

Abstract
Streptococcus pneumoniae (pneumococcus) remains a significant health threat worldwide, especially to the young and old. While some of the biomolecules involved in pneumococcal pathogenesis are known and understood in mechanistic terms, little is known about the molecular details of bacterium/host interactions. We report here the solution structure of the ‘repeated' adhesion domains (domains R1 and R2) of the principal pneumococcal adhesin, choline binding protein A (CbpA). Further, we provide insights into the mechanism by which CbpA binds its human receptor, polymeric immunoglobulin receptor (pIgR). The R domains, comprised of 12 imperfect copies of the leucine zipper heptad motif, adopt a unique 3-α-helix, raft-like structure. Each pair of α-helices is antiparallel and conserved residues in the loop between Helices 1 and 2 exhibit a novel ‘tyrosine fork' structure that is involved in binding pIgR. This and other structural features that we show are conserved in most pneumococcal strains appear to generally play an important role in bacterial adhesion to pIgR. Interestingly, pneumococcus is the only bacterium known to adhere to and invade human cells by binding to pIgR.
Keywords: bacterial pathogenesis, calorimetry, protein–protein interactions, solution NMR, surface plasmon resonance
Introduction
Streptococcus pneumoniae (pneumococcus) remains the most common invasive bacterial agent leading to hospitalization in all age groups (Schuchat et al, 2001), with the majority of these cases affecting either children or the elderly. Penicillin remains the primary mode of treatment, but the emergence of antibiotic resistance has intensified the search for new therapeutic approaches (Whitney et al, 2000). While disease prevention through vaccination is partially effective, this approach is less effective in the youngest and oldest patients (Butler et al, 1993). Hence, studies of disease mechanisms may provide insights into new antibacterial therapies. The pneumococcus invades human nasopharyngeal epithelial (NE) cells and enters the blood stream through a process termed reverse transcytosis mediated by polymeric immunoglobulin receptor (pIgR) (Zhang et al, 2000). The normal function of pIgR is to transport secretory IgA (sIgA) from the basolateral to the apical surface of NE cells (Mostov and Kaetzel, 1999). A protein on the bacterial surface, choline binding protein A (CbpA), binds specifically to an extracellular domain of pIgR and hijacks the endocytosis machinery to translocate pneumococci across NE cells into the blood stream. While the participation of CbpA in pneumococcal adhesion and invasion is well established (Rosenow et al, 1997), the molecular details of these processes are not understood.
CbpA (also referred to as PspC, SpsA and PbcA) is one of 15 proteins identified in the genome of the TIGR4 strain (Tettelin et al, 2001) that exhibit multiple C-terminal repeats of an ∼19-amino-acid motif that binds choline moieties present on the bacterial cell wall (Gosink and Tuomanen, 2000). The mechanism of bacterial surface attachment by CbpA can be understood on the basis of structural studies of another choline binding protein (Cbp), LytA. (Note: We use the generic term CbpA to refer to the CbpA protein from the TIGR4 strain.) The C-terminal domain of LytA contains seven repeats of the choline binding motif and adopts a unique β-solenoid structure, with choline groups binding between β-hairpin ‘steps' of the staircase-like structure (Fernandez-Tornero et al, 2001). Similarly, CbpA has eight repeats of this motif in its C-terminus (Figure 1A) and is predicted to bind surface-exposed choline groups in a similar manner (Fernandez-Tornero et al, 2001). The N-terminal domains of the 15 TIGR4 Cbps differ widely, with functions ranging from bacterial autolysis (LytA) and glucosaminidase activity (LytB) to adhesion to human NE cells (CbpA) (Gosink et al, 2000; Tettelin et al, 2001). The sequence of the CbpA N-terminus (residues 39–514; Tettelin et al, 2001) exhibits numerous repeats of the leucine zipper (LZ) motif (Landschulz et al, 1988) that cluster within five domains termed A, B, R1, R2 and C (Figure 1A). Domains A, B and C are 21–25 amino acids in length and are predicted to form coiled-coil dimers (Figure 1B) (Lupas et al, 1991). The ∼110-amino-acid-long, ‘repeated' domains, R1 and R2 (78% identical) (Zhang et al, 2000), are also predicted to form self-associated, coiled-coil structures. We refer to these as ‘R' domains. The segments that connect these domains are predicted to lack secondary structure. In general, LZ motifs mediate intra- or intermolecular α-helix/α-helix interactions, and are well understood as the basis for parallel self-association into α-helical dimers, trimers and tetramers (Lupas, 1996). LZ motifs also mediate antiparallel α-helix/α-helix interactions (Oakley and Hollenbeck, 2001). To date, however, the R domains of CbpA have not been structurally characterized.
Figure 1.
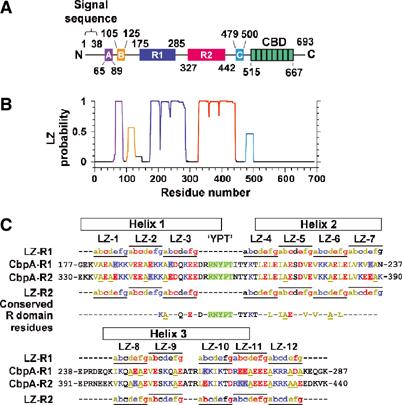
Domain structure of CbpA (from the TIGR4 strain of S. pneumoniae). (A) Domains labeled A, B, R1, R2 and C exhibit multiple repeats of the LZ motif, and the domain labeled CBD (choline binding domain) contains eight repeats of the choline binding motif. (B) LZ probability for CbpA determined using COILS (Lupas et al, 1991). (C) Sequences of domains R1 and R2. The letters a–g and horizontal lines indicate the locations of the 12 LZ heptad motifs. Within the LZ motifs, hydrophobic residues are colored brown (L, V, I and A), acidic residues red (D and E) and basic residues blue (K and R). Sites in the R1 and R2 sequences where an acidic residue is swapped for a basic residue, or vice versa, are indicated by red and blue shading, respectively. Residues of the conserved RNYPT motif are colored green and the three α-helices of CbpA-R2 are indicated by black rectangles. Residues that are conserved in 87 R domain sequences from 47 pneumococcal strains are also given.
CbpA R domains bind the extracellular, immunoglobulin-like (Ig-like) domains of pIgR termed secretory component (SC) (Hammerschmidt et al, 1997; Zhang et al, 2000). SC covalently binds to IgA dimers and, after transcytosis from the basolateral to apical surface of NE cells, the SC/IgA2 complex is proteolytically cleaved from pIgR to release sIgA (Mostov, 1994). A conserved hexapeptide motif within the R domains is required for binding to sIgA (Hammerschmidt et al, 2000). Here we present the solution structure of domain R2 of CbpA containing this motif determined using NMR spectroscopy. Further, we used this structure to model that for domain R1. Surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC) were used to gain insight into the mechanism by which CbpA binds components of pIgR. Our results provide the first molecular insights into the structurally novel mechanism by which pneumococcus binds pIgR and subsequently invades human cells.
Results
Conserved features of CbpA sequences from many pneumococcal strains
The R domains of CbpA from the TIGR4 strain of S. pneumoniae are highly conserved in CbpA sequences from other pneumococcal strains (Iannelli et al, 2002). We compared CbpA sequences (excluding the choline binding motifs) from 47 pneumococcal strains and quantified R domain conservation (Figure 2A and B). A total of 39 sequences exhibit R1 and R2 domains that are ⩾50% identical to the TIGR4 domains, six exhibit one R2-like (⩾50% identical) domain and one exhibits one R1-like (93% identical) domain. Residues at 22 positions are identical in these R1 and R2 domains, 87 in total, including five residues within the hexapeptide motif discussed earlier. Thus, our structural and biophysical results for the R domains of the TIGR4 strain are relevant to CbpA from virtually all known pneumococcal strains.
Figure 2.
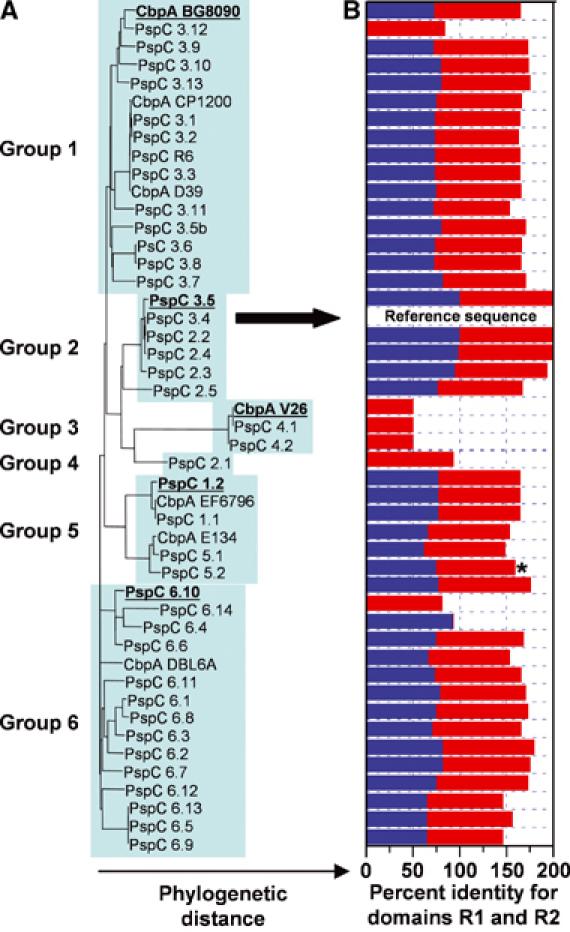
R domain structure is conserved in CbpA sequences from most pneumococcal strains. (A) Phylogenetic tree showing the relationships between 47 CbpA sequences, which cluster into six groups. The TIGR4 sequence is also termed PspC 3.4 (marked by arrow). (B) The histogram illustrates the percentage of amino-acid identity (relative to TIGR4 CbpA) for the R1 and/or R2 domains (R1, blue bars; R2, red bars). Details of this analysis are found in Supplementary data.
Solution structure of CbpA, domains R1 and R2
The secondary structure of N-terminal CbpA domains was elucidated using circular dichroism (CD) and NMR spectroscopy. Domains R1 and R2 (78% identical) each contain 12 imperfect copies of the LZ motif and span residues 175–285 and 327–442, respectively. Eight of the 24 differences between R1 and R2 correspond to substitution of Glu by Lys, or vice versa (Figure 1C). The CbpA R domain LZ motifs are similar to those found in coiled-coil proteins (Figure 1C) (Lupas et al, 1991); however, the amino acid at position d of the heptad motif, which is usually Leu in the classical LZ motif, is most frequently Ala in the R domains.
CbpA-R1 and -R2 possess extensive α-helical structure on the basis of CD spectra (Figure 3B) and we used NMR spectroscopy to determine the CbpA-R2 structure in solution. Secondary 13Cα chemical shifts for CbpA-R2 (Figure 4A), which are indicative of secondary structure, clearly revealed three α-helices: Helix 1 (residues 330–357), Helix 2 (residues 366–390) and Helix 3 (residues 396–425). These adopt an unusual three-α-helix, raft-like structure through antiparallel α-helix/α-helix interactions (Figure 4B). The α-helices, while antiparallel, are not exactly coaxial due to slight coiling (Figure 4C). The crossing angle for Helices 1 and 2 is −178° and that for Helices 2 and 3 is −176°. In addition, we generated a homology model of CbpA-R1 based on the structure of CbpA-R2. The structures of CbpA-R2 (Figure 4B) and -R1 indicate that many hydrophobic residues are buried at the two α-helix/α-helix interfaces and that the protein surfaces are dominated by polar and charged residues (Figure 4D). CbpA-R2 exhibits a bilobed electrostatic potential, with one face of the structure highly electronegative (red contours in Figure 4D, left) and the other electropositive (blue contours). The differences in charged residues between CbpA-R2 and -R1 cause increased electronegative character on one face of CbpA-R1 and reduced electropositive character on the other (Figure 4D, right).
Figure 3.
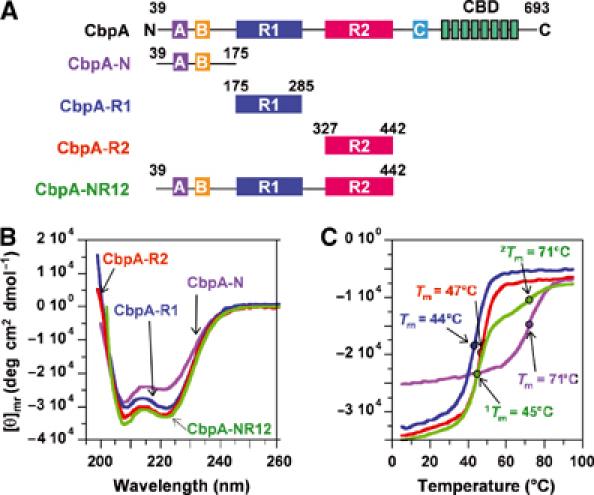
Secondary structure of CbpA domains. (A) CbpA constructs used in this study. (B) CD spectra for CbpA-N (violet trace), -R1 (blue trace), -R2 (red trace) and -NR12 (green trace). (C) Thermal denaturation traces obtained by measuring CD ellipticity at 222 nm at different temperatures. The coloring scheme is as in (B).
Figure 4.
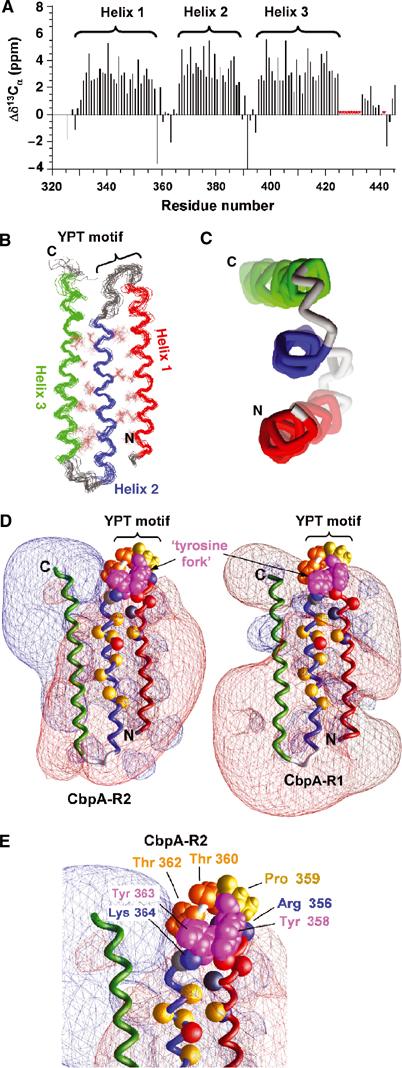
Solution structure of CbpA R domains. (A) Secondary 13Cα chemical shift values for CbpA-R2 showing the three α-helices. Resonances for residues marked by red asterisks are unassigned. (B) Superposed ensemble of 20 lowest-energy structures of CbpA-R2 obtained from solution NMR data; the backbone and select hydrophobic residues (in brown color) at the two α-helix/α-helix interfaces are illustrated. Helix 1 is colored red, Helix 2 blue and Helix 3 green. The location of the YPT motif is noted. (C) End-on view of the three α-helices of domain R2, colored as in (B). (D) Contour maps of electrostatic potentials (±1kt; red, negative potential; blue, positive potential) for CbpA-R2 (left) and homology model of CbpA-R1 (right) generated using GRASP (Nicholls et al, 1991). The α-helices are colored as in (B). Tyr 358 and 363 (magenta; labeled ‘tyrosine fork'), Pro 359 (yellow) and Thr 360 and 362 (orange) are also illustrated. The Cα atoms of other conserved residues are illustrated as colored spheres (Lys 346, Arg 356 and Lys 364 (blue); Glu 352, Asp 354 and Glu 372 (red); Gln 350 and Thr 365 (gray); Ala 347, Ile 368, Ile 370, Ala 371, Val 375, Val 377, Ala 80 and Leu 382 (yellow)). (E) Close-up view of conserved residues in loop between Helix 1 and Helix 2 of CbpA-R2 that protrude into a region of neutral electrostatic potential. Key residues are illustrated as in (D).
Peptides with the sequence RNYPT bind to sIgA and SC, and mutations in this motif in SpsA, a CbpA variant with one R domain, abolish binding to sIgA (Hammerschmidt et al, 2000). The YPT motif, which occurs once in each of the two TIGR4 CbpA R domains (Figure 1C), is found in the loop between Helix 1 and Helix 2 in the CbpA-R2 structure (Figure 4B and E). Interestingly, these and other conserved residues in the loop protrude into solvent from an electrostatically neutral region of CbpA-R2, with two partially stacked Tyr residues forming a ‘tyrosine fork' structure (Figure 4D, left, and Figure 4E). Most residues in the Helix 1/Helix 2 loop are conserved in CbpA-R1 and thus the homology model of domain R1 exhibits a similar tyrosine fork feature. While the differences in charged residues mentioned above affect the electrostatic properties of domain R1, the potentials in the vicinity of the YPT motif in this domain are very similar to those of the R2 domain (Figure 4D, right), suggesting that the negative charge of this region and the protruding YPT motif are involved in binding to pIgR. Many of the residues in this region have significant hydrophobic character, including Tyr 358 and 363, Pro 359, and Thr 360 and 362 (Figure 4E) (Nozaki and Tanford, 1971; Creighton, 1993), suggesting that hydrophobic interactions are involved in receptor binding.
Proteins that contain multiple LZ motifs often self-associate. However, the results of equilibrium analytical ultracentrifugation (AUC) experiments showed that CbpA-R1 and -R2 are predominantly monomers in solution (Supplementary Figure 1C and D). Analysis of sedimentation data for both domains did, however, provide evidence for very weak self-association at high protein concentration (Supplementary Table 1). Further, we showed using equilibrium AUC that CbpA-R1 and Cbp-R2 do not interact to form hetero-oligomers (data not shown). Importantly, these results show that the principal role of the CbpA LZ motifs is to mediate the folding, not oligomerization, of the R domains.
Structural properties of CbpA-N (residues 39–174)
The role of the N-terminal domain of CbpA (containing LZ domains A and B; Figure 1A) in adhesion and invasion is not known. To provide preliminary insights into function, we characterized the structure and self-association properties of CbpA-N (residues 39–174). CD results show that CbpA-N adopts α-helical secondary structure (Figure 3B) and that it has a stable fold (Figure 3C). As observed for CbpA-R1 and CbpA-R2, AUC analysis showed that CbpA-N is monodisperse; however, a small degree of oligomerization was observed at 0 and 50 mM NaCl (Supplementary Table 1).
The full CbpA N-terminus (residues 39–442) is comprised of three α-helical domains
We used CD to compare the structure of a 404-amino-acid fragment of CbpA (CbpA-NR12; Figure 3A) with that of the three domains within this fragment (CbpA-N, -R1 and -R2). CD spectra for CbpA-N, -R1, -R2 and -NR12 (Figure 3B) showed that all constructs are highly α-helical. Further, we monitored ellipticity at 222 nm for each construct as the temperature was raised from 5 to 95°C to characterize thermal stability. CbpA-R1 and -R2 exhibited thermal unfolding transitions at 44 and 47°C, respectively (Figure 3C). CbpA-N also exhibits a single thermal transition but at a much higher temperature, 71°C (Figure 3C). Based on these data, we conclude that CbpA-N, -R1 and -R2 are individually folded, stable α-helical domains. The thermal melting curve for CbpA-NR12 is a composite of those for the individual domains, exhibiting two thermal transitions, one at 45°C and a second at 71°C. These data suggest strongly that the structure of the individual CbpA domains is preserved in the multidomain construct CbpA-NR12.
We also compared the structure of CbpA-R2 with that of the R2 domain within CbpA-NR12 by analyzing 2D 1H-15N TROSY spectra. The spectrum of CbpA-R2 (Figure 5A) exhibited the appropriate number of resonances, most of which appeared outside the crowded central region. The resonance pattern of CbpA-R2 was well reproduced in the spectrum of CbpA-NR12. The similarity between the two spectra is illustrated by the blue overlays in Figure 5A and B; the blue layer identifies resonances that appeared at identical positions in the two spectra. In Figure 5A, the spectrum of CbpA-R2 is the red-colored layer; however, most resonances appear blue because they also appeared in Figure 5B. In Figure 5B, approximately 40 isolated resonances (excluding obvious side-chain resonances) are colored blue, indicating that they also appeared in Figure 5A. CbpA-R2 is comprised of 119 amino acids and the fact that resonances for one-third of these appeared at identical chemical shift values in the two spectra suggests that the structure of CbpA-R2 is the same in the two constructs. These data provide further support for the conclusion that the N-terminus of CbpA (residues 39–442) is comprised of three independent, α-helical domains.
Figure 5.
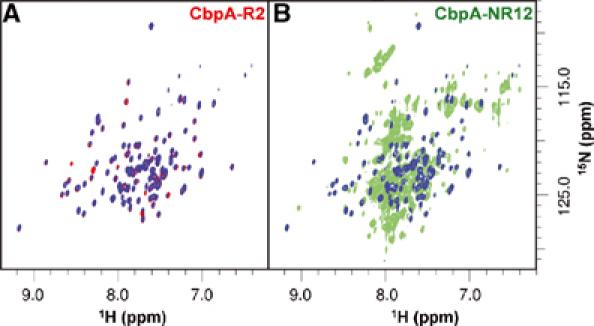
The 800 MHz 2D 1H-15N TROSY spectra of 2H/13C/15N-labeled CbpA-R2 (A) and -NR12 (B). The individual spectra are illustrated in red (A) and green (B) ink. The blue overlays in panels A and B identify resonances that appear at identical positions in the two spectra.
Role of CbpA ‘R' domains in pIgR binding
We studied the binding of domains of CbpA to components of pIgR using several approaches to understand the underlying interaction mechanism. First, we used ELISA to measure the extent to which CbpA constructs bind to sIgA. In agreement with previous studies (Hammerschmidt et al, 2000; Zhang et al, 2000), our results show that multidomain fragments of CbpA, including CbpA-R12 and -NR12, bind to immobilized sIgA (Supplementary Figure 2). However, this assay detected only a weak interaction between sIgA and CbpA-N, -R1 and -R2. These results suggested that multiple CbpA domains cooperate in binding sIgA.
To clarify the role of domains R1 and R2 in pIgR binding, we performed binding experiments using SPR and ITC. We immobilized either sIgA or SC expressed in Sf9 insect cells (SC-D15) on SPR biosensor surfaces and monitored CbpA binding. The experiments were performed at several different concentrations of each CbpA construct (Supplementary Figure 2B) and a global fitting procedure was used to determine association (ka) and dissociation (kd) rate constants (Figure 6A and Supplementary Table 2). Further, these rate constants were used to calculate the apparent Gibbs free energy of binding (ΔGSPR) for each reaction (ΔGSPR=−RT ln(kd/ka); Figure 6A). SPR data for CbpA fragments binding sIgA and SC-D15 showed similar trends and, therefore, only those for binding to sIgA are illustrated in Figure 6A and Supplementary Figure 2B. Kinetic constants for all reactions (sIgA and SC-D15) are given in Supplementary Table 2. Because CbpA-NR12 and -R12 each contain two ‘R' domains, each with one YPT motif, we determined whether the individual R domains bound to components of pIgR. CbpA-R1 and -R2 bound to the sIgA surface more rapidly than did those containing two R domains (CbpA-R12 and -NR12) (Figure 6A and Supplementary Table 2), as would be expected if association was controlled by diffusion. These constructs dissociated from the sIgA surface with rates that depended on the number of R domains; CbpA-R1 and -R2 dissociated more rapidly than did CbpA-R12 and -NR12. CbpA-R1 and -R2 exhibited similar ΔGSPR values (−13.4 and −12.8 kcal mol−1, respectively), and the value for CbpA-NR12 was only slightly more negative (−14.0 kcal mol−1). In this surface-based assay, the presence of two R domains in CbpA-NR12 appears to enhance binding to sIgA only slightly.
Figure 6.
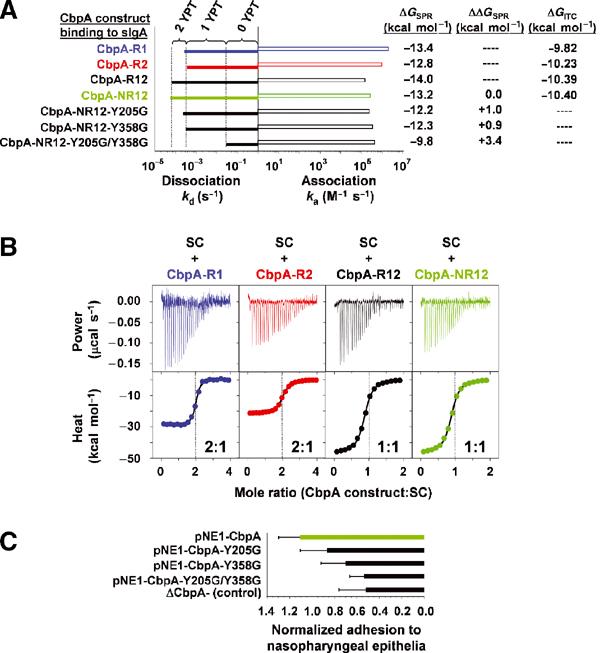
Insights into the molecular mechanism of CbpA/pIgR interactions. (A) Histogram illustrating the association (ka) and dissociation (kd) rate constants derived from SPR data for different CbpA constructs interacting with immobilized sIgA. The rate constants were obtained by fitting equations for a 1:1 binding model to raw data like those illustrated in Supplementary Figure 2B. (B) Raw ITC data (top) and binding curves (bottom) for CbpA-R1 (blue), -R2 (red), -R12 (black) and -NR12 (green) binding to SC-D15. In the bottom panels, the colored circles show raw data points and the black lines show the fit of equations for a single binding site model to the raw data. Only every second data point is illustrated although all points were included in the analysis. The CbpA:SC-D15 mole ratios at the reaction end points are noted in each bottom panel. (C) Adhesion of pneumococci to NE (Detroit) cells. ΔCbpA− pneumococci were transformed with pNE1 plasmids that encoded full-length CbpA or CbpA with one or two mutations within the YPT motif. ΔCbpA− (control) corresponds to results for untransformed ΔCbpA− bacteria.
While the SPR data provided important insights into CbpA:sIgA (and SC) interactions, due to the surface-based format of this assay, ΔGSPR values may not reflect binding behavior in solution. Further, the analysis of binding stoichiometry from SPR data can be problematic. To overcome these limitations, we used ITC to monitor binding of the CbpA constructs to SC-D15. Solutions of CbpA-R1, CbpA-R2, CbpA-R12 or CbpA-NR12 were injected into a solution of SC-D15 and the evolved heat was measured (Figure 6B and Table I). These data were fit using standard equations to give values of the enthalpy (ΔHITC), Gibbs free energy (ΔGITC) and entropy (−TΔSITC) of binding. In addition, the CbpA:SC-D15 mole ratio (N) for each construct was determined (Table I). Binding to SC-D15 was studied because only this protein could be obtained with sufficient purity and in sufficient quantities. The absolute values of ΔGITC are between 3 and 4 kcal mol−1 smaller than those determined from SPR-derived rate constants; however, only a small difference was observed between the values for the different CbpA constructs, which parallels the results from SPR (Table I and Figure 6A). Surface immobilization of sIgA and SC-D15 in the SPR experiments may promote association relative to the situation in solution, and give rise to anomalously large values of ΔGSPR. Despite this caveat, ΔGSPR values can be compared to give an accurate measure of relative binding affinities for different CbpA constructs. The ITC results show that 2 mol of CbpA-R1 or CbpA-R2, or 1 mol of CbpA-R12 or CbpA-NR12, bind to 1 mol of SC-D15. This latter result, along with the similarity of the ΔGITC values and kinetic constants for CbpA-R1 and -R2 binding SC-D15, strongly suggests that there are two thermodynamically indistinguishable binding sites in SC-D15 for CbpA R domains. ΔGITC values for CbpA-R12 and -NR12, each with two R domains, are only marginally larger in absolute magnitude than those for CbpA-R1 and -R2, which, at first, may seem inconsistent with the presence of two binding sites in SC-D15. However, the values of the entropy parameter (−T ΔS) for the former constructs are twice those for the latter (Table I), which largely nullifies the enthalpic benefits (in CbpA-R12 and -NR12) associated with having two R domains. The large entropic penalty of binding for constructs with two R domains may arise from restriction upon binding of a highly flexible polypeptide linker between domains R1 and R2.
Table 1.
Thermodynamic parameters obtained from analysis of ITC data for CbpA fragments binding to SC-D15 in solution
| Construct | ΔG (kcal mol−1) | ΔH (kcal mol−1) | −T ΔS (kcal mol−1) | Nnormalizeda (moles of CbpA per moles of SC) |
|---|---|---|---|---|
| CbpA-R1 | −9.82±0.01 | −26.60±0.05 | +16.78±0.10 | 1.86±0.01 |
| CbpA-R2 | −10.23±0.02 | −21.19±0.07 | +10.96±0.10 | 2.19±0.01 |
| CbpA-R12 | −10.39±0.02 | −46.30±0.14 | +35.91±0.10 | 0.87±0.01 |
| CbpA-NR12 |
−10.40±0.01 |
−38.88±0.07 |
+28.48±0.10 |
1.07±0.01 |
| aNnormalized values were determined using 1.42 μM as the effective SC-D15 protein concentration (see Materials and methods for details). ΔG, ΔH and −T ΔS values are in units of kcal per mole of CbpA construct. | ||||
Mutation of residues within the YPT motif of CbpA interferes with sIgA binding (Hammerschmidt et al, 2000). To further confirm our binding model, we prepared three CbpA-NR12 constructs in which the Tyr residue of the YPT motif was mutated to Gly in the R1 domain (CbpA-NR12-Y205G), the R2 domain (CbpA-NR12-Y358G) or both domains (CbpA-NR12-Y205G/Y358G) and used SPR to determine ka, kd and ΔGSPR values (Figure 6A). Similar mutant CbpA constructs in which the Pro residue of the YPT motif was mutated to Gly were also prepared and analyzed. We reasoned that Gly would readily mimic the backbone conformation of Tyr and Pro residues and, therefore, other than eliminating a side chain, this substitution would not disrupt the structure of the R domains, as was shown by CD studies of CbpA-NR12-Y205G/Y358G (data not shown). The results for the GPT and YGT mutants binding to sIgA and SC-D15 were similar (Supplementary Table 2), and only those for the GPT mutants binding sIgA are discussed here. The ka values for the mutant constructs binding to immobilized sIgA were similar to that of CbpA-NR12. However, the kd values were different and depended on the number of intact YPT motifs. These rates are on average five-fold larger (faster) than that observed for CbpA-NR12. Mutation of both YPT motifs increased the dissociation rate further to a value that is approximately 500-fold larger than that for CbpA-NR12. The value of ΔGSPR for CbpA-NR12 binding to sIgA is −13.2 kcal mol−1, while those for the single GPT mutants are −12.2 and −12.3 kcal mol−1 (for CbpA-NR12-Y205G and -Y358G, respectively). The ΔGSPR value for the double GPT mutant, CbpA-NR12-Y205G/Y358G, is −9.8 kcal mol−1. These results show that one R domain, either R1 or R2, is sufficient for CbpA to bind sIgA (and SC) with high affinity. Affinity is diminished by about 1 kcal mol−1 if one residue in the YPT motif in either R1 or R2 in CbpA-NR12 is mutated and by about 3.5 kcal mol−1 if one residue in each YPT motif in CbpA-NR12 is mutated (Figure 6A). CbpA-N failed to bind immobilized sIgA or SC-D15 in SPR experiments at concentrations up to 1.5 μM (data not shown), showing that the R domains within CbpA-NR12 are the principal determinants of interactions with sIgA (and SC). Further, the similarity of binding data for CbpA-R1 and -R2, and the two single GPT mutants of CbpA-NR12, indicates that the R1 and R2 domains interact similarly with the two R domain binding sites within sIgA and SC. It has been shown recently that CbpA binds to Ig-like domains 3 and 4 of SC (Lu et al, 2003; Elm et al, 2004a). It is possible that the two binding sites we have identified are found within these two domains.
Role of CbpA ‘R' domains in adhesion to nasopharyngeal epithelia
To understand whether there were parallels between our in vitro findings on the effects of mutations within the YPT motifs on CbpA binding to pIgR and bacterial adhesion to cellular receptors, we performed adhesion assays with pneumococci and human NE cells (Detroit cells). We expressed wild-type and mutant CbpA proteins in a CbpA null pneumococcal strain (ΔCbpA−) and measured adhesion of these bacteria to Detroit cells (Figure 6C). While the reproducibility of results from this biological assay is inferior to that associated with our SPR results, the adhesion results clearly show the same trends as do results from SPR. For example, mutation of the Tyr residue in one of the two YPT motifs does not have a statistically significant effect on the number of pneumococci that adhere to epithelial cells. However, mutation of Tyr within both YPT motifs reduces adhesion to the level associated with pneumococci that lack CbpA. These results show that, while CbpA is not the sole determinant of adhesion to human target cells, incorporation of mutations in two out of 693 amino acids in the CbpA sequence that significantly decrease affinity for sIgA and SC in vitro (Figure 6A) has similar effects on adhesion to human cells.
Discussion
Novel structural features of CbpA domains
The mode of attachment of pneumococci to the surface of human epithelial cells is uniquely characterized by the absence of pili or fibrillar bacterial protrusions. Rather, the pneumococcus utilizes a novel mechanism to adhere to and invade human cells; this mechanism is mediated in part by CbpA, which is secreted and recaptured onto the bacterial surface. Among pathogenic bacteria, the use of choline moieties on the bacterial surface as binding sites for functional proteins is uniquely essential to pneumococci (Tomasz, 1967; Rosenow et al, 1997). We show here that domains R1 and R2 of CbpA adopt a strikingly simple structure comprised of three α-helices that bundle together through antiparallel interactions into a flat, raft-like structure. While we have not experimentally determined the structure of CbpA-R1, the similarity of its primary sequence, CD spectra and thermodynamic and kinetic properties to those of the R2 domain strongly suggests that our homology model is accurate. The R domains are unusual in their helical topology and in the highly polar and charged nature of their molecular surfaces. A prominent feature with mixed polar and hydrophobic character—the tyrosine fork—near the loop between Helices 1 and 2 contains several conserved residues that are likely to play a key role in binding to Ig-like domains of pIgR. Most other conserved residues (Figures 1C and 4D) are hydrophobic and are involved in α-helix/α-helix interactions. These may be conserved to preserve the helical topology of the R domains. Other conserved residues are charged and/or polar and are generally surface exposed; these residues may also play roles in interactions with pIgR.
The arrangement of the three α-helices of CbpA-R2 is different from that of other LZ proteins in that the antiparallel α-helices are nearly coaxial, with crossing angles near −180° (−178° for Helices 1 and 2, −176° for Helices 2 and 3). We identified four proteins with one pair of antiparallel α-helices comprised of LZ sequences that exhibit structural similarity to CbpA-R2 (Pdb file 1ROP, Dali Z-score, 5.4; 1GRJ, 4.9; 1CXZ, 4.8; and 1AQT, 4.2) (Holm and Sander, 1993; Oakley and Hollenbeck, 2001). The α-helix crossing angles in these proteins are −161, −153, −160 and −159°, respectively. These crossing angles arise due to the bulky nature of the hydrophobic side chains of Ile, Leu and Val at positions a and d in the heptad repeats, which forces the helices to twist in order to pack ‘knobs-into-holes' (Crick, 1953). The LZ motifs in CbpA-R1 and -R2 differ from the classical motif in that alanine is often found at position d (Figure 1C). The presence of a limited number of small alanine side chains at the α-helix/α-helix interface may allow nearly perfect antiparallel packing of α-helices into the flat, raft-like structure of CbpA-R2.
Phylogenetic analysis reveals conserved structural features of CbpA
Our CbpA mutagenesis results showed that preservation of the Tyr and Pro residues within at least one YPT motif is essential for high-affinity binding to sIgA. Our results are consistent with the findings of others. In particular, Elm et al (2004b) showed that a fragment of SpsA (residues 37–283) with a single R2-like domain (SpsA is also termed PspC 2.1, which constitutes Group 4 in Figure 2) was a competitive inhibitor of pneumococcal adherence to human cells that express pIgR. Importantly, this group also showed that a related CbpA fragment in which Tyr 201 (within the YPT motif) was mutated to Asp was not an inhibitor of adherence. We also observed that the isolated domains, CbpA-R1 and -R2, bind sIgA and SC with similar kinetic and thermodynamic constants. This suggests that the conserved features of the R domains are associated with receptor binding. Further, we reasoned that the residues in these domains that are most highly conserved among different CbpA sequences are involved in receptor binding. Our phylogenetic analysis showed that 22 residues that are conserved in all sequences (Figures 1C and 4D) are found in the C-terminal portion of Helix 1, in the loop connecting Helices 1 and 2 and in the N-terminal portion of Helix 2. These residues, many of which are hydrophobic, are evenly distributed on the two faces and within the two α-helix/α-helix interfaces of the CbpA-R2 structure. The majority are found in Helix 2. For the R2 domain of the TIGR4 strain, an abundance of acidic residues in the midst of the conserved residues gives rise to a strong, negative electrostatic potential on one face of CbpA. However, the electronegative character near the loop containing the YPT motif is neutralized, in part due to the influence of two conserved basic residues, Arg 356 and Lys 364 (Figure 4E). This causes the side chains of the two Tyr residues (Tyr 358 and 363) of the tyrosine fork to protrude from one face of the raft-like structure into a region of low electrostatic potential (<∣±1kt∣). In addition, several other conserved residues (Pro 359, Thr 360 and Thr 362) protrude prominently from the loop on the opposite face of the raft-like structure. Together, our results from mutagenesis and binding experiments and this structure-based analysis strongly suggest that these conserved residues play important roles in binding to SC. While these residues sit in a node in the electrostatic potential map, it is possible that the SC surface has electropositive character that serves, in the initial stages of binding, to attract the electronegative surface of CbpA-R2 and possibly other CbpA R domains.
Materials and methods
Construction of CbpA expression plasmids and expression and purification of CbpA proteins
Expression plasmids for CbpA proteins were prepared using standard methods, as discussed in detail in Supplementary Materials. CbpA proteins for structural and biophysical studies were expressed in Escherichia coli and purified using standard procedures, as given in Supplementary Materials.
Expression of SC-D15 in Sf9 cells and purification
A gene encoding pIgR D1–D5 (residues 1–589 of the mature protein with a C-terminal 6xHis tag) was subcloned into the baculovirus transfer vector pAcGP67b (Pharmingen) in-frame with the gp67 hydrophobic secretion signal. Recombinant virus was generated by cotransfection of the transfer vector with linearized viral DNA (Baculogold; Pharmingen). SC-D15 was harvested from the supernatants of baculovirus-infected High 5 Cells (Invitrogen), which were concentrated and buffer exchanged into 20 mM Tris pH 8.0 and 150 mM NaCl. SC-D15 was purified on Ni-NTA resin (Qiagen) followed by gel filtration chromatography (Superdex 200, Amersham-Pharmacia).
Adhesion experiments
Pneumococci were grown in C+Y media (Lacks and Hotchkiss, 1960) to early stationary phase. All strains were grown in the presence of chloramphenicol (5 μg ml−1), the ΔCbpA− mutant strain with erythromycin (1 μg ml−1) and those transformed with the pNE1/CbpA vectors with spectinomycin (500 μg ml−1). Cells were pelleted and resuspended in fluorescein isothiocyanate (FITC; 1 mg ml−1; Sigma) (Rosenow et al, 1997; Gosink et al, 2000). After incubation at 25°C for 30 min, the bacteria were washed with PBS containing Ca2+ and Mg2+ (Mediatech), and diluted to 1 × 107 CFU ml−1. Confluent monolayers of Detroit 562 epithelial cells (ATCC) were established in 96-well Terasaki trays and activated prior to infection with human TNFα (10 ng well−1) for 2 h. The cells were infected with 105 CFU of labeled bacteria for 30 min at 37°C. The cells were washed four times with PBS containing Ca2+ and Mg2+ and fixed with 2.5% (vol/vol) glutaraldehyde. The number of adherent bacteria was determined by counting using fluorescence microscopy. We used the following procedure to determine that pneumococci transformed with pNE1/CbpA vectors expressed equal amounts of wild-type and mutant CbpA on the bacterial surface. The total bacterial CbpA and surface-bound CbpA (extracted from the bacterial surface with 10% choline) were quantified by Western analysis using a polyclonal anti-CbpA antibody generated against the CbpA N-terminus (residues 39–443) in rabbits. The antibody was purified using affinity chromatography before use.
NMR spectroscopy
Isotope labeled samples for NMR studies were prepared by culturing BL21(DE3) cells in isotope-labeled MOPS-based minimal media (Neidhardt et al, 1974). For 15N-labeling, 15N-labeled ammonium chloride was used; for 13C/15N-labeling, 13C-labeled glucose was also used; and for 2H/13C/15N-labeling, 2H2O was also used. NMR samples (1 mM) were prepared in 10 mM potassium phosphate, 50 mM NaCl, pH 6.5, 5% (v/v) 2H2O and 0.02% (w/v) sodium azide. Gradient-, sensitivity-enhanced 2D 1H-15N TROSY spectra (Weigelt, 1998; Rance et al, 1999; Zhu et al, 1999) for CbpA-R2 and -NR12 were recorded using identical acquisition parameters: 1H: spectral width, 14 368 Hz; data size, 1k complex points; 15N: spectral width, 2433 Hz; data size, 128 complex points. A Cosine window function and zero-filling were applied prior to Fourier transformation in each dimension. Backbone resonance assignments for 2H/13C/15N-labeled CbpA-R2 were determined through the analysis of multiple 3D and 4D spectra, including 3D constant time- (CT-) HNCA, CT-HN(CO)CA (Yamazaki et al, 1994b), CT-HN(CA)CB and CT-HN(COCA)CB (Yamazaki et al, 1994a), and 4D HNCOCA and HNCACO (Mulder et al, 2000). In addition, a 4D 1H-15N HSQC-NOESY-1H-15N HSQC spectrum was analyzed to confirm backbone assignments and to obtain local backbone helical distance restraints. Side-chain assignments were made through the analysis of 3D C(CO)NH-TOCSY, H(CCO)NH-TOCSY, HBHA(CBCACO)NH, HCCH-COSY and HCCH-TOSCY spectra. Additional 3D and 4D NOESY spectra, including 3D NOESY-1H-15N HSQC, 3D NOESY-1H-13C HSQC, 4D 1H-15N HSQC-NOESY-1H-13C HSQC and 4D 1H-13C HSQC-NOESY-1H-13C HSQC, were used to obtain distance restraints for structure calculations. Spectra were recorded at 25°C using several spectrometers: a Varian Inova 600, a Varian INOVA 900 and a Bruker AVANCE 800. Spectra were processed using NMRPipe software (Delaglio et al, 1995) and analyzed using Felix software (Accelerys Inc.). The 1H dimensions of spectra were referenced to external TSP and the 13C and 15N dimensions were referenced indirectly using the appropriate ratios of gyromagnetic ratios (Cavanagh et al, 1996). Secondary 13Cα chemical shift values (Δδ13Cα) were calculated as described (Schwarzinger et al, 2001).
Structure calculation, refinement and analysis
CbpA-R2 structures were calculated using interproton distance restraints estimated from 3D and 4D NOESY cross-peak volumes. Restraint lower bounds were set to 1.8 Å and upper bounds to 2.5, 3.5 and 6.0 Å for large, medium and small volumes, respectively. The program TALOS (Cornilescu et al, 1999) was used to estimate backbone dihedral angles psi (ψ) and phi (φ) based on 1Hα, 15N, 13Cα, 13Cβ and 13C′ chemical shift values within the α-helical segments of CbpA-R2. In addition, hydrogen-bond (H-bond) restraints were used within α-helices. Amide protons involved in H-bonds were identified on the basis of slow exchange with 2H2O. Restraints are summarized in Table II. Structures were calculated using torsion angle dynamics (TAD) (Stein et al, 1997) within CNS (Brunger et al, 1998). The TAD protocol was performed in stages: (1) 75 ps high-temperature TAD (50 000 K) followed by (2) cooling to 1000 K over the course of 75 ps and ramping of the van der Waals scaling term from 0.1 to 1.0. (3) The molecules were further cooled to 300 K over the course of 20 ps using conventional Cartesian dynamics followed finally by (4) 10 000 steps of conjugate gradient energy minimization. The NOE (dihedral) restraint energy term was 150 kcal mol−1 (100 kcal mol−1) for stages 1–3 and 100 kcal mol−1 (300 kcal mol−1) for the last. A total of 200 structures were calculated, and the 40 lowest-energy structures were further refined by using the SANDER module of AMBER 8.0 (Case et al, 2004). Solvent was represented by the generalized-Born (GB) model (Xia et al, 2002). Structures were first energy minimized for 1 ps without restraints followed by 40 ps of simulated annealing from 400 to 0 K with all restraints. The distance and angle restraint force constants were 20 and 2 kcal mol−1 Å−2, respectively. Statistics for the 20 lowest-energy structures are given in Table II. The program Define_S was used to determine the α-helix crossing angles for CbpA-R2 and other LZ-containing proteins (Richards and Kundrot, 1988). Further, we compared the structure of CbpA-R2 with those of other proteins using the Dali server (http://www.ebi.ac.uk/dali/Interactive.html). The coordinates for the 20 lowest-energy CbpA-R2 structures have been deposited under PDB ID code 1w9r.
Table 2.
Statistics of 20 lowest-energy structures of CbpA-R2 based on solution NMR data
| Total number of NOEs | 2289 |
| Intraresidue | 922 |
| Inter-residue | 1367 |
| Sequential | 622 |
| Medium range (i, i up to ±4) | 596 |
| Long range (i, i ±5 or larger) | 149 |
| Total number of dihedral restraints | 166 |
| ψ | 83 |
| φ | 83 |
| R.m.s.d. from mean structure (Å) | |
| All residues within structural region | |
| Backbone heavy atoms (328–425) | 1.0±0.2 |
| All heavy atoms | 1.5±0.4 |
| Helixes 1–3 (residues 330–357, 365–390, 395–425) | |
| Backbone heavy atoms | 0.9±0.1 |
| All heavy atoms | 1.4±0.3 |
| Distance restraint violations (average number per structure) | |
| Restraints violated by >0.50 Å | 1.5 |
| Maximum distance restraint violation (Å) | 1.6 |
| Torsion angle restraint violations (average number per structure) | |
| ψ restraints violated by >5° | 1 |
| φ restraints violated by >5° | 0 |
| Ramachandran ψ, φ statistics | |
| Angles in most favored region | 84.5% |
| Angles in allowed region | 13.5% |
| Angles in generously allowed region | 1.6% |
| Angles in disallowed region | 0.4% |
Homology modeling of CbpA-R1
We used the program MOE (Chemical_Computing_Group) to calculate a homology model of CbpA-R1 using the experimental structure of CbpA-R2 as the template. Briefly, MOE first aligned the sequence of the target (R1) with that of the template (R2). Next, the side chains of nonidentical residues were built and side-chain conformations were adjusted by reference to a rotomer library followed by energy minimization using the AMBER '94 forcefield.
SPR measurements
SPR experiments were performed at 25°C using a BIACORE 3000 instrument (Biacore Inc.). sIgA and SC-D15 were covalently attached to different carboxymethyl-dextran-coated gold surfaces (CM-5 Chip, Biacore) using the manufacturer's procedures. Binding was measured by flowing CbpA proteins in 10 mM HEPES, 150 mM NaCl, 3 mM EDTA and 0.005% Surfactant P20 (pH 7.4) (HBS-EP buffer, Biacore) at a flow rate of 20 μl min−1 through the reference and sIgA-containing flow cells in sequence. A blank was also run consisting of only buffer. Following the injection, dissociation of CbpA proteins was measured by flowing only buffer through the cells. The chip surfaces were regenerated by injecting 20 μl of 0.1% SDS through the cells. Data reported are the difference in SPR signal between the flow cell containing sIgA and the reference cell. Contributions to the signal from buffer were removed by subtraction of the blank (buffer only) injection from the reference-subtracted signal (Morton and Myszka, 1998). Triplicate injections were made and the average taken. Data were analyzed by simultaneously fitting association and dissociation phases at all concentrations using the BiaEvaluation software (Biacore Inc.). The model used for fitting was the simplest that was able to account for the observed binding. The concentrations of CbpA constructs used in these experiments are given in the legend to Supplementary Figure 2B.
ITC experiments
ITC experiments were performed at 25°C using a VP-ITC (Microcal) calorimeter. A preliminary 2 μl injection of either CbpA-R1, CbpA-R2, CbpA-R12 or CbpA-NR12 (all at 40 μM) was followed by multiple injections of 5 μl at 300 s intervals into a solution of 4 μM SC-D15. All proteins were dissolved in 20 mM sodium phosphate (pH 6.5) and 100 mM NaCl. Thermodynamic parameters were obtained using Origin software (OriginLab) using a single binding site model after correcting heat values for the effect of dilution. The results of titrations with the CbpA constructs and SC-D15 indicated that the effective concentration of SC-D15 was 1.42 μM, indicating that only 36% of SC molecules were competent for binding CbpA. This conclusion was confirmed using gel filtration chromatography, as follows. In the absence of CbpA, SC-D15 migrated as a single species. Addition of an excess of CbpA-R1 caused only ∼40% of the parent SC peak intensity to shift to a more rapidly migrating species (corresponding to the CbpA-R1:SC-D15 complex). Formation of this complex was complete upon addition of approximately 0.8 molar equivalents of CbpA-R1. We used this effective concentration of SC-D15 (1.42 μM) when analyzing raw heat data to obtain CbpA:SC-D15 mole ratios (Table I).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Material
Acknowledgments
We acknowledge Weixing Zhang for assistance with NMR experiments, Daniel Stokes and David Cardwell for assistance with protein production, Abby Parrill for generating the homology model of CbpA-R1, Cheon-Gil Park and Ross Hilliard for help with preparation of SC-D15 and ITC experiments, Charles Ross for computer support and Charles Galea for helpful comments on the manuscript. Ranjith Muhandiram and Lewis Kay are acknowledged for graciously providing Varian NMR pulse programs. We thank Dr Eriks Kupce of Varian for recording the 900 MHz 2D TROSY spectrum of CbpA-R2, and Peter M Snow and the Caltech Protein Expression Facility for expression of SC-D15. AEH was supported by the Whitehead Institute for Biomedical Research. We acknowledge support from the American Lebanese Syrian Associated Charities (ALSAC), the National Cancer Institute (CA82491 to RWK), National Center for Research Resources (RR014675 for a Biacore 3000 instrument) and a Cancer Center (CORE) Support Grant (CA 21765). We dedicate this paper to the memory of Peter M Snow who died tragically in a bicycling accident in Maine, USA, on Wednesday, August 4th.
References
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Butler JC, Breiman RF, Campbell JF, Lipman HB, Broome CV, Facklam RR (1993) Pneumococcal polysaccharide vaccine efficacy. An evaluation of current recommendations. JAMA 270: 1826–1831 [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham ITE, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA (2004) AMBER 8. University of California, San Francisco, http://amber.scripps.edu/doc8/amber 8.pdf
- Cavanagh J, Fairbrother WJ, Palmer AG III, Skelton NJ (1996) Protein NMR Spectroscopy. New York: Academic Press [Google Scholar]
- Chemical_Computing_Group MOE version 2004.02. Montreal, Quebec, Canada
- Cornilescu G, Delaglio F, Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13: 289–302 [DOI] [PubMed] [Google Scholar]
- Creighton TE (1993) Proteins: Structures and Molecular Properties. New York, NY: WH Freeman & Co. [Google Scholar]
- Crick FHC (1953) The packing of alpha-helices: simple coiled-coils. Acta Crystallogr 6: 689–697 [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMR Pipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6: 277–293 [DOI] [PubMed] [Google Scholar]
- Elm C, Braathen R, Bergmann S, Frank R, Vaerman JP, Kaetzel CS, Chhatwal GS, Johansen FE, Hammerschmidt S (2004a) Ectodomains 3 and 4 of human polymeric immunoglobulin receptor (hpIgR) mediate invasion of Streptococcus pneumoniae into the epithelium. J Biol Chem 279: 6296–6304. Epub 2003 Dec 6293 [DOI] [PubMed] [Google Scholar]
- Elm C, Rohde M, Vaerman JP, Chhatwal GS, Hammerschmidt S (2004b) Characterization of the interaction of the pneumococcal surface protein SpsA with the human polymeric immunoglobulin receptor (hpIgR). Indian J Med Res 119: 61–65 [PubMed] [Google Scholar]
- Fernandez-Tornero C, Lopez R, Garcia E, Gimenez-Gallego G, Romero A (2001) A novel solenoid fold in the cell wall anchoring domain of the pneumococcal virulence factor LytA. Nat Struct Biol 8: 1020–1024 [DOI] [PubMed] [Google Scholar]
- Gosink K, Tuomanen E (2000) Streptococcus pneumoniae: invasion and Inflammation. In Gram-positive Pathogens, Fischetti VA (ed) Washington, DC: American Society for Microbiology [Google Scholar]
- Gosink KK, Mann ER, Guglielmo C, Tuomanen EI, Masure HR (2000) Role of novel choline binding proteins in virulence of Streptococcus pneumoniae. Infect Immun 68: 5690–5695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerschmidt S, Talay SR, Brandtzaeg P, Chhatwal GS (1997) SpsA, a novel pneumococcal surface protein with specific binding to secretory immunoglobulin A and secretory component. Mol Microbiol 25: 1113–1124 [DOI] [PubMed] [Google Scholar]
- Hammerschmidt S, Tillig MP, Wolff S, Vaerman JP, Chhatwal GS (2000) Species-specific binding of human secretory component to SpsA protein of Streptococcus pneumoniae via a hexapeptide motif. Mol Microbiol 36: 726–736 [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C (1993) Protein structure comparison by alignment of distance matrices. J Mol Biol 233: 123–138 [DOI] [PubMed] [Google Scholar]
- Iannelli F, Oggioni MR, Pozzi G (2002) Allelic variation in the highly polymorphic locus pspC of Streptococcus pneumoniae. Gene 284: 63–71 [DOI] [PubMed] [Google Scholar]
- Lacks S, Hotchkiss RD (1960) A study of the genetic material determining an enzyme in Pneumococcus. Biochim Biophys Acta 39: 508–518 [DOI] [PubMed] [Google Scholar]
- Landschulz WH, Johnson PF, McKnight SL (1988) The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science 240: 1759–1764 [DOI] [PubMed] [Google Scholar]
- Lu L, Lamm ME, Li H, Corthesy B, Zhang JR (2003) The human polymeric immunoglobulin receptor binds to Streptococcus pneumoniae via domains 3 and 4. J Biol Chem 278: 48178–48187. Epub 4 Sep 2003, 48117 [DOI] [PubMed] [Google Scholar]
- Lupas A (1996) Coiled coils: new structures and new functions. Trends Biochem Sci 21: 375–382 [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J (1991) Predicting coiled coils from protein sequences. Science 252: 1162–1164 [DOI] [PubMed] [Google Scholar]
- Morton TA, Myszka DG (1998) Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Methods Enzymol 295: 268–294 [DOI] [PubMed] [Google Scholar]
- Mostov KE (1994) Transepithelial transport of immunoglobulins. Annu Rev Immunol 12: 63–84 [DOI] [PubMed] [Google Scholar]
- Mostov KE, Kaetzel CS (1999) Immunoglobulin transport and the polymeric immunoglobulin receptor. In Mucosal Immunology, Lamm ME, Mestecky J, Michael Lamm M, McHgee JR (eds) New York: Academic Press
- Mulder FA, Ayed A, Yang D, Arrowsmith CH, Kay LE (2000) Assignment of 1H(N), 15N, 13C(alpha), 13CO and 13C(beta) resonances in a 67 kDa p53 dimer using 4D-TROSY NMR spectroscopy. J Biomol NMR 18: 173–176 [DOI] [PubMed] [Google Scholar]
- Neidhardt FC, Bloch PL, Smith DF (1974) Culture medium for enterobacteria. J Bacteriol 119: 736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls A, Sharp KA, Honig B (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281–296 [DOI] [PubMed] [Google Scholar]
- Nozaki Y, Tanford C (1971) The solubility of amino acids and two glycine peptides in aqueous ethanol and dioxane solutions. Establishment of a hydrophobicity scale. J Biol Chem 246: 2211–2217 [PubMed] [Google Scholar]
- Oakley MG, Hollenbeck JJ (2001) The design of antiparallel coiled coils. Curr Opin Struct Biol 11: 450–457 [DOI] [PubMed] [Google Scholar]
- Rance M, Loria JP, Palmer AGR (1999) Sensitivity improvement of transverse relaxation-optimized spectroscopy. J Magn Reson 136: 92–101 [DOI] [PubMed] [Google Scholar]
- Richards FM, Kundrot CE (1988) Identification of structural motifs from protein coordinate data: secondary structure and first-level supersecondary structure. Proteins 3: 71–84 [DOI] [PubMed] [Google Scholar]
- Rosenow C, Ryan P, Weiser JN, Johnson S, Fontan P, Ortqvist A, Masure HR (1997) Contribution of novel choline-binding proteins to adherence, colonization and immunogenicity of Streptococcus pneumoniae. Mol Microbiol 25: 819–829 [DOI] [PubMed] [Google Scholar]
- Schuchat A, Hilger T, Zell E, Farley MM, Reingold A, Harrison L, Lefkowitz L, Danila R, Stefonek K, Barrett N, Morse D, Pinner R (2001) Active bacterial core surveillance of the emerging infections program network. Emerg Infect Dis 7: 92–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzinger S, Kroon GJA, Foss TR, Chung J, Wright PE, Dyson HJ (2001) Sequence-dependent correction of random coil NMR chemical shifts. J Am Chem Soc 123: 2970–2978 [DOI] [PubMed] [Google Scholar]
- Stein EG, Rice LM, Brunger AT (1997) Torsion-angle molecular dynamics as a new efficient tool for NMR structure calculation. J Magn Reson 124: 154–164 [DOI] [PubMed] [Google Scholar]
- Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, Durkin AS, Gwinn M, Kolonay JF, Nelson WC, Peterson JD, Umayam LA, White O, Salzberg SL, Lewis MR, Radune D, Holtzapple E, Khouri H, Wolf AM, Utterback TR, Hansen CL, McDonald LA, Feldblyum TV, Angiuoli S, Dickinson T, Hickey EK, Holt IE, Loftus BJ, Yang F, Smith HO, Venter JC, Dougherty BA, Morrison DA, Hollingshead SK, Fraser CM (2001) Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293: 498–506 [DOI] [PubMed] [Google Scholar]
- Tomasz A (1967) Choline in the cell wall of a bacterium: novel type of polymer-linked choline in Pneumococcus. Science 157: 694–697 [DOI] [PubMed] [Google Scholar]
- Weigelt J (1998) Single scan, sensitivity- and gradient-enhanced TROSY for multidimensional NMR experiments. J Am Chem Soc 120: 10778–10779 [Google Scholar]
- Whitney CG, Farley MM, Hadler J, Harrison LH, Lexau C, Reingold A, Lefkowitz L, Cieslak PR, Cetron M, Zell ER, Jorgensen JH, Schuchat A (2000) Increasing prevalence of multidrug-resistant Streptococcus pneumoniae in the United States. N Engl J Med 343: 1917–1924 [DOI] [PubMed] [Google Scholar]
- Xia B, Tsui V, Case DA, Dyson HJ, Wright PE (2002) Comparison of protein solution structures refined by molecular dynamics simulation in vacuum, with a generalized Born model, and with explicit water. J Biomol NMR 22: 317–331 [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Lee W, Arrowsmith CH, Muhandiram DR, Kay LE (1994a) A suite of triple resonance NMR experiments for the backbone assignment of 15N, 13C, 2H labeled proteins with high sensitivity. J Am Chem Soc 116: 11655–11666 [Google Scholar]
- Yamazaki T, Lee W, Revington M, Mattiello DL, Dahlquist FW, Arrowsmith CH, Kay LE (1994b) An HNCA pulse scheme for the backbone assignment of 15N, 13C, 2H-labeled proteins: application to a 37-kDa Trp repressor–DNA complex. J Am Chem Soc 116: 6464–6465 [Google Scholar]
- Zhang JR, Mostov KE, Lamm ME, Nanno M, Shimida S, Ohwaki M, Tuomanen E (2000) The polymeric immunoglobulin receptor translocates pneumococci across human nasopharyngeal epithelial cells. Cell 102: 827–837 [DOI] [PubMed] [Google Scholar]
- Zhu G, Kong X, Sze K (1999) Gradient and sensitivity enhancement of 2D TROSY with water flip-back, 3D NOESY-TROSY and TOCSY-TROSY experiments. J Biomol NMR 13: 77–81 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Material