluorescence staining
Immunofluorescence was performed as described previously (Kesavapany et al, 2004). Fluorescent images were obtained with a Zeiss LSM-510 laser-scanning confocal microscope and images were managed with Adobe Photoshop.
In situ cell death detection (TUNEL and cleaved caspase-3 staining assays)
After primary cortical neurons were cultured and transfected using Lipofectamine 2000 (Invitrogen), TUNEL staining was performed according to the manufacturer's instructions using the In situ cell death detection kit TMR red (Roche). TUNEL staining and cleaved caspase-3 fluorescent images were captured with a Zeiss LSM-510 laser-scanning confocal microscope and images were managed with Adobe Photoshop. Cell counts were performed as described in figure legends.
Supplementary Material
Supplementary figure
Acknowledgments
We thank Dr Carolyn Smith of the NINDS Light Imaging Facility for his assistance with confocal microscopy studies. We thank Dr Toshio Ohshima (RIKEN, Japan) for the gift of the p35 promoter plasmid and Dr Li Huei Tsai (Harvard Medical School, Boston, MA) for p35 and Cdk5 cDNA constructs.
References
- Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD (2000) Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci USA 97: 2910–2915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez A, Toro R, Caceres A, Maccioni RB (1999) Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death. FEBS Lett 459: 421–426 [DOI] [PubMed] [Google Scholar]
- Amin ND, Albers W, Pant HC (2002) Cyclin-dependent kinase 5 (cdk5) activation requires interaction with three domains of p35. J Neurosci Res 67: 354–362 [DOI] [PubMed] [Google Scholar]
- Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, Tsai LH, Kwon YT, Girault JA, Czernik AJ, Huganir RL, Hemmings HC Jr, Nairn AC, Greengard P (1999) Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature 402: 669–671 [DOI] [PubMed] [Google Scholar]
- Bu B, Li J, Davies P, Vincent I (2002) Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann-Pick type C murine model. J Neurosci 22: 6515–6525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busciglio J, Lorenzo A, Yeh J, Yankner BA (1995) beta-Amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 14: 879–888 [DOI] [PubMed] [Google Scholar]
- Busser J, Geldmacher DS, Herrup K (1998) Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci 18: 2801–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH (2003) Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40: 471–483 [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME (1999) Cellular survival: a play in three Akts. Genes Dev 13: 2905–2927 [DOI] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH (2001) A decade of CDK5. Nat Rev Mol Cell Biol 2: 749–759 [DOI] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L (1998) A third-generation lentivirus vector with a conditional packaging system. J Virol 72: 8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J (2003) The relationship between amyloid and tau. J Mol Neurosci 20: 203–206 [DOI] [PubMed] [Google Scholar]
- Hashiguchi M, Saito T, Hisanaga S, Hashiguchi T (2002) Truncation of CDK5 activator p35 induces intensive phosphorylation of Ser202/Thr205 of human tau. J Biol Chem 277: 44525–44530 [DOI] [PubMed] [Google Scholar]
- Julien JP, Mushynski WE (1998) Neurofilaments in health and disease. Prog Nucleic Acid Res Mol Biol 61: 1–23 [DOI] [PubMed] [Google Scholar]
- Kerokoski P, Suuronen T, Salminen A, Soininen H, Pirttila T (2002) Cleavage of the cyclin-dependent kinase 5 activator p35 to p25 does not induce tau hyperphosphorylation. Biochem Biophys Res Commun 298: 693–698 [DOI] [PubMed] [Google Scholar]
- Kesavapany S, Amin N, Zheng YL, Nijhara R, Jaffe H, Sihag R, Gutkind JS, Takahashi S, Kulkarni A, Grant P, Pant HC (2004) p35/cyclin-dependent kinase 5 phosphorylation of ras guanine nucleotide releasing factor 2 (RasGRF2) mediates Rac-dependent extracellular signal-regulated kinase 1/2 activity, altering RasGRF2 and microtubule-associated protein 1b distribution in neurons. J Neurosci 24: 4421–4431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S (2000) Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem 275: 17166–17172 [DOI] [PubMed] [Google Scholar]
- Kwon YT, Gupta A, Zhou Y, Nikolic M, Tsai LH (2000) Regulation of N-cadherin-mediated adhesion by the p35–Cdk5 kinase. Curr Biol 10: 363–372 [DOI] [PubMed] [Google Scholar]
- Lau LF, Seymour PA, Sanner MA, Schachter JB (2002) Cdk5 as a drug target for the treatment of Alzheimer's disease. J Mol Neurosci 19: 267–273 [DOI] [PubMed] [Google Scholar]
- Lee KY, Clark AW, Rosales JL, Chapman K, Fung T, Johnston RN (1999) Elevated neuronal Cdc2-like kinase activity in the Alzheimer disease brain. Neurosci Res 34: 21–29 [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405: 360–364 [DOI] [PubMed] [Google Scholar]
- Li BS, Ma W, Jaffe H, Zheng Y, Takahashi S, Zhang L, Kulkarni AB, Pant HC (2003a) Cyclin-dependent kinase-5 is involved in neuregulin-dependent activation of phosphatidylinositol 3-kinase and Akt activity mediating neuronal survival. J Biol Chem 278: 35702–35709 [DOI] [PubMed] [Google Scholar]
- Li BS, Sun MK, Zhang L, Takahashi S, Ma W, Vinade L, Kulkarni AB, Brady RO, Pant HC (2001) Regulation of NMDA receptors by cyclin-dependent kinase-5. Proc Natl Acad Sci USA 98: 12742–12747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Faibushevich A, Turunen BJ, Yoon SO, Georg G, Michaelis ML, Dobrowsky RT (2003b) Stabilization of the cyclin-dependent kinase 5 activator, p35, by paclitaxel decreases beta-amyloid toxicity in cortical neurons. J Neurochem 84: 347–362 [DOI] [PubMed] [Google Scholar]
- Love S (2003) Neuronal expression of cell cycle-related proteins after brain ischaemia in man. Neurosci Lett 353: 29–32 [DOI] [PubMed] [Google Scholar]
- Michaelis ML, Dobrowsky RT, Li G (2002) Tau neurofibrillary pathology and microtubule stability. J Mol Neurosci 19: 289–293 [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Lariviere RC, Julien JP (2001) Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron 30: 135–147 [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Sayyaparaju MR, Smulson ME, Yamin TT, Yu VL, Miller DK (1995) Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376: 37–43 [DOI] [PubMed] [Google Scholar]
- Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna, Nixon R, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K (2003) Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38: 555–565 [DOI] [PubMed] [Google Scholar]
- Ohshima T, Gilmore EC, Longenecker G, Jacobowitz DM, Brady RO, Herrup K, Kulkarni AB (1999) Migration defects of cdk5(−/−) neurons in the developing cerebellum is cell autonomous. J Neurosci 19: 6017–6026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402: 615–622 [DOI] [PubMed] [Google Scholar]
- Pei JJ, Braak H, Gong CX, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF (2002) Up-regulation of cell division cycle (cdc) 2 kinase in neurons with early stage Alzheimer's disease neurofibrillary degeneration. Acta Neuropathol (Berl) 104: 369–376 [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Brandon EP, Kootstra N, Gage FH, Verma IM (2001) Delivery of the Cre recombinase by a self-deleting lentiviral vector: efficient gene targeting in vivo. Proc Natl Acad Sci USA 98: 11450–11455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales JL, Nodwell MJ, Johnston RN, Lee KY (2000) Cdk5/p25(nck5a) interaction with synaptic proteins in bovine brain. J Cell Biochem 78: 151–159 [DOI] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Rao BS, Chattarji S, Kelleher RJ III, Kandel ER, Duff K, Kirkwood A, Shen J (2004) Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42: 23–36 [DOI] [PubMed] [Google Scholar]
- Sharma P, Veeranna, Sharma M, Amin ND, Sihag RK, Grant P, Ahn N, Kulkarni AB, Pant HC (2002) Phosphorylation of MEK1 by cdk5/p35 down-regulates the mitogen-activated protein kinase pathway. J Biol Chem 277: 528–534 [DOI] [PubMed] [Google Scholar]
- Tanaka T, Veeranna, Ohshima T, Rajan P, Amin ND, Cho A, Sreenath T, Pant HC, Brady RO, Kulkarni AB (2001) Neuronal cyclin-dependent kinase 5 activity is critical for survival. J Neurosci 21: 550–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon A, Yu H, Wang L, Rogaeva E, Sato C, Chishti MA, Kawarai T, Hasegawa H, Chen F, Davies P, Fraser PE, Westaway D, St George-Hyslop PH (2003) Brain levels of CDK5 activator p25 are not increased in Alzheimer's or other neurodegenerative diseases with neurofibrillary tangles. J Neurochem 86: 572–581 [DOI] [PubMed] [Google Scholar]
- Town T, Zolton J, Shaffner R, Schnell B, Crescentini R, Wu Y, Zeng J, DelleDonne A, Obregon D, Tan J, Mullan M (2002) p35/Cdk5 pathway mediates soluble amyloid-beta peptide-induced tau phosphorylation in vitro. J Neurosci Res 69: 362–372 [DOI] [PubMed] [Google Scholar]
- Veeranna, Amin ND, Ahn NG, Jaffe H, Winters CA, Grant P, Pant HC (1998) Mitogen-activated protein kinases (Erk1,2) phosphorylate Lys-Ser-Pro (KSP) repeats in neurofilament proteins NF-H and NF-M. J Neurosci 18: 4008–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I, Jicha G, Rosado M, Dickson DW (1997) Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci 17: 3588–3598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I, Pae CI, Hallows JL (2003) The cell cycle and human neurodegenerative disease. Prog Cell Cycle Res 5: 31–41 [PubMed] [Google Scholar]
- Vincent I, Rosado M, Davies P (1996) Mitotic mechanisms in Alzheimer's disease? J Cell Biol 132: 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BC, Lubec G (2001) p25 protein in neurodegeneration. Nature 411: 763–764, discussion 764–765 [DOI] [PubMed] [Google Scholar]
- Zheng YL, Li BS, Amin ND, Albers W, Pant HC (2002) A peptide derived from cyclin-dependent kinase activator (p35) specifically inhibits Cdk5 activity and phosphorylation of tau protein in transfected cells. Eur J Biochem 269: 4427–4434 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure