

Abstract
Retinoblastoma gene product (pRB) plays critical roles in regulation of the cell cycle and tumor suppression. It is known that downregulation of pRB can stimulate carcinogenesis via abrogation of the pRB pathway, although the mechanism has not been elucidated. In this study, we found that Mdm2, a ubiquitin ligase for p53, promoted ubiquitin-dependent degradation of pRB. pRB was efficiently ubiquitinated by wild-type Mdm2 in vivo as well as in vitro, but other RB family proteins were not. Mutant Mdm2 with a substitution in the RING finger domain showed dominant-negative stabilization of pRB. Both knockout and knockdown of Mdm2 caused accumulation of pRB. Moreover, Mdm2 inhibited pRB-mediated flat formation of Saos-2 cells. Downregulation of pRB expression was correlated with a high level of expression of Mdm2 in human lung cancers. These results suggest that Mdm2 regulates function of pRB via ubiquitin-dependent degradation of pRB.
Keywords: Mdm2, p53, RB protein, tumor suppressor, ubiquitin ligase
Introduction
The ubiquitin–proteasome system controls the abundance of a number of cellular proteins (Hershko, 1997), particularly short-lived regulatory proteins such as IκBα (Yaron et al, 1998; Hatakeyama et al, 1999), β-catenin (Kitagawa et al, 1999), p27Kip1 (Carrano et al, 1999; Nakayama et al, 2000) and cyclins (Koepp et al, 2001). Polyubiquitinated proteins conjugated as a result of collaboration among a ubiquitin-activating enzyme (E1), a ubiquitin-conjugation enzyme (E2) and a ubiquitin ligase (E3) are selectively recognized and hydrolyzed by the 26S proteasome (Hershko, 1997). The specificity of target protein selection is defined by ubiquitin ligases (Deshaies, 1999; Joazeiro and Weissman, 2000). The tumor suppressor gene product p53 is differentially regulated by two distinct types of ubiquitin ligase: one is a HECT-type ligase and the other is a RING finger-type ligase. In the presence of papilloma virus E6 protein, p53 is ubiquitinated by E6AP, a HECT-type ubiquitin ligase (Scheffner et al, 1993). In the absence of E6, Mdm2, a RING finger-type ubiquitin ligase, ubiquitinates p53 (Honda et al, 1997; Fang et al, 2000; Honda and Yasuda, 2000). Recently, it has been reported that Pirh2 (Leng et al, 2003) and COP1 (Dornan et al, 2004) are also involved in ubiquitin-dependent degradation of p53. p53 transcriptionally regulates Pirh2 and COP1 as well as Mdm2. The Mdm2 gene is an oncogene found to be amplified in 30–40% of human sarcomas without mutations in the p53 gene (Oliner et al, 1992). Degradation of p53 protein is thought to be accelerated by the overexpression of Mdm2 in these tumors. In addition, ARF, one of the alternative products of the INK4/ARF locus, binds to Mdm2 and thereby prevents it from antagonizing p53 (Pomerantz et al, 1998; Honda and Yasuda, 1999). The INK4/ARF, which is frequently mutated or deleted in human cancers, is a typical tumor suppressor gene (Quelle et al, 1995). Another product of this locus is the CDK inhibitor p16ink4a, which regulates the retinoblastoma protein (pRB) pathway to inhibit cyclin D–Cdk4/6 kinases. At the late G1 phase, cyclin D–Cdk4/6 kinases phosphorylate and inactivate pRB, which plays a central role in the pRB pathway (Kamb, 1995; Weinberg, 1995; Kitagawa et al, 1996). Then, the target transcription factors are released from pRB and activate the transcription of growth-promoting genes such as Cyclin E and DHFR, thus promoting cell cycle progression from G1 to S phase. Downregulation of pRB should induce abrogation of cell cycle regulation followed by acceleration of the transformation of normal cells to malignant cells. Thus, proteins that promote the degradation of pRB, such as ubiquitin ligase, are thought to exhibit oncogenic activity. We attempted to identify ubiquitin ligase(s) for pRB. pRB binds to many nuclear proteins, including Mdm2 and BRCA1, which have ubiquitin ligase activities. However, the physiological significance of their association with pRB has not been clarified. Here we provide in vivo evidence that Mdm2 is a ubiquitin ligase for pRB. We also provide evidence of downregulation of pRB by Mdm2.
Results
Mdm2 facilitates ubiquitination of pRB in vivo
It has been reported that Mdm2 is one of the binding proteins of pRB (Xiao et al, 1995), although the physiological significance was not clarified. First, to investigate whether endogenous pRB can bind to Mdm2, we performed immunoprecipitation following immunoblotting assays (IP/IB). As shown in Figure 1A, endogenous pRB was detected in the immunoprecipitate with anti-Mdm2 antibody. Alternatively, endogenous Mdm2 was detected in the immunoprecipitate with anti-pRB antibody (Figure 1B). These results confirm that endogenous pRB binds to endogenous Mdm2. We next investigated whether Mdm2 is a ubiquitin ligase for pRB. We previously reported that Mdm2 shows ubiquitin ligase activity via the RING finger domain and promotes p53 ubiquitination (Honda et al, 1997; Honda and Yasuda, 1999; 2000). In that study, we showed that mutant Mdm2 (mt-Mdm2) with a substitution of alanine for cysteine 438 in the RING finger motif lacked ubiquitin ligase activity for p53. As shown in Figure 2A, wild-type human Mdm2 (wt-Mdm2) or mt-Mdm2 expression plasmid was transfected into NIH3T3 cells with a full-length RB plasmid and an HA-tagged ubiquitin plasmid. Immunoprecipitated pRB was clearly ubiquitinated in the presence of wt-Mdm2 but not in the presence of mt-Mdm2 (Figure 2A, lanes 1–5), whereas both wt- and mt-Mdm2 were bound to pRB (Figure 2A, lanes 6–10). The pRB-bound Mdm2 activity was monitored by autoubiquitination of Mdm2 (Figure 2A, lanes 6–10). On the other hand, other known ubiquitin ligases such as Fbw1/FWD1, MD6 and Skp2 could not promote ubiquitination of pRB (data not shown). BRCA1 is also a ubiquitin ligase that is known to bind to pRB. However, even BRCA1-NT (N-terminal region of BRCA1) and BARD1, another ubiquitin ligase functioning as a cofactor for BRCA1 (Hashizume et al, 2001), did not ubiquitinate pRB when these proteins and HA-Ub were expressed together with pRB in HEK293 cells (Figure 2B, lanes 7 and 12). BRCA1-NT retained BARD1-dependent ubiquitin ligase activity (Supplementary Figure 1). Thus, only Mdm2 enhanced the ubiquitination of pRB. In vivo ubiquitination of pRB by Mdm2 was also detected in HEK293 cells (Figure 2B) and HCT116 cells (data not shown). Importantly, ubiquitination of endogenous pRB was observed as shown in Figure 2C. Its ubiquitination was facilitated by transfection of wt-Mdm2 but not mt-Mdm2. These results suggest that Mdm2 is a ubiquitin ligase of pRB.
Figure 1.
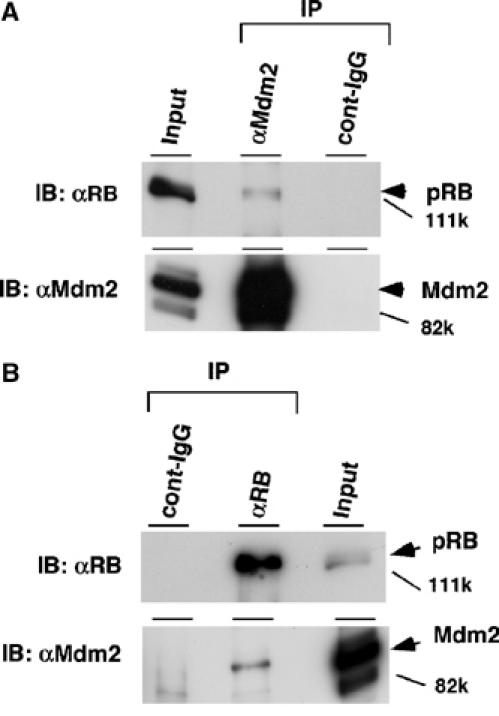
Endogenous Mdm2 is associated with endogenous pRB. Exponentially growing HCT116 cells were treated with 20 μM MG115, a proteasome inhibitor, for 8 h. The cell lysates were subjected to IP with anti-Mdm2 (SMP-14) (A) or anti-pRB (G3-245) (B) and control immunoglobulin. The immunoprecipitates were separated by SDS–6% PAGE and then subjected to IB with anti-Mdm2 (SMP-14) or anti-pRB (G3-245) for detection of endogenous pRB and Mdm2. In all, 1% of the total lysate for IP was loaded in an input lane.
Figure 2.
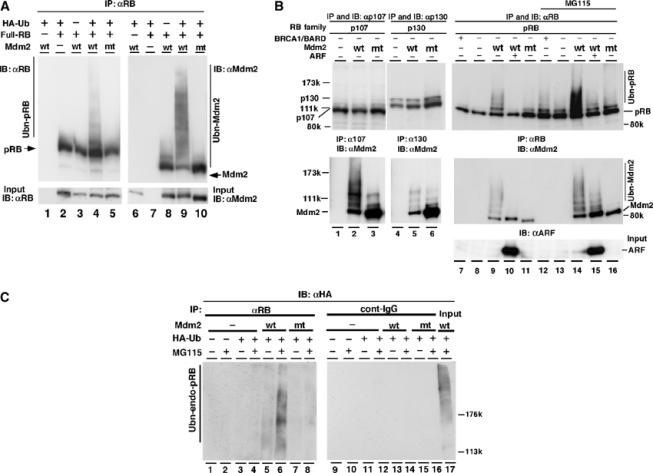
Mdm2 facilitates ubiquitination of pRB in vivo. (A) The full-length human RB expression plasmid pcDNA4-HisMax-hRB (full-RB) was transfected together with pcDNA4-HisMax wild-type (wt) or pcDNA4-HisMax-RING-finger mutant Mdm2C438A (mt) and pCGN-HA-ubiquitin (HA-Ub) into NIH3T3 cells. At 40 h after transfection, cells were incubated with or without 20 μM MG115 for 8 h. The immunoprecipitates obtained with anti-pRB antibody (G3-245) from cell lysates were separated by SDS–7% PAGE and detected by IB with anti-pRB (lanes 1–5) or anti-Mdm2 (lanes 6–10). In all, 3% of the cell lysate for IP was used in an input sample. wt-Mdm2 possessing self-ubiquitination activity promoted ubiquitination of pRB. (B) Full-length RB family expression plasmids of pRB, p107 or p130 were transfected into HEK293 cells with or without the plasmids of Mdm2 (wt or mt), BRCA1-NT (BRCA1) and myc-ARF (ARF). BRCA1-NT, an N-terminal region of BRCA1, is known to retain the ubiquitin ligase activity (Hashizume et al, 2001). To activate BRCA1-NT, BARD1, a cofactor for BRCA1, was coexpressed. Plasmid of HA-Ub was cotransfected in all lanes. The immunoprecipitate obtained with anti-pRB (C-15), anti-p107 or anti-p130 was subjected to IB with anti-pRB (C-15), anti-p107 or anti-p130. Co-immunoprecipitated Mdm2 was monitored with anti-Mdm2 antibodies. In all, 2% of the lysate used for IP was loaded in an input lane and analyzed with anti-ARF antibody. (C) To detect ubiquitination of endogenous pRB, HCT116 cells were transfected with or without the plasmids of HA-Ub or Mdm2 (wt or mt). At 40 h after transfection, cells were incubated with or without 20 μM MG115 for 10 h. The cell lysates were immunoprecipitated (IP) with anti-pRB antibody (G3-245) or control immunoglobulin (cont-IgG) and separated by SDS–6% PAGE. Ubiquitination of endogenous pRB was detected by IB with anti-HA (12CA5) antibody.
Specificities of ubiquitination of pRB by Mdm2
To evaluate the specificity of ubiquitination of pRB by Mdm2, we performed an in vivo ubiquitination assay using RB family proteins p107 and p130 (Ashizawa et al, 2001), which have not been shown to interact with Mdm2. Interestingly, both p107 and p130 recruited Mdm2 but were not ubiquitinated by Mdm2 (Figure 2B, lanes 1–6). In addition, both knockout and knockdown of Mdm2 caused accumulation of pRB but not of p107 and p130 proteins (Figures 5A and 6A). Thus, these results suggest that Mdm2 selectively ubiquitinates pRB among RB family tumor suppressor proteins.
Figure 5.
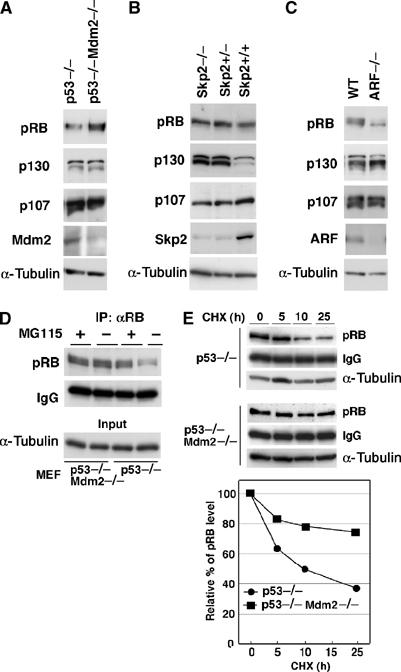
Accumulation of pRB in Mdm2−/− MEFs. Lysates prepared from p53−/− or p53−/−Mdm2−/− (A), Skp2+/+, Skp2+/− or Skp2−/− (B) or wild-type (WT) or ARF−/− (C) MEFs were subjected to IB with antibodies against p130, p107, Mdm2 and α-tubulin. pRB was immunoprecipitated with anti-pRB antibody and analyzed with the antibody. (D) MEFs were treated with or without 20 μM MG115 for 10 h. pRB was immunoprecipitated with anti-pRB antibody and analyzed by immunoblotting with the antibody. α-Tubulin levels in 6% of the total material used are shown in input lanes. (E) MEFs were treated with 10 μg/ml of cycloheximide for the indicated time, and the endogenous pRB levels were analyzed as described in Materials and methods.
Figure 6.
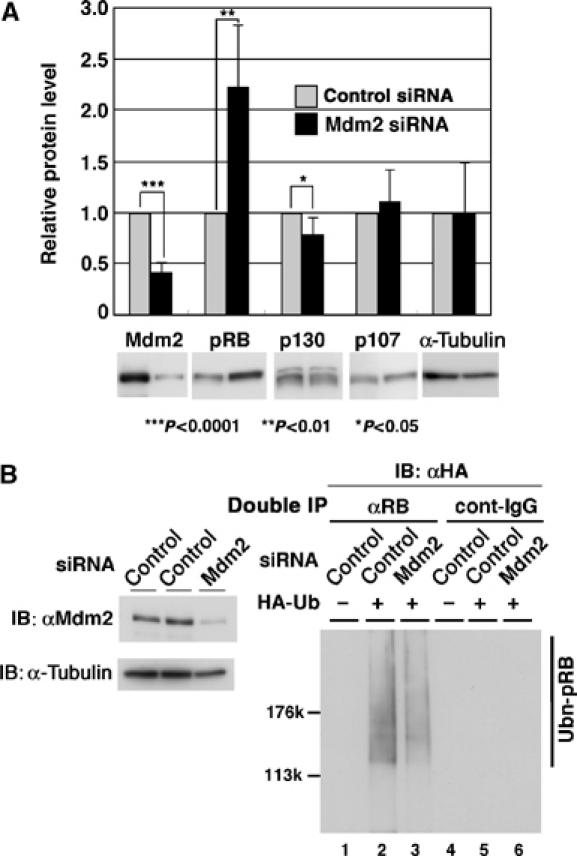
Effects of Mdm2 siRNA on ubiquitination of pRB. (A) Knockdown of Mdm2 results in accumulation of pRB. U2OS cells were multiply transfected with 40 nM siRNAs targeting human Mdm2 (Mdm2 siRNA) or control oligo RNA (control siRNA) as described in Materials and methods. Cell lysates were subjected to IB with antibodies as indicated. Quantitative data are represented as fold values of control siRNA and means±s.e. of 3–4 separate experiments. (B) Knockdown of Mdm2 interferes with ubiquitination of pRB. In vivo ubiquitination of pRB in U2OS cells, in which Mdm2 is downregulated by RNAi, was performed. At 24 h after the third transfection of siRNA, the expression plasmids of pRB and HA-Ub were transfected into the cells. The levels of Mdm2 in siRNA-treated cells were analyzed by IB with anti-Mdm2 or anti-α-tubulin antibody (left panel). Double IP with anti-pRB antibody (αRB) or control immunoglobulin (cont-IgG) was performed as described in Materials and methods. The precipitates were then subjected to IB with anti-HA antibody for the detection of ubiquitinated pRB (right panel).
ARF inhibits Mdm2-mediated ubiquitination of pRB
It has been reported that ARF protein inhibits both in vitro ubiquitination and in vivo downregulation of p53 protein via abrogation of Mdm2 activity (Pomerantz et al, 1998; Honda and Yasuda, 1999). We found that ARF also inhibited ubiquitination of pRB by Mdm2 (Figure 2B, top panel, lanes 10 and 15). Autoubiquitination of Mdm2 bound to pRB was also inhibited by ARF (Figure 2B, middle panel, lanes 10 and 15). Ubiquitination of p53 by Mdm2 was also negatively regulated by ARF in vivo (data not shown).
In vitro ubiquitination of pRB by Mdm2
We next tested whether Mdm2 ubiquitinates pRB in an in vitro ubiquitination system. As shown in Figure 3A, ubiquitination of recombinant pRB was facilitated by lysates of Sf9 cells expressing wt-Mdm2 (lanes 1 and 3), but not mt-Mdm2 (lane 4). Furthermore, pRB bound to glutathione S-transferase (GST)-wt-Mdm2, which was purified with glutathione beads after in vitro ubiquitination reaction, was ubiquitinated as well as p53, as shown in Figure 3B. In contrast, pRB bound to GST-mt-Mdm2 was scarcely ubiquitinated. We then tried to reconstitute the in vitro ubiquitination of pRB using purified components. Purified GST-Mdm2 proteins and recombinant full-length pRB were incubated with E1, 6xHis-UbcH5 (E2) and biotinylated ubiquitin in an in vitro ubiquitination buffer. Immunoprecipitated pRB was ubiquitinated by wt-Mdm2 but not by mt-Mdm2 in this reconstitution assay (Figure 3C, right). These findings suggest that Mdm2 is a ubiquitin ligase of pRB as well as p53. Interestingly, ubiquitination of pRB by Mdm2 was decreased in the presence of p53 in vitro (Figure 3C, lanes 2 and 4) as well as in vivo (Supplementary Figure 2). On the other hand, ubiquitination of p53 was diminished in the presence of pRB (Supplementary Figure 2). Thus, p53 and pRB may compete with each other for the Mdm2-mediated ubiquitination.
Figure 3.
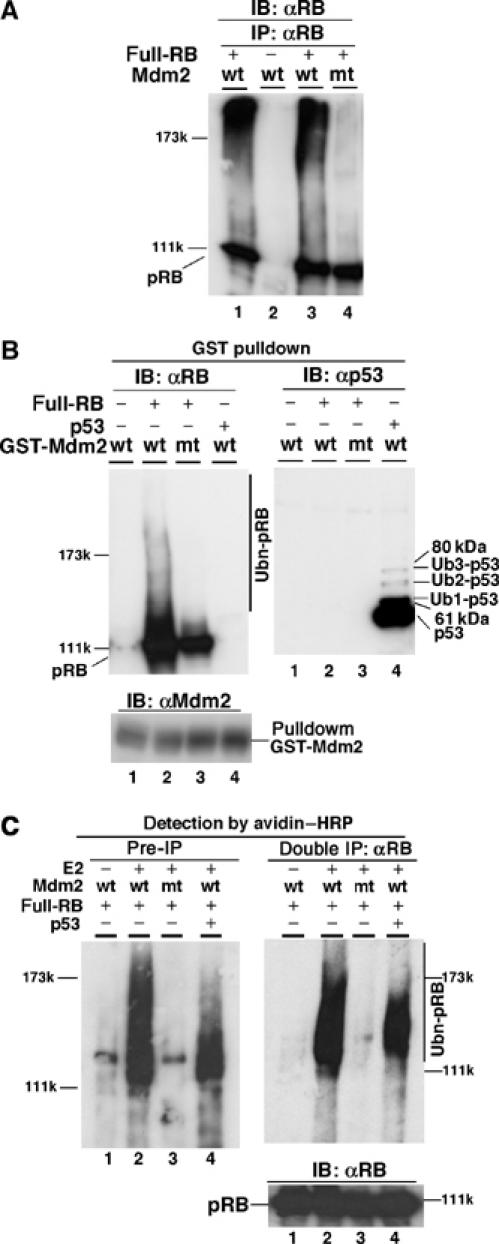
In vitro ubiquitination of recombinant pRB by Mdm2. (A) Ubiquitination of pRB by Mdm2 in vitro. Lysates of Sf9 cells expressing GST-wt-Mdm2 or mt-Mdm2 were incubated with recombinant full-length pRB together with E1, recombinant 6xHis-UbcH5, ubiquitin and 10 mM ATP in an in vitro ubiquitination buffer. pRB was immunoprecipitated with anti-pRB (C-15) antibody and analyzed by IB with anti-pRB antibody. (B) Ubiquitination of Mdm2-bound pRB. In vitro ubiquitination assay was performed as described in (A). GST-Mdm2-pRB or -p53 complex was then pulled down with glutathione beads, and analyzed by IB with anti-pRB (C-15), anti-Mdm2 (SMP14) or anti-p53 (Pab240) antibodies. (C) Reconstitution of pRB ubiquitination using purified proteins. Purified GST-Mdm2 and recombinant full-length pRB were incubated with E1, UbcH5 (E2) and biotinylated ubiquitin with or without purified p53 in the in vitro ubiquitination buffer. After denaturation, pRB was immunoprecipitated two times with anti-pRB antibody (3H9, MBL) and separated by SDS–6% PAGE. Ubiquitination of pRB in both the reaction mixture (Pre-IP) and immunoprecipitates (Double IP) was detected by avidin–HRP. Immunoprecipitated pRB was analyzed with the anti-pRB antibody.
Facilitation of pRB degradation by Mdm2
We next examined the effect of a proteasome inhibitor on the levels of endogenous pRB in HCT116 cells (Figure 4A). The proteasome inhibitor MG115 accumulated endogenous pRB as well as endogenous Mdm2. We then carried out pulse-chase experiments to determine whether Mdm2 affects the half-life of pRB (Figure 4B). RB-RI, an N-terminal region-truncated pRB, was used in this experiment since the expression of full-length pRB and incorporation of [35S]methionine were not sufficient for the signals of full-length pRB to be detectable. As shown in Supplementary Figure 3A, RB-RI retains both the pocket domain of full-length pRB and the C-terminal Mdm2-binding domain (Xiao et al, 1995). It has also been reported that RB-RI has the same activity as that of full-length pRB, such as flat formation activity in Saos cells (Qin et al, 1992). Since the downregulation of pRB by Mdm2 required the C-terminal region of pRB (Supplementary Figure 3B), RB-RI should be degraded in almost the same manner as that of full-length pRB. Figure 4B shows that the introduction of wt-Mdm2 increased the rate of turnover of RB-RI in vivo. In contrast, MG115 treatment suppressed the downregulation of RB-RI by Mdm2. Furthermore, mt-Mdm2 exhibited a dominant-negative effect on the degradation of RB-RI. Consistent with this result, the results of chase experiments with cycloheximide treatment revealed that degradation of full-length pRB in SRB1-mt-Mdm2 cells, pRB-inducible and mt-Mdm2-expressing cells, was slower than in control SRB1 cells (Figure 4C). The induced levels of pRB in SRB1 cells were also increased in mt-Mdm2-overexpressing cells but were decreased in wt-Mdm2-overexpressing cells. pRB in the wt-Mdm2-overexpressing cells was stabilized in the presence of MG115 (Miwa et al, manuscript in preparation). These findings suggest that Mdm2 promotes degradation of pRB through the ubiquitin–proteasome system.
Figure 4.
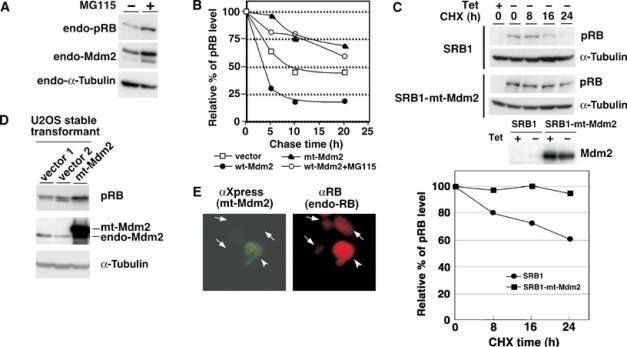
Mdm2-mediated degradation of pRB. (A) Accumulation of pRB by treatment with a proteasome inhibitor. Exponentially growing HCT116 cells were treated with 20 μM MG115 for 8 h. Endogenous proteins were detected by IB with antibodies as indicated. (B) Enhancement of the degradation of pRB by Mdm2. Pulse-chase experiments were performed as described in Materials and methods. The amount of pRB (RB-RI) at 0 h was defined as 100%. RB-RI, an N-terminus-truncated pRB, was used in this experiment since the expression of full-length pRB and incorporation of [35S]methionine were not sufficient for the signals of full-length pRB to be detectable. (C) Increase by mt-Mdm2 in the half-life of pRB. Full-length pRB was induced in control SRB1 (SRB1) and SRB1-mt-Mdm2 (SRB1-mt-Mdm2) cells by depletion of tetracycline for 48 h from the maintaining medium. Then, the cells were treated with 10 μg/ml of cycloheximide (CHX) for the indicated time. Cell lysates were subjected to IB with anti-pRB or anti-α-tubulin antibody. The amount of full-pRB at 0 h was defined as 100%. (D) Accumulation of endogenous pRB in U2OS cells expressing mt-Mdm2. The amount of pRB in U2OS cells stably expressing mt-Mdm2 (mt-Mdm2) or an empty vector (vector 1 and 2) was examined by IB. (E) The level of endogenous pRB was increased by mt-Mdm2. Human glioblastoma T98G cells, in which p53 is mutated, were transfected with pcDNA4-HisMax-mt-Mdm2. After 48 h, cells were subjected to immunostaining with anti-Xpress antibody for transfected Mdm2 (green) and anti-RB (C-15) for endogenous pRB (red).
Inhibition of Mdm2 activity results in accumulation of endogenous pRB
To confirm that Mdm2 contributes to degradation of pRB in physiological conditions, we examined whether the endogenous levels of pRB are accumulated in mt-Mdm2-expressing cells. We generated U2OS cells stably expressing mt-Mdm2 or an empty vector. As shown in Figure 4D, endogenous pRB was accumulated in mt-Mdm2-expressing cells but not in control cells (vector 1 and 2). The levels of pRB in the control cells were similar to those in wild-type U2OS cells (data not shown). In addition, endogenous pRB (αRB; red) in T98G cells, which were transiently transfected with the plasmid pcDNA4-HisMax-mt-Mdm2, was stabilized in mt-Mdm2 (αXpress; green)-expressing cells (Figure 4E).
For further confirmation that Mdm2 contributes to degradation of pRB, we investigated endogenous pRB levels in Mdm2-knockout mouse embryonic fibroblasts (MEFs). We used p53−/−Mdm2−/− MEFs, since Mdm2−/− MEFs have not been established because Mdm2-knockout mice die during the early embryonic stage. As shown in Figure 5A, pRB was accumulated more in p53−/−Mdm2−/− MEFs than in p53−/− MEFs, whereas the levels of p130 and p107 were unchanged, consistent with the results showing that p130 and p107 were not targets of Mdm2 (Figure 2B). The levels of pRB and p107 were not affected by depletion of Skp2 (Skp2−/− MEF) (Nakayama et al, 2000) (Figure 5B), but p130 was stabilized in Skp2−/− MEFs, supporting a recent study that Skp2 targets p130, leading to proteasome-dependent degradation of p130. In ARF−/− MEFs (Kamijo et al, 1997), pRB levels were lower than those in wild-type cells, suggesting that enhancement of Mdm2 activity caused facilitated down-regulation of pRB (Figure 5C). MG115 caused accumulation of pRB in p53−/− MEFs, whereas it did not increase the levels of pRB in p53−/−Mdm2−/− MEFs (Figure 5D). This indicates that pRB was stabilized in p53−/−Mdm2−/− cells escaping from ubiquitination followed by proteasome-dependent degradation.
We then tested whether the half-life of pRB is prolonged in p53−/−Mdm2−/− MEFs. A chase experiment by treatment of cells with cycloheximide revealed that the degradation rate of pRB in p53−/−Mdm2−/− MEFs was slower than that in p53−/− MEFs (Figure 5E). Taken together, these results suggest that endogenous Mdm2 is involved in degradation of endogenous pRB.
Abrogation of Mdm2 by siRNA stabilizes endogenous pRB
We further investigated the effects of knockdown of Mdm2 by siRNA on pRB levels. U2OS cells were transfected with Mdm2 siRNA or control siRNA oligo, and the cell lysates were subjected to IB with antibodies against pRB, p130, p107 and α-tubulin. As shown in Figure 6A, Mdm2 levels were decreased, whereas pRB was accumulated in Mdm2-knock down cells. In contrast, the levels of p130, p107 and α-tubulin were unchanged. To determine whether knockdown of Mdm2 interfered with ubiquitination of pRB, we measured pRB ubiquitination activity in Mdm2-knockout cells using the double IP method as described in Materials and methods. In vivo ubiquitination of pRB was reduced in Mdm2-knockdown cells (Figure 6B, right panel). Thus, Mdm2 functions in ubiquitination and degradation of pRB.
Given the contribution of Mdm2 to degradation of pRB, downregulation of Mdm2 could alter the rate of proliferation of affected cells. We examined the effects of Mdm2 siRNA on the rate of cell proliferation in U2OS cells or H1299 cells, a p53-null cell line (Supplementary Figure 4). The rate of proliferation of U2OS cells was decreased in Mdm2-knockdown cells. A decrease in the rate of proliferation by knockdown of Mdm2 was also observed in H1299 cells. Furthermore, the downregulation of Mdm2 altered the cell cycle profile, causing reduction of population at S phase of H1299 cells.
Mdm2 interferes with the pRB pathway
To investigate whether Mdm2 regulates pRB function in vivo, we introduced a wt- or a mt-Mdm2 expression vector into SRB-1 cells, pRB-inducible Saos-2 cells (Figure 7). It is known that ectopic expression of pRB in Saos-2 cells, an RB-deficient osteosarcoma cell line, causes flat cell formation (Ookawa et al, 2001). SRB-1 cells are stable transformants containing human RB cDNA driven by a tetracycline-regulated promoter (Ookawa et al, 1997; 2001). Therefore, SRB-1 cells in which pRB expression is induced by depletion of tetracycline exhibit flat cell formation. To identify the cells expressing Mdm2 and detect pRB-mediated flat cell formation, we used an expression vector that permits both Mdm2 and EGFP cDNA to be translated from a single bicistronic mRNA, which should then make Mdm2-expressing cells identifiable by EGFP fluorescence. Depletion of tetracycline caused flat cell formation in approximately 65% of EGFP-positive cells with mock transfection (Figure 7Ba–d and C), whereas expression of wt-Mdm2 significantly prevented formation of the enlarged flat cell phenotype (Figure 7Be, g and C). In contrast, mt-Mdm2 permitted flat cell formation in 70% of the cells (Figure 7Bf, h and C). In EGFP-negative cells, which had not been efficiently transfected with the expression vector, 70% of the cells showed the flat phenotype (data not shown). In these experiments, pRB levels were lower in wt-Mdm2-expressing cells than in nontransfected cells (Figure 7A, left). On the contrary, pRB levels were high in mt-Mdm2-transfected cells compared to the levels in nontransfected cells (Figure 7A, right), consistent with the results showing that dominant-negative effect of mt-Mdm2 resulted in pRB accumulation (Figure 4). These observations suggest that Mdm2 may inhibit pRB function in vivo via its ubiquitin ligase activity against pRB, although the possibility that Mdm2 indirectly regulates pRB function cannot be ruled out.
Figure 7.
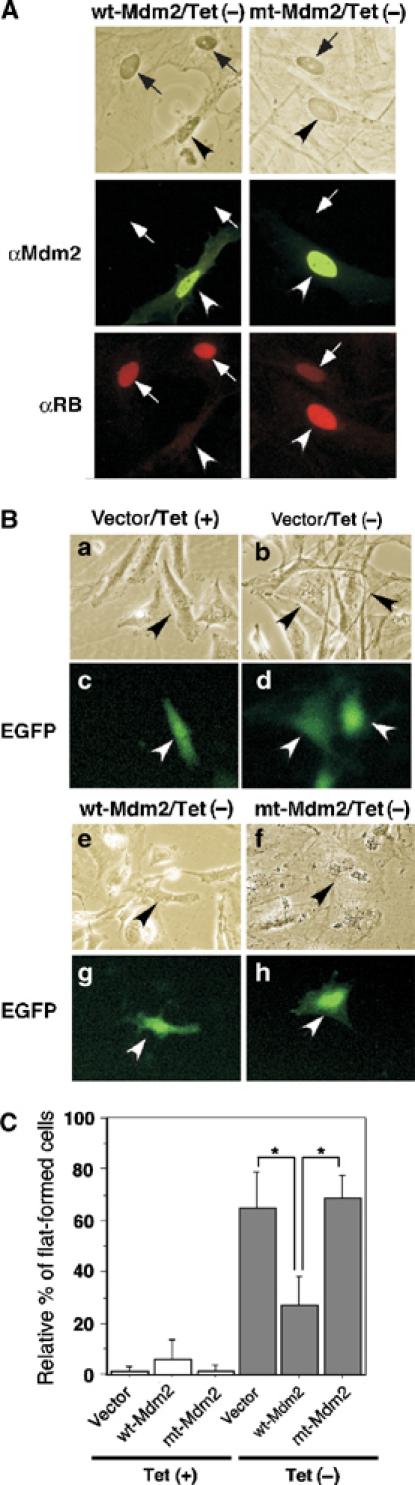
Mdm2 inhibits pRB-mediated flat cell formation. (A) Cellular pRB levels were downregulated by Mdm2. SRB1 cells were transfected with pcDNA4-HisMax-Mdm2 (wt or mt). After transfection, the cells were cultured for 7 days in 10% FBS–DMEM medium without (Tet−) 1 μg/ml of tetracycline for induction of pRB. Cells were subjected to immunostaining with anti-Mdm2 (green) or anti-RB (red). (B) Mdm2 inhibited pRB-mediated flat formation of osteosarcoma cells. SRB1 cells were transfected with pIRES2-EGFP-wtMdm2 (wt-Mdm2), pIRES2-EGFP-mtMdm2 (mt-Mdm2) or pIRES2-EGFP (vector) as described in Materials and methods. After transfection, the cells were cultured in 10% FBS–DMEM medium with (Tet+) or without (Tet−) 1 μg/ml of tetracycline for induction of pRB. Cells were photographed at day 7 or 8 after transfection using a phase-contrast microscope (a, b, e, f) and a fluorescent microscope (c, d, g, h) at × 200 magnification. The arrows indicate EGFP-positive cells. (C) The ratio of the number of flat cells among EGFP-positive cells to the total number of EGFP-positive cells (100%) was calculated. The number of cells was counted in 6–10 fields for each experiment. All values are means±s.e. of 8–10 separate experiments. *P<0.001.
Effects of Mdm2 on pRB expression in human non-small lung cancers
In order to evaluate the effects of Mdm2 on pRB expression in human clinical tumors, immunohistochemical analysis of the expressions of both pRB and Mdm2 in non-small cell lung carcinoma tissues from 30 patients was carried out. Two representative patterns of immunostaining with anti-pRB and anti-Mdm2 antibodies are shown in Figure 8A. In sample no. 18, an adenocarcinoma tissue sample, Mdm2 signal was negative and pRB signal was clearly positive. On the other hand, Mdm2 signal was positive and pRB signal was negative in sample no. 2, a squamous cell carcinoma tissue sample. We graded the levels of Mdm2 in the 30 tissue samples as low, moderate and high. Eight of the 30 tissue samples showed signs of Mdm2 overexpression, and Mdm2 expression level was low in eight samples. The other 14 samples showed a moderate expression level of Mdm2. Statistical analysis of all samples using Fisher's method indicated that expression levels of pRB were significantly decreased in Mdm2-high tissues compared to those in Mdm2-low tissues (P=0.0013) (Figure 8B). pRB expression in Mdm2-moderate tissues was in the middle level of that in Mdm2-high and Mdm2-low tissues. In normal tissues of the same patients, overexpression of Mdm2 was not observed. These results suggest that Mdm2 promotes downregulation of pRB in human lung cancer. We then determined LOH of pRB and the status of p53 in these tumors by SSCP analysis among six Mdm2-high tumors described above (Miwa et al, manuscript in preparation). Expression levels of p16ink4a proteins were also determined by immunohistochemical analysis among six Mdm2-high tumors and six Mdm2-low tumors described above (Miwa et al, manuscript in preparation). It has been shown that the levels of p16ink4a and pRB are high and low, respectively, in many human cervical cancers (Klaes et al, 2001). LOH of pRB was found in only one case, the p53 genes were mutants in four cases and the p16ink4a levels were high in three cases of the Mdm2-high tumors. In contrast, the p16ink4a levels were at the background level in all of the Mdm2-low tumors. The p16ink4a levels were also at the background level in normal tissues.
Figure 8.
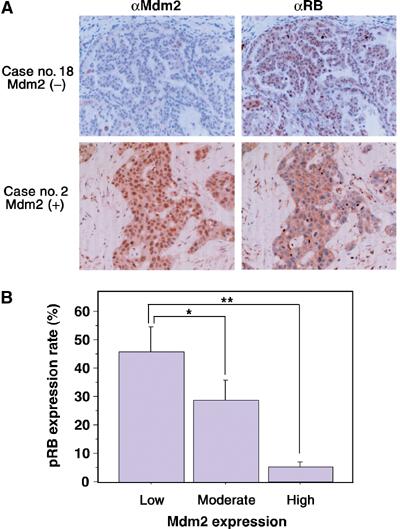
Inverse correlation between pRB and Mdm2 expressions in human lung cancers. (A) Immunohistochemical staining for pRB and Mdm2 in human non-small cell lung cancer tissues was carried out using streptavidin–biotin–peroxidase complex methods as described in Materials and methods. Original magnification is × 400. (B) The graph shows the levels of pRB expression of non-small cell lung carcinomas (n=30). pRB expressions were classified according to Mdm2 expression (low, moderate or high). Eight of the 30 tissue samples showed signs of Mdm2 overexpression, and Mdm2 expression was low in eight samples. The other 14 samples showed a moderate expression level of Mdm2. pRB expression levels were significantly decreased in Mdm2-high samples. *P<0.05, **P<0.005.
Discussion
pRB, which plays a major role in the pRB pathway, negatively regulates cell cycle progression from G1 to S phase. As shown by many previous studies, pRB is mainly regulated via the phosphorylation of pRB by G1 cyclin-dependent kinases at restriction points (Weinberg, 1995). In this study, we found that Mdm2 promoted ubiquitination and degradation of pRB. Gene disruption of Mdm2 and downregulation of Mdm2 by siRNA caused accumulation of endogenous pRB in cells (Figures 5 and 6). A series of our results implies that ubiquitin-dependent degradation by Mdm2 is another mechanism of downregulation of pRB function.
Our collaborator Yasuda et al found that Mdm2 is a ubiquitin ligase of p53 (Honda et al, 1997). There have been many reports on Mdm2-mediated regulation of p53 (Michael and Oren, 2003). Alternatively, it has been suggested that Mdm2 has p53-independent functions in cell cycle regulation, differentiation, DNA repair, basal transcription and other biological processes (Ganguli and Wasylyk, 2003). Mdm2 has been shown to be a pRB-binding protein (Xiao et al., 1995), but the physiological function of this binding has not been elucidated. As shown in Figure 1, we confirmed that endogenous Mdm2 bound with endogenous pRB. In Supplementary Figure 3, it is shown that Mdm2-mediated degradation of pRB required the C-terminal region of pRB, which was bound to Mdm2. In addition, the pRB-binding domain in Mdm2 was required for ubiquitination of pRB (data not shown). Therefore, binding of pRB with Mdm2 is necessary for degradation of pRB. In contrast, RB family proteins, p107 and p130, were not ubiquitinated by Mdm2 but they were associated with Mdm2 (Figure 2B). Moreover, knockout or knockdown of Mdm2 by siRNA caused accumulation of endogenous pRB but not p107 and p130 in cells (Figure 5A and B). Disruption of Skp2 caused accumulation of p130, supporting the previous finding that a ubiquitin ligase of p130 is SCFSkp2 as reported by Tedesco et al (2002). These data suggest that the Mdm2-mediated ubiquitination of both pRB and p53 is specific. Dubs-Poterszman et al (1995) reported that Mdm2 abrogates p107-mediated G1 arrest in Saos-2 cells. Because Mdm2 has no ubiquitin ligase activity for p107 and since both p53 and RB were inactivated in Saos-2 cells, another function of Mdm2 might also be involved in the p107-mediated arrest.
Activity of pRB is mainly regulated via phosphorylation of pRB by cyclin-dependent kinases at the restriction point in the late G1 phase of the normal cell cycle. The proteolytic degradation of pRB by Mdm2 may play a role in the regulation of the amounts of active pRB in cells to maintain the threshold level of active pRB. When the Mdm2 levels or its activities increase, by the gene amplification of Mdm2 or inactivation of ARF, for example, the threshold level of pRB might become lower, thereby facilitating progression from G1 to S phase and causing accelerated proliferation. Activation of Mdm2 through phosphorylation of serine residues in Mdm2 (Zhang and Prives, 2001; Zhou et al, 2001) via some signal transduction pathways, including the Akt and MAP kinase pathways, might induce degradation of pRB, thereby accelerating the G1–S transition. The decrease in cellular pRB, caused by loss or mutation of the gene or by its degradation, would perturb the pRB pathway, one of two key pathways for tumor suppression. The results shown in Figure 7 suggest that the overexpression of Mdm2 suppressed the pRB function in SRB1 (Saos2 with inducible pRB) cells, because inhibition of flat formation of these cells indicates interference with the pRB pathway (Ookawa et al, 1997). Since Saos-2 cells have no normal p53 protein, it is likely that Mdm2 facilitates degradation of pRB and suppresses the pRB pathway in these cells. Besides, downregulation of Mdm2 by siRNA reduced proliferation of U2OS cells (wild-type p53-positive cells) and H1299 cells (p53-negative cells) regardless of p53 status. These observations suggest that Mdm2 inhibits pRB function in vivo via its ubiquitin ligase activity against pRB, although the possibility that Mdm2 indirectly regulates pRB function cannot be ruled out. Taking into consideration that Mdm2 has been known to abrogate p107-mediated G1 arrest in Saos-2 cells as described above (Dubs-Poterszman et al, 1995), the effect of Mdm2 knockdown on the cell cycle profile might also be explained by interference with p107 activity.
There are several reports of Mdm2 being a possible regulator of both p53 and pRB. Yap et al (1999) speculated in their review that pRB may protect p53 from Mdm2-mediated degradation. As shown in Figure 3C, ubiquitination of pRB was suppressed in the presence of p53. Reciprocally, in vivo ubiquitination of p53 was inhibited in the presence of pRB (Supplementary Figure 2). p53 and pRB may compete with each other for their ubiquitination mediated by Mdm2, whereas Mdm2 binds to p53 and pRB through different regions on Mdm2 protein. These findings may support the results obtained by Hsieh et al (1999) and Yap et al (2000) showing that pRB overcomes the antiapoptotic function of Mdm2 against p53. On the other hand, it is also likely that p53 negatively regulates pRB via Mdm2-dependent degradation, since Mdm2 is one of the target genes of p53. Taken together, these results suggest that Mdm2 is a key molecule that bridges pRB and p53 as reported in previous reports (Yap et al, 1999; Ganguli and Wasylyk, 2003).
The simultaneous abrogation of the activities of two major tumor suppressor pathways, the pRB and the p53 pathways, might cooperatively induce transformation of cells. Our pathological study showed the reciprocal correlation of pRB expression with Mdm2 expression in human non-small cell lung cancers. In normal tissues of the same patients, overexpression of Mdm2 was not observed. LOH of pRB was not found in most of Mdm2-overexpressing tumors. Moreover, some Mdm2-overexpressing tumors with mutant p53 also suppress the pRB levels (Miwa et al, manuscript in preparation). These results suggest that Mdm2 contributes to degradation of pRB in human cancer cells. As Mdm2 is known to be an oncogene product, our findings might imply that downregulation of pRB by Mdm2 is also involved in carcinogenesis. Further study will be required to clarify whether downregulation of pRB by Mdm2 contributes to cell transformation and carcinogenesis.
Materials and methods
Plasmids
A clone of the full-length human pRB cDNA was recloned into pcDNA4-HisMax (Invitrogen). pcDNA4-HisMax-hRB-RI encoding 6xHis-Xpress-tagged N-terminus-truncated pRB, designated RB-RI, was constructed as follows. The full-length human pRB cDNA was recloned into pBluescript II. The resultant pBluescript II-RB was digested with EcoRI and NotI to yield the region of pRB cDNA encoding amino acids 301–949, and the DNA fragment was then ligated into the EcoRI–NotI site of the pcDNA4/HisMax vector, which has a 6xHis tag and an Xpress tag at the N-terminus. Plasmid of 3xFlag pRB was generated by inserting the full length of pRB cDNA in pcDNA3.1 containing 3xFlag tag sequences. Expression vectors of BRCA1-NT (N-terminal) and BARD1 were kindly provided by T Ohta (Hashizume et al, 2001). Expression vectors of p107 and p130 were kindly provided by M Hatakeyama (Ashizawa et al, 2001). To construct the plasmids pcDNA4/HisMax-wt-Mdm2 and pcDNA4/HisMax-mt-Mdm2 (C438A), the BamHI–EcoRI fragment of each cDNA was subcloned into the BamHI–EcoRI site of pcDNA4/HisMax plasmid.
Cell lines
The SRB1 cell line, which is a full-pRB-inducible clone of Saos-2 cells regulated by tetracycline (Tet-Off system) (Ookawa et al, 1997; 2001), and Skp2−/−, Skp2+/− and Skp2+/+ MEFs (Nakayama et al, 2000) were kindly provided by Dr Ookawa and Dr Nakayama, respectively. p53−/−, p53−/−Mdm2−/− (Jones et al, 1996) and ARF−/− MEFs (Kamijo et al, 1997) were previously reported. A U2OS stable cell line expressing mt-Mdm2 was selected with zeocine. The SRB1-mt-Mdm2 cell line, a stable transfectant with pcDNA4/HisMax-mt-Mdm2, was established from SRB1 by screening with zeocine. SRB1 and SRB1-mt-Mdm2 cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 0.5 mg/ml of G418, 0.3 mg/ml of hygromycin, 1 μg/ml of tetracycline and 0.3 mg/ml of zeocine (for SRB1-mt-Mdm2). HEK293 cells, HCT116 cells and U2OS cells were cultured in DMEM containing 10% FBS. NIH3T3 cells were cultured in DMEM containing 10% calf serum.
In vitro ubiquitination
Recombinant wt- and mt-Mdm2 proteins fused with GST were produced in insect cells using a baculovirus system. The cell lysates were used in the in vitro ubiquitination assay in Figure 3A and B. For the reconstitution assay in Figure 3C, the GST proteins were purified using glutathione beads. Purified GST-Mdm2 and recombinant full-length pRB (QED Bioscience) were incubated with E1 (Boston Biochem.), recombinant 6xHis-UbcH5, ubiquitin or biotinylated ubiquitin in an in vitro ubiquitination buffer (50 mM Tris–HCl, pH 8.3, 2 mM dithiothreitol, 5 mM MgCl2, 10 mM ATP, 1 mM phosphocreatinine, 0.2 U/ml phosphocreatine kinase, 4 mM calpain inhibitor I (LLnL), 25 μg/ml ubiquitin aldehyde and 10 μg/ml protease inhibitors) at 30°C for 30 min. The reactions were terminated and the proteins were denatured in SDS sample buffer at 100°C for 10 min.
In vivo ubiquitination
In vivo ubiquitination of pRB was assayed as follows. All plasmids were transfected into HCT116 cells in 10-cm dishes by lipofection with Lipofectamine-plus reagent (Invitrogen) or FuGENE6 (Roche Applied Science) or into HEK293 cells and NIH3T3 cells in 10-cm dishes by the calcium phosphate method. The cell lysates were prepared with lysis buffer A (50 mM Tris–HCl, pH 7.5, 300 mM NaCl, 0.5% Triton X-100, 10 μg/ml each of antipain, pepstatin, E-64, leupeptin and chymostatin, 400 μM Na3VO4, 10 mM NaF and 10 mM sodium pyrophosphate) 48 h after transfection. To induce accumulation of polyubiquitinated pRB, cells were treated with a proteasome inhibitor, MG115 (20 μM), for 8–10 h starting at 38–40 h after transfection and then harvested. The cell lysates were incubated with 2 μg of anti-pRB antibody (C-15, Santa Cruz, or G3-245, BD Pharmingen) and protein G–Sepharose 4FF (Amersham Biochem) at 4°C for 2 h. The immunocomplexes were washed four times with lysis buffer A. In the case of double IP, the first immunocomplex was denatured by treatment with SDS sample buffer at 100°C for 10 min. Then, ubiquitinated pRB was reimmunoprecipitated with anti-pRB antibody (C-15 or G3-245), separated by SDS–PAGE (6 or 7%) and transferred from the gel onto a PVDF membrane. Proteins were detected by IB with the antibodies anti-HA (12CA4, Roche), anti-pRB (C15, IF8 or G3-245) or anti-Xpress antibody followed by treatment with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit antibody (Promega) and visualized using an enhanced chemiluminescence system (PerkinElmer).
Pulse-chase experiment
A 7 μg portion of RB-RI plasmid was transfected with 14 μg of an empty vector, wt-Mdm2 or mt-Mdm2 into HEK293 cells. At 42 h after transfection, cells were metabolically labeled with Trans[35S] (ICN Pharmaceuticals Inc.) at 87.5 m Ci/ml for 1 h and chased for the indicated times with or without MG115 (20 μM). Cell lysates were subjected to IP with anti-pRB antibody (C-15) followed by purification with protein G–Sepharose, separated by SDS–7% PAGE, exposed to an imaging plate and analyzed quantitatively using BAS-1000 (Fuji Film).
For the chase experiment with cycloheximide, MEFs were treated with 10 μg/ml of cycloheximide for the indicated time. SRB1 and SRB1-mt-Mdm2 cells were treated in the same way 48 h after depletion of tetracycline. pRB in MEFs was immunoprecipitated with anti-pRB antibody (G3-245), and the amounts of precipitated pRB were examined by IB analysis. Other cell lysates were subjected to IB with anti-RB or anti-α-tubulin antibody (Sigma).
RNA interference
U2OS cells grown in six-well plates at 80–90% confluence were incubated in 1.8 ml/well of DMEM containing 10% FBS. An 80 pmol portion of dsRNA oligos (40 nM/well) in 0.2 ml of Opti-MEM I medium (Invitrogen) containing 3.6 μl of Lipofectamine 2000 (Invitrogen) was added to the medium in each well. After transfection for 12 h, the medium was replaced with 2 ml of fresh DMEM containing 10% FBS and the culture was continued for another 24 h. The cells were then treated with trypsin–EDTA to be regrown in new six-well plates for 12 h, following the next transfection in the same way as described above. Transfection was carried out repeatedly three times in a single assay. The nucleotide sequences of the siRNA for Mdm2 were 5′-r(AAGGAAUAAGCCCUGCCCA)d(TT)-3′.
Flat cell formation assay
wt- or mt-Mdm2 cDNA was ligated into pIRES2-EGFP plasmid (Clontech), which contains an internal ribosome entry site that permits both the gene of interest and the EGFP gene to be translated from a single bicistronic mRNA, to generate the plasmid pIRES2-EGFP-wtMdm2 or pIRES2-EGFP-mtMdm2. SRB1 cells were transfected with pIRES2-EGFP-wtMdm2 (wt-Mdm2), pIRES2-EGFP-mtMdm2 (mt-Mdm2) or pIRES2-EGFP (Vector) with FuGENE6 transfection reagent. After transfection, the cells were cultured in a six-well plate with (Tet+) or without (Tet−) 1 μg/ml of tetracycline for 7 days.
Immunohistochemistry
For immunohistochemical analysis, 5-μm-thick formalin-fixed, paraffin-embedded sections were deparaffinized, dehydrated and placed in a microwave oven for 40 min in citric acid solution (10 mM, pH 6). The slides were incubated with 0.3% H2O2 in absolute methanol for 30 min to quench endogenous peroxidase activity. The sections were incubated with primary antibodies against Mdm2 (1B10, Novocastra) or RB (3H9, MBL) overnight at 4°C. A biotin-conjugated secondary antibody was added to the sections for 30 min. A streptavidin–biotin–peroxidase complex and a chromogen solution containing diaminobenzidine were applied to each section for 10 s to 2 min according to the manufacturer's instructions (Histofine SAB kit, Nichirei). The sections were lightly counterstained with hematoxylin and mounted with a permanent mounting medium. For the evaluation of pRB and Mdm2 expression, the number of stained nuclei was counted. At least 10 high-power fields were chosen randomly and scored for the percentage of cells in each section in which more than 100 cells were stained.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Acknowledgments
We thank Shigetsugu Hatakeyama, Masanori Hatakeyama, Keiichi Nakayama, Keiko Nakayama, Hideaki Higashi, Michiko Shirane, Mariko Mihara and Tomohiko Ohta for gifts of reagents, cell lines, plasmids and helpful discussions, and Sayuri Suzuki, Yuichi Sakairi, Nao Inami, Eri Kinoshita, Yosuke Honda, Mamoru Kobayashi and Daisuke Yamada for technical assistance. This work was supported in part by grants from the Ministry of Education, Science, Sports, Culture and Technology of Japan (MK and KK), by a COE program of Hamamatsu University School of Medicine from the Ministry of Education, Science, Sports, Culture and Technology of Japan (MK), Astra Zeneca Research Grant (MK) and by grants from Novartis Foundation (MK) and Uehara Foundation (MK).
References
- Ashizawa S, Nishizawa H, Yamada M, Higashi H, Kondo T, Ozawa H, Kakita A, Hatakeyama M (2001) Collective inhibition of pRB family proteins by phosphorylation in cells with p16INK4a loss or cyclin E overexpression. J Biol Chem 276: 11362–11370 [DOI] [PubMed] [Google Scholar]
- Carrano AC, Eytan E, Hershko A, Pagano M (1999) SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1: 193–199 [DOI] [PubMed] [Google Scholar]
- Deshaies RJ (1999) SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol 15: 435–467 [DOI] [PubMed] [Google Scholar]
- Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O'Rourke K, Koeppen H, Dixit VM (2004) The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 429: 86–92 [DOI] [PubMed] [Google Scholar]
- Dubs-Poterszman MC, Tocque B, Wasylyk B (1995) MDM2 transformation in the absence of p53 and abrogation of the p107 G1 cell-cycle arrest. Oncogene 11: 2445–2449 [PubMed] [Google Scholar]
- Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM (2000) Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem 275: 8945–8951 [DOI] [PubMed] [Google Scholar]
- Ganguli G, Wasylyk B (2003) p53-independent functions of MDM2. Mol Cancer Res 1: 1027–1035 [PubMed] [Google Scholar]
- Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, Ogata H, Ohta T (2001) The RING heterodimer BRCA1–BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem 276: 14537–14540 [DOI] [PubMed] [Google Scholar]
- Hatakeyama S, Kitagawa M, Nakayama K, Shirane M, Matsumoto M, Hattori K, Higashi H, Nakano H, Okumura K, Onoe K, Good RA, Nakayama K-i (1999) Ubiquitin-dependent degradation of IκBα is mediated by a ubiquitin ligase Skp1/Cul 1/F-box protein FWD1. Proc Natl Acad Sci USA 96: 3859–3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A (1997) Roles of ubiquitin-mediated proteolysis in cell cycle control. Curr Opin Cell Biol 9: 788–799 [DOI] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H (1997) Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 420: 25–27 [DOI] [PubMed] [Google Scholar]
- Honda R, Yasuda H (1999) Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J 18: 22–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Yasuda H (2000) Activity of MDM2, a ubiquitin ligase, toward p53 or itself is dependent on the RING finger domain of the ligase. Oncogene 19: 1473–1476 [DOI] [PubMed] [Google Scholar]
- Hsieh JK, Chan FS, O'Connor DJ, Mittnacht S, Zhong S, Lu X (1999) RB regulates the stability and the apoptotic function of p53 via MDM2. Mol Cell 3: 181–193 [DOI] [PubMed] [Google Scholar]
- Joazeiro CA, Weissman AM (2000) RING finger proteins: mediators of ubiquitin ligase activity. Cell 102: 549–552 [DOI] [PubMed] [Google Scholar]
- Jones SN, Sands AT, Hancock AR, Vogel H, Donehower LA, Linke SP, Wahl GM, Bradley A (1996) The tumorigenic potential and cell growth characteristics of p53-deficient cells are equivalent in the presence or absence of Mdm2. Proc Natl Acad Sci USA 93: 14106–14111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamb A (1995) Cell-cycle regulators and cancer. Trends Genet 11: 136–140 [DOI] [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ (1997) Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 91: 649–659 [DOI] [PubMed] [Google Scholar]
- Klaes R, Friedrich T, Spitkovsky D, Ridder R, Rudy W, Petry U, Dallenbach-Hellweg G, Schmidt D, von Knebel Doeberitz M (2001) Overexpression of p16(INK4A) as a specific marker for dysplastic and neoplastic epithelial cells of the cervix uteri. Int J Cancer 92: 276–284 [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Hatakeyama S, Shirane M, Matsumoto M, Ishida N, Hattori K, Nakamichi I, Kikuchi A, Nakayama K-i, Nakayama K (1999) An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of β-catenin. EMBO J 18: 2401–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Ikeda M, Tamai K, Kato J, Segawa K, Yoshida E, Nishimura S, Taya Y (1996) The consensus motif for phosphorylation by cyclin D1–Cdk4 is different from that for phosphorylation by cyclin A/E–Cdk2. EMBO J 15: 7060–7069 [PMC free article] [PubMed] [Google Scholar]
- Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ (2001) Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 294: 173–177 [DOI] [PubMed] [Google Scholar]
- Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, Parant JM, Lozano G, Hakem R, Benchimol S (2003) Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 112: 779–791 [DOI] [PubMed] [Google Scholar]
- Michael D, Oren M (2003) The p53–Mdm2 module and the ubiquitin system. Semin Cancer Biol 13: 49–58 [DOI] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, Shirane M, Tsunematsu R, Tsukiyama T, Ishida N, Kitagawa M, Nakayama K-i, Hatakeyama S (2000) Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J 19: 2069–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B (1992) Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 358: 80–83 [DOI] [PubMed] [Google Scholar]
- Ookawa K, Tsuchida S, Adachi J, Yokota J (1997) Differentiation induced by RB expression and apoptosis induced by p53 expression in an osteosarcoma cell line. Oncogene 14: 1389–1396 [DOI] [PubMed] [Google Scholar]
- Ookawa K, Tsuchida S, Kohno T, Yokota J (2001) Alterations in expression of E2F-1 and E2F-responsive genes by RB, p53 and p21(Sdi1/WAF1/Cip1) expression. FEBS Lett 500: 25–30 [DOI] [PubMed] [Google Scholar]
- Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee HW, Cordon-Cardo C, DePinho RA (1998) The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell 92: 713–723 [DOI] [PubMed] [Google Scholar]
- Qin X-Q, Chittenden T, Livingston DM, Kaelin WG Jr (1992) Identification of growth suppression domain within the retinoblastoma gene product. Genes Dev 6: 953–964 [DOI] [PubMed] [Google Scholar]
- Quelle DE, Zundy F, Ashmun RA, Sherr CJ (1995) Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 83: 993–1000 [DOI] [PubMed] [Google Scholar]
- Scheffner M, Huibregtse JM, Vierstra RD, Howly PM (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75: 495–505 [DOI] [PubMed] [Google Scholar]
- Tedesco D, Lukas J, Reed SI (2002) The pRb-related protein p130 is regulated by phosphorylation-dependent proteolysis via the protein-ubiquitin ligase SCF(Skp2). Genes Dev 16: 2946–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA (1995) The retinoblastoma protein and cell cycle control. Cell 81: 323–330 [DOI] [PubMed] [Google Scholar]
- Xiao Z-X, Chen J, Levine AJ, Modjtahedi N, Xing J, Sellers WR, Livingston DM (1995) Interaction between the retinoblastoma protein and the oncoprotein MDM2. Nature 375: 694–698 [DOI] [PubMed] [Google Scholar]
- Yap DB, Hsieh JK, Chan FS, Lu X (1999) mdm2: a bridge over the two tumour suppressors, p53 and Rb. Oncogene 18: 7681–7689 [DOI] [PubMed] [Google Scholar]
- Yap DB, Hsieh JK, Lu X (2000) Mdm2 inhibits the apoptotic function of p53 mainly by targeting it for degradation. J Biol Chem 275: 37296–37302 [DOI] [PubMed] [Google Scholar]
- Yaron A, Davis M, Lavon I, Amit S, Manning AM, Andersen JS, Mann M, Mercurio F, Ben-Neriah Y (1998) Identification of the receptor component of the IκBα-ubiquitin ligase. Nature 396: 590–594 [DOI] [PubMed] [Google Scholar]
- Zhang T, Prives C (2001) Cyclin A–CDK phosphorylation regulates MDM2 protein interactions. J Biol Chem 276: 29702–29710 [DOI] [PubMed] [Google Scholar]
- Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC (2001) HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol 3: 973–982 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4