

Abstract
Protein phosphatase 5 (Ppp5) is a serine/threonine protein phosphatase comprising a regulatory tetratricopeptide repeat (TPR) domain N-terminal to its phosphatase domain. Ppp5 functions in signalling pathways that control cellular responses to stress, glucocorticoids and DNA damage. Its phosphatase activity is suppressed by an autoinhibited conformation maintained by the TPR domain and a C-terminal subdomain. By interacting with the TPR domain, heat shock protein 90 (Hsp90) and fatty acids including arachidonic acid stimulate phosphatase activity. Here, we describe the structure of the autoinhibited state of Ppp5, revealing mechanisms of TPR-mediated phosphatase inhibition and Hsp90- and arachidonic acid-induced stimulation of phosphatase activity. The TPR domain engages with the catalytic channel of the phosphatase domain, restricting access to the catalytic site. This autoinhibited conformation of Ppp5 is stabilised by the C-terminal αJ helix that contacts a region of the Hsp90-binding groove on the TPR domain. Hsp90 activates Ppp5 by disrupting TPR–phosphatase domain interactions, permitting substrate access to the constitutively active phosphatase domain, whereas arachidonic acid prompts an alternate conformation of the TPR domain, destabilising the TPR–phosphatase domain interface.
Keywords: Hsp90, protein phosphatase 5, protein phosphorylation, TPR
Introduction
Reversible phosphorylation of proteins on serine and threonine residues contributes to the regulation of virtually all functions of the eukaryotic cell. Protein phosphatase 5 (Ppp5), a ubiquitous member of the PPP family of serine/threonine protein phosphatases, participates in several stress-activated cellular signalling pathways that regulate growth arrest, apoptosis and response to ionising radiation-induced DNA damage (Zuo et al, 1998; Morita et al, 2001; Ali et al, 2004; Wechsler et al, 2004; Zeke et al, 2004). Similar to other PPP family members (PP1, PP2A, Ppp4), Ppp5 is potently inhibited by the tumour promoters okadaic acid, microcystin, cantharadin, calyculin A and tautomycin (Chen et al, 1994; Borthwick et al, 2001). However, whereas PP1, PP2A and Ppp4 exist as dimers or trimers with a catalytic subunit bound to regulatory subunit(s), Ppp5 uniquely comprises a regulatory N-terminal tetratricopeptide repeat (TPR) domain fused to a C-terminal phosphatase catalytic domain (reviewed in Cohen, 1997; Andreeva and Kutuzov, 1999; Chinkers, 2001).
The discoveries that Ppp5 forms complexes with glucocorticoid receptors and the Ask1 protein kinase implicated the phosphatase as a regulator of signalling networks initiated by glucocorticoids (Chen et al, 1996; Zuo et al, 1999) and oxidative stress (Morita et al, 2001). Ppp5 interacts with the TPR acceptor site of the heat shock protein Hsp90 and competes with TPR containing immunophilins in binding to some glucocorticoid receptor complexes (Chen et al, 1996; Silverstein et al, 1997). Suppression of Ppp5 expression with antisense oligonucleotides enhances glucocorticoid-mediated phosphorylation of p53 on Ser15 with consequent expression of p21WAF1/Cip1 and concomitant G1 growth arrest in a lung carcinoma cell line (Zuo et al, 1998, 1999; Urban et al, 2003). Oxidative stress-mediated activation of apoptosis signal-regulating kinase 1 (Ask1), which promotes apoptosis via the JNK and p38 MAP kinase cascades, was inhibited by overexpression of Ppp5 (Morita et al, 2001). This negative regulation of Ask1 by Ppp5 is likely to involve dephosphorylation of a phospho-threonine residue within the activation loop of Ask1. Ppp5 also suppresses the transient increase in Ask1 activity produced by hypoxia (Zhou et al, 2004). In a further link between Ppp5 and Ask1, rapamycin, an inhibitor of the mammalian target of rapamycin (mTOR), induces sustained activation of Ask1, associated with inhibition of Ppp5 activity (Huang et al, 2004).
Recently, two important studies have demonstrated roles for Ppp5 in the DNA damage response. Ppp5 associates with both ataxia telangiectasia mutated (ATM) and DNA-PK (Ali et al, 2004; Wechsler et al, 2004), two PI3K-related kinases whose activation in response to genotoxic stress initiates DNA damage-induced checkpoints and DNA repair, respectively. Ppp5 regulates the phosphorylation states of these kinases and their downstream signalling events. Significantly, a fragment of DNA-PK containing an autophosphorylation site (Thr2609) interacts with the TPR domain of Ppp5, suggesting that DNA-PK–Ppp5 interactions may be dependent on DNA-PK phosphorylation states (Wechsler et al, 2004). A variety of other proteins have also been reported to associate with Ppp5. These include the atrial natriuretic peptide receptor (Chinkers, 1994), the TPR containing Cdc16 and Cdc27 subunits of the anaphase promoting complex (Ollendorf and Donoghue, 1997), the human homologue of Arabidopsis thaliana blue-light photoreceptor, cryptochrome 2 (Zhao and Sancar, 1997), the A subunit of PP2A (Lubert et al, 2001) and the Hsp90-dependent haem-regulated eIF2α kinase (Shao et al, 2002). The Gα12/Gα13 subunits of heterotrimeric G proteins also interact with Ppp5, suggesting that Ppp5 is a downstream effector of Gα12/Gα13 signalling to ion channels (Yamaguchi et al, 2002).
Unlike PP1 and PP2A, the in vitro basal activity of Ppp5 is extremely low. Numerous studies have demonstrated that the TPR domain and C-terminal 13 residues cooperate to maintain the phosphatase in an autoinhibited state, with suppressed phosphatase activity. Removal of either the TPR domain or C-terminal 13 residues by limited proteolysis or deletion mutagenesis potently activates the enzyme some 10- to 50-fold (Chen and Cohen, 1997; Sinclair et al, 1999; Kang et al, 2001). These findings indicate that the isolated phosphatase domain is constitutively active, and that the TPR domain and C-terminal segment coordinately suppress catalytic activity. How Ppp5 is stimulated in vivo remains unclear, although a number of Ppp5 activators have been identified from in vitro studies. For example, polyunsaturated fatty acids, such as arachidonic acid, and saturated and unsaturated fatty acyl CoA esters stimulate the phosphatase activity of Ppp5 through interaction with its TPR domain (Chen and Cohen, 1997; Skinner et al, 1997; Ramsey and Chinkers, 2002). Furthermore, proteins that interact with the TPR domain also stimulate phosphatase activity, notably the C-terminal domain of Hsp90 (Ramsey and Chinkers, 2002) and Gα12/Gα13 subunits (Yamaguchi et al, 2002).
The crystal structure of the TPR domain of Ppp5 revealed how the three TPR motifs, each comprising a pair of antiparallel α-helices, are organised to form a superhelical structure presenting an amphipathic groove that provides a binding site for interacting proteins (Das et al, 1998). Basic residues within this groove interact with acidic residues in the TPR acceptor site at the C-terminus of Hsp90 (Russell et al, 1999; Ramsey et al, 2000). More recently, the crystal structure of the catalytic domain of Ppp5 (residues 169–499) was reported (Swingle et al, 2004). The tertiary structure of the catalytic core and proposed catalytic mechanism are very similar to those of PP1 (Egloff et al, 1995; Goldberg et al, 1995) and calcineurin/PP2B (Griffith et al, 1995; Kissinger et al, 1995) with which Ppp5 shares approximately 40% identity (Barton et al, 1994).
An understanding of how the TPR domain and C-terminal segment of Ppp5 cooperate to suppress catalytic activity requires structural information of the full-length protein. Here we describe the crystal structure of the autoinhibited conformation of human Ppp5. The structure reveals an extensive interface between the TPR and phosphatase domains, augmented by the C-terminus of the protein, that blocks access to the catalytic site, thereby inhibiting activity. The TPR–phosphatase domain interface includes a region of the Hsp90-binding groove of the TPR domain, suggesting that TPR–Hsp90 interactions would dissociate the TPR domain from the phosphatase catalytic site, activating the phosphatase. In contrast, long-chain fatty acids activate Ppp5 by stabilising an alternate conformation of the TPR domain that disrupts contacts with the phosphatase domain.
Results and discussion
Structure determination
The structure of Ppp5 was solved by means of molecular replacement with the initial phasing obtained by determining the position of the catalytic domain of the molecule using the structure of the catalytic subunit of PP1 as a search object. Details of the structure determination procedure are described in Materials and methods. Four Ppp5 molecules exist within the asymmetric unit and three are well defined, except for the N-terminal six residues and the TPR–phosphatase domain linker residues (152–157) that are disordered. The fourth Ppp5 molecule has a much less ordered structure, the electron density for its TPR domain being particularly weak, with breaks in density corresponding to both side-chain and main-chain features. In the crystal, Ppp5 molecules are assembled into dimers, with the dimer interface formed by the antiparallel association of the edge β-strand (β14) of the catalytic domain β-sandwich, creating a continuous β-sheet shared between two molecules. The TPR domain does not participate in the dimer interactions. The physiological relevance of this dimer is unknown. In solution, the protein behaves as a monomer, eluting at the expected position for a monomeric 58 kDa protein on an S200 size exclusion column. Moreover, the crystal structure of the isolated catalytic domain of Ppp5 is a monomer (Swingle et al, 2004).
Description of overall structure
Ppp5 is organised into two distinct domains: the N-terminal TPR domain is connected via a partially disordered linker region to the phosphatase domain (Figure 1). Immediately C-terminal to the phosphatase domain, the polypeptide forms a subdomain of 20 residues terminating in a short two-turn α-helix (αJ) that is unique to Ppp5. The TPR domain is composed of three tandem TPR motifs arranged in parallel (Das et al, 1998). As each TPR motif of 34 residues is generated from a pair of antiparallel α-helices, the TPR domain is composed of a series of antiparallel α-helices (termed αA and αB, connected by short turns) rotated relative to one another by a constant 24°. This topology creates a right-handed superhelical structure generating a pronounced concave surface on one side with a contrasting convex surface on the other. The architecture of the catalytic phosphatase domain of Ppp5 is essentially identical to that of PP1 and PP2B, other members of the PPP Ser/Thr-specific protein phosphatase family (Egloff et al, 1995; Goldberg et al, 1995; Griffith et al, 1995; Kissinger et al, 1995). The architecture of these proteins is characterised by a central β-sandwich surrounded on one side by seven α-helices and on the other by a subdomain consisting of three α-helices and a three-stranded mixed β-sheet. Loops connecting β-stands from the β-sandwich with the flanking α-helices contribute conserved catalytic residues that function to coordinate two metal ions at the centre of the catalytic site. This binuclear metal centre is responsible for mediating the dephosphorylation reaction by coordinating the scissile phosphate group of the phosphorylated substrate adjacent to a metal-activated nucleophilic water molecule. The catalytic centre lies within a pronounced surface channel created by the association of the two β-sheets of the β-sandwich, which presumably allows engagement with the phosphoprotein substrate (Figure 1). Such a feature is common to the catalytic domains of PP1 and PP2B. Inhibitory toxins of PP1, and the intrasteric inhibitory domain of PP2B, associate with the equivalent channel in these phosphatases. In Ppp5, the C-terminal αJ helix packs onto one side of the β-sandwich and functions to deepen the catalytic site channel. Pairwise comparison of the four independent copies of Ppp5 within the asymmetric unit indicates that the catalytic domains of the four molecules have essentially identical conformations (mean pairwise r.m.s.d. between equivalent Cα atoms is 0.15 Å). However, there are small differences in the relative orientations of the TPR domains relative to the phosphatase domains, suggesting flexibility at the domain interface.
Figure 1.
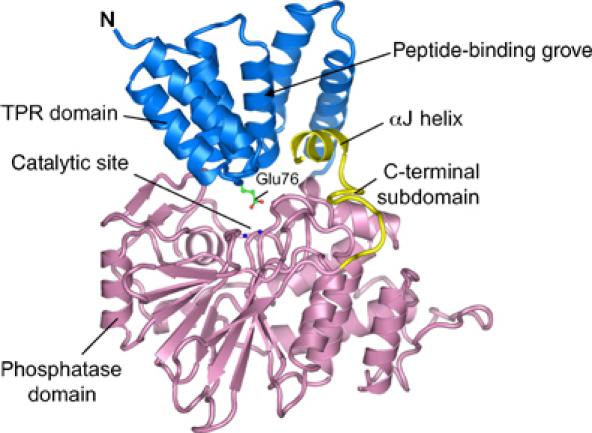
Structure of human Ppp5. Ribbon representation of Ppp5 with the TPR and phosphatase domains coloured blue and pink, respectively. The C-terminal subdomain including the αJ helix is in yellow. Metal ions of the binuclear centre are shown as blue spheres. The figures were produced using PYMOL (http://www.pymol.org).
Previously determined structures of the isolated TPR (Das et al, 1998) and catalytic domains of Ppp5 (Swingle et al, 2004) indicated that these domains form autonomously folded structures. Within the context of the autoinhibited state of Ppp5, there are small changes in conformation of these individual domains. In the catalytic domain, these differences are confined to a small readjustment of the C-terminal αJ helix. Otherwise, the structures are identical and equivalent Cα atoms superimpose with an r.m.s.d. of 0.4 Å (Figure 2A). Similarly, the conformations of the three core TPR motifs of the TPR domain are the same between the isolated domain and the full-length protein (equivalent Cα atoms superimpose with an r.m.s.d. of 0.6 Å). However, the seventh helix (α7) of the isolated TPR domain, which forms an extended helix that packs antiparallel to the third TPR domain (Das et al, 1998), is rotated by ∼5° in Ppp5, and the C-terminal five turns of α7 unwind to form the linker connecting the TPR and phosphatase domains, together with the N-terminus of the phosphatase domain (Figure 2B).
Figure 2.
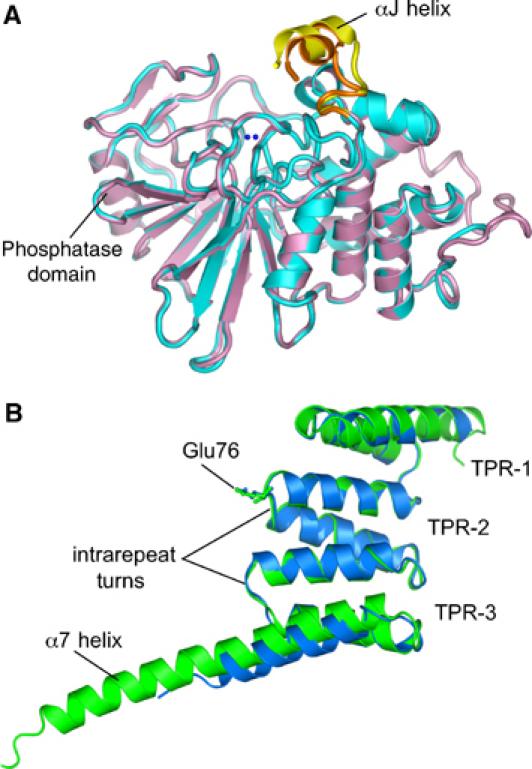
The TPR domain does not alter the conformation of the phosphatase domain. (A) Superimposition of the isolated phosphatase domain of Ppp5 (cyan) onto the Ppp5 phosphatase domain (salmon) within full-length Ppp5 shows a small shift of the C-terminal subdomain αJ helix (yellow: full-length Ppp5; orange: isolated Ppp5 domain). (B) Superimposition of the TPR domain of full-length (blue) and isolated protein (green).
TPR domain inhibits Ppp5 by blocking the catalytic channel
We crystallised Ppp5 in the absence of activating ligands where Ppp5 is between 5 and 15% of the activity of either the fully activated protein or the constitutively active isolated catalytic phosphatase domain, implying that our structure represents the autoinhibited conformation of Ppp5. In this state, the TPR and phosphatase domains engage to form an extensive interface, which blocks access to the phosphatase catalytic site, thereby explaining the ability of the TPR domain to inhibit Ppp5 phosphatase activity (Figures 1 and 3A). Comparison with the structure of the isolated catalytic domain shows that there are no changes in conformation of the phosphatase catalytic site on association with the TPR domain, indicating that TPR-induced inhibition of catalytic activity is not due to distortion of the phosphatase catalytic domain. Specifically, the interactions between the TPR and phosphatase domains involve contacts between the turns connecting the αA and αB helices of TPR motifs 1–3 (intrarepeat turns), which form a contiguous ridge that inserts into the catalytic channel of the phosphatase domain, reminiscent of how toxins inhibit PP1 (Goldberg et al, 1995; Maynes et al, 2001: Kita et al, 2002) (Figures 1 and 3A). Specific contacts between the TPR and phosphatase domains are relatively few. Of particular importance, however, are hydrogen bonds formed between the carboxylate side chain of Glu76, which projects from the intrarepeat turn of TPR-2 (second TPR motif of the TPR domain), and two catalytic site residues, Arg275 and Tyr451 (Figure 3B). Both Arg275 and Tyr451 are conserved in all members of the PPP Ser/Thr phosphatase family (Barton et al, 1994). Arg275 coordinates the phosphate group of the phosphoprotein substrate, whereas Tyr451 may also be involved in substrate recognition and/or metal ion coordination (Egloff et al, 1995). Consistent with our structural data that Glu76 participates in stabilising the autoinhibited state of Ppp5, a previous study found that substituting Ala for Glu76 activates Ppp5 some 10-fold (Kang et al, 2001). Apart from a hydrogen bond between the phenolic hydroxyl group of Tyr313 of the phosphatase domain, and a main-chain carbonyl oxygen atom of Leu108 of αA of TPR-3, other TPR–phosphatase domain interactions are predominantly nonpolar in character. The most extensive set of nonpolar interactions are formed between the aliphatic side chains of two Leu residues, Leu493 and Leu494, lying on one face of the C-terminal αJ helix, which pack into a pocket on the TPR domain generated by the αA and αB helices of TPR-3 and TPR domain α7 helix. These interactions are likely to be responsible for the small shift in conformation of the αJ helix in the autoinhibited state compared with the constitutively active isolated phosphatase domain (Figures 2A and 3C).
Figure 3.
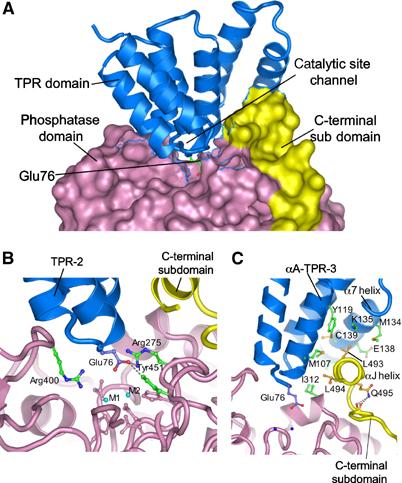
Phosphatase–TPR domain interactions. (A) The phosphatase domain and C-terminal subdomain are represented with a molecular surface, and the TPR domain as ribbon. Intra-TPR turns form a ridge that inserts into the phosphatase domain catalytic channel, with Glu76 of TPR-2 projecting towards the binuclear metal centre. (B) Glu76 of the TPR-2 interacts with Arg275 and Tyr451 at the catalytic site. Metal ions are indicated as M1 and M2 and side chains of metal ion-binding residues are coloured pink. (C) The αJ helix forms hydrophobic contacts with the TPR domain. Detailed interactions involving Leu493 and Leu494 of the αJ helix with TPR-3 and α7 of the TPR domain are shown. The amide side chain of Gln495 donates hydrogen bonds to the main-chain carbonyls of 489 and 490, stabilising the position of the αJ helix.
Previous biochemical data have revealed that both the TPR domain and the extreme C-terminal 10 residues function in a coordinated manner to maintain the autoinhibited state of Ppp5 (Sinclair et al, 1999; Kang et al, 2001). Removal of either the TPR domain or the C-terminal 13 residues leads to a constitutively active phosphatase that is essentially insensitive to fatty acid stimulation. Our structure shows that the C-terminal residues do not restrict access to the catalytic site themselves, but function to promote the autoinhibited conformation of Ppp5 by stabilising the interactions between the TPR and phosphatase domains. Consistent with this notion, removal of the C-terminal 13 residues renders Ppp5 more susceptible to trypsin cleavage within the linker region connecting the TPR and phosphatase domains, suggesting that the αJ helix functions to maintain Ppp5 in a constrained conformational state (Sinclair et al, 1999). In a detailed analysis of the contribution of C-terminal residues to the repressed state, Kang et al (2001) found that deletion of five or more residues is required to destabilise the autoinhibited state, leading to a 10-fold activation of the enzyme. Examination of the Ppp5 structure indicates that the amide side chain of Gln495 (the fifth residue from the C-terminus) stabilises the tight turn immediately preceding the αJ helix by donating hydrogen bonds to the carbonyl oxygen atoms of Tyr489 and Ala490 (Figure 3C).
TPR-mediated inhibition resembles inhibition of PP1 by toxins and PP2B by its autoinhibitory domain
The mechanism of TPR-mediated inhibition of the Ppp5 phosphatase domain bears striking resemblance to mechanisms of inhibition of PP1 by natural product toxins and the autoinhibition of PP2B by its C-terminal regulatory segment. Superimposition of the phosphatase domains of the PP1–toxin complexes and PP2B onto the Ppp5 phosphatase domain exemplifies the nature of these similarities (Figure 4). Inhibition of phosphatase activity is achieved by blocking access to the catalytic binuclear metal centre. In Ppp5, the carboxylate group of Glu76 of the TPR domain contacts the side chain of one of the catalytic Arg residues (Arg275 in Ppp5) and Tyr451, being within 4.2 Å of the binuclear metal centre, although direct contacts between the negatively charged group and the two metals are not made. A similar structural feature is observed in the PP1–toxin complexes and in the autoinhibited state of PP2B. In the PP1–microcystin complex, a Glu residue of microcystin contacts Arg96 of the PP1 catalytic site (equivalent to Arg275 of Ppp5) (Goldberg et al, 1995), whereas in PP2B, Glu481 of the C-terminal autoinhibitory segment is shifted slightly relative to the position of Glu76 of Ppp5, and contacts the neighbouring catalytic Arg site residue, Arg254 (equivalent to Arg400 of Ppp5) (Kissinger et al, 1995). The carboxylic acid in the C1 position of okadaic acid contacts Arg96 of the PP1 catalytic site (Holmes et al, 2002). Finally, the anionic group of the calyculin A toxin is a phosphate moiety, and in the PP1–calyculin A structure this group contacts both catalytic site Arg residues of PP1 (Kita et al, 2002). Anionic groups on inhibitory toxins of PP1 and autoinhibitory segments of Ppp5 and PP2B interact with their respective phosphatase catalytic sites in a manner that partially mimics the mode of interaction between the phosphate group of a phosphorylated substrate and the catalytic site.
Figure 4.
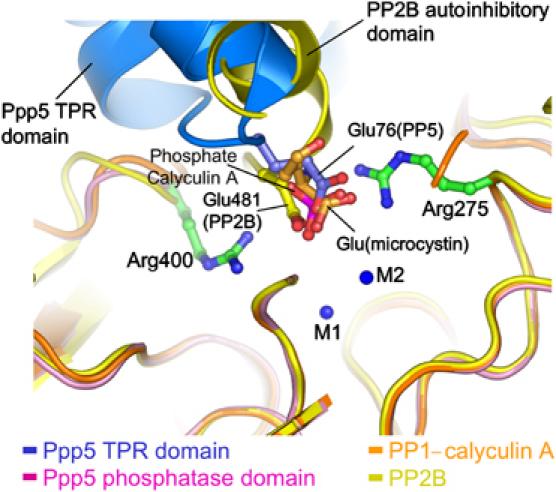
Inhibition of Ppp5 by the TPR domain Glu76 residue resembles inhibition of PP1 by toxins and autoinhibition of PP2B. Superimposition of the phosphatase domains of Ppp5, PP1 in complex with toxins microcystin and calyculin A and the autoinhibited state of PP2B shows a conserved anionic group that contacts the invariant catalytic site Arg-phosphate binding and Tyr residues of these phosphatases. For clarity, only the glutamate residue of microcystin and the phosphate group of calyculin A are shown.
Bacterially expressed and native mammalian Ppp5 are potently inhibited by both okadaic acid and microcystin (IC50 values of 1–7 and 1–15 nM, respectively) (Chen et al, 1994; Borthwick et al, 2001; Dean et al, 2001; Honkanen and Golden, 2002), and microcystin coupled to Sepharose was found to deplete 80% of Ppp5 from cauliflower extracts (Meek et al, 1999). These findings indicate that both toxins interact with the catalytic site of Ppp5 and, by analogy to PP1–toxin structures, are likely to engage the catalytic channel, which interacts with the TPR domain in the autoinhibited Ppp5 structure. Inhibition of Ppp5 by toxins will therefore be mutually exclusive with the suppression of phosphatase activity mediated by the TPR domain, and inhibition of Ppp5 by toxins can only be measured once the TPR domain has been displaced. Indeed Borthwick et al (2001) and Dean et al (2001) assayed the okadaic acid-mediated inhibition of Ppp5 by stimulating the enzyme with 100–200 μM polyunsaturated fatty acid, whereas Chen et al (1994) determined the toxin sensitivity of basal Ppp5 activity that results from limited proteolysis of Ppp5 causing removal of the inhibitory TPR domain and/or the C-terminal region. Plant Ppp5 is stimulated by arachidonic acid (Meek et al, 1999), suggesting that it has an autoinhibited structure similar to that of mammalian Ppp5. The efficient capture of plant Ppp5 on microcystin–Sepharose indicates that microcystin can displace the TPR domain from the phosphatase domain catalytic channel.
Activation of Ppp5 by TPR-mediated Hsp90 interactions
Ppp5 has been isolated from cells as a complex including both Hsp90 and Hsp70 (Chen et al, 1996; Silverstein et al, 1997; Zeke et al, 2004), and interactions between Ppp5 and Hsp90 are mediated by the Ppp5 TPR domain and C-terminus of Hsp90 (Russell et al, 1999; Ramsey et al, 2000). Interaction of Hsp70 with Ppp5 is also via the TPR domain of Ppp5 (Zeke et al, 2004) and therefore likely to involve the four C-terminal residues (EEVD) of Hsp70, which are identical to those in Hsp90. Biochemical and mutagenesis studies of Ppp5–TPR domain interactions with Hsp90 (Russell et al, 1999; Ramsey et al, 2000), together with the crystal structures of the TPR domains of Hop (Hsp70 and Hsp90 organising protein) with Hsp70 and Hsp90-based peptides (Scheufler et al, 2000) have provided insights into how the Ppp5 TPR domain interacts with Hsp70/90. In the structure of the TPR1 domain (which comprises three TPR motifs) of Hop in complex with a peptide based on the C-terminal seven residues of Hsp70 (Hsp70 peptide), the Hsp70 peptide interacts with the Hop TPR domain in an extended conformation (Scheufler et al, 2000). Peptide–protein interactions are dominated by hydrogen bonds involving the carboxylate groups of acidic residues and the C-terminus of the Hsp70/90 EEVD motif interacting with conserved Arg and Lys residues lining the basic peptide-binding channel of Hop.
Arg, Lys and Asn residues responsible for Hop–Hsp70/90 interactions are conserved in other Hsp70/90-binding TPR domains, including Ppp5 (Scheufler et al, 2000), and studies from Chinkers and co-workers (Russell et al, 1999) demonstrated that Lys32, Lys97 and Arg101 are required forHsp90–Ppp5 interactions. Lys97 and Arg101 are situated on adjacent helical turns of the αA helix of TPR-3, in close proximity to Lys32 on αA of TPR-1 (Figure 5). These residues together with Arg74 of αA of TPR-2 create a basic region at one side of the TPR peptide-binding channel for engagement of the Hsp70/90 acidic C-terminal region, and it is reasonable to assume that the Ppp5 TPR domain engages Hsp70/90 in a manner similar to that observed in the Hop–peptide complexes. The Hsp70 peptide binds to the Hop TPR domain such that the sequence of Hsp70 runs antiparallel with the αA helices of the TPR motifs. This places the C-terminus of the Hsp70 peptide close to the turns connecting adjacent TPR motifs (inter-repeat turns), whereas amino acids 6–7 residues N-terminal to the C-terminal Asp of Hsp70 (termed P-6 and P-7) are positioned adjacent to the intrarepeat turns (Scheufler et al, 2000) (Figure 5). The Ppp5 TPR domain engages the catalytic channel of the phosphatase domain mainly via these turns, whereas the basic region within the TPR peptide-binding channel formed from Lys32, Lys97 and Arg101 does not participate in the TPR–catalytic domain interface, but remains solvent exposed. Interestingly, in a site-directed mutagenesis study of Ppp5, Chinkers and co-workers (Kang et al, 2001) found that mutation of basic residues within the TPR peptide-binding groove had no affect on the basal activity of Ppp5, indicating that residues required (although not necessarily sufficient) to mediate TPR–Hsp90 interactions are distinct from those mediating autoinhibitory interactions with the phosphatase domain. The αJ helix, C-terminal to the phosphatase domain, however, interacts with the concave channel of the TPR domain, close to the intrarepeat turns. This association would partially block one side of the peptide-binding channel, preventing optimal peptide–TPR domain interactions. Superimposing the Hop–Hsp70 peptide complex onto the TPR domain of Ppp5 reveals that the C-terminal region of the peptide-binding groove of the Ppp5 TPR domain is accessible to the peptide, but that the αJ helix occludes the binding site for peptide residues P-5 to P-7, with these residues sterically overlapping the phosphatase αJ helix (Figure 5). Acidic residues of the EEVD motif are required for Ppp5–Hsp90 interactions, supporting the notion that the C-termini of Hsp90 and Hsp70 interact with the TPR domains of Ppp5 and Hop by similar mechanisms (Ramsey et al, 2000). In addition to these residues, an acidic region (residues −9 to −13 relative to the Hsp90 C-terminus) is also required for Hsp90–Ppp5 association (Ramsey et al, 2000), demonstrating that optimal interactions between Hsp90 and Ppp5 overlap the αJ helix–TPR contacts.
Figure 5.
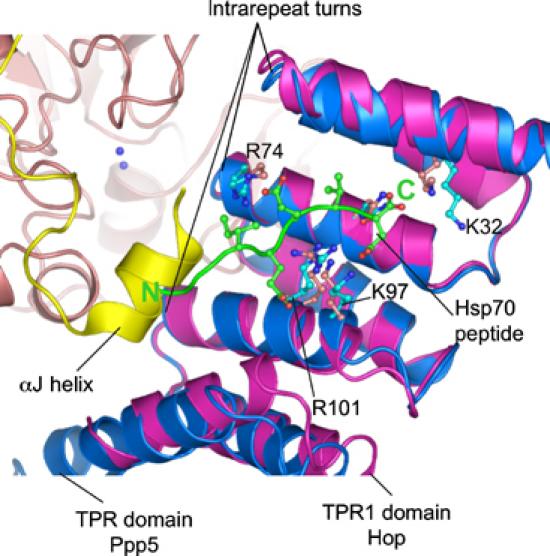
Model of Hsp90 activation of Ppp5 based on the structure of the Hop–Hsp70 peptide. The TPR domain (purple) of the Hop–Hsp70 peptide complex is superimposed onto the TPR domain of Ppp5 (blue). Peptide-binding basic residues are conserved in the TPR domains of both proteins, suggesting a related mechanism of peptide binding. The region of the Hsp70 peptide N-terminal to the IEEVD motif overlaps the position of the αJ helix, suggesting that optimal peptide binding to the Ppp5 TPR domain is mutually exclusive with TPR–phosphatase domain interactions. The N- and C-termini of the Hsp70 peptide are indicated by N and C, respectively. The C-terminus of Hsp90 (MEEVD) is predicted to bind to the Ppp5 TPR domain with a similar conformation as seen in the Hop–Hsp70 complex (see text for details).
Hsp90 activates Ppp5 by disrupting phosphatase–TPR domain interactions
Our structure of Ppp5 suggests that high-affinity TPR–Hsp90 interactions are mutually exclusive with TPR-mediated autoinhibition of the phosphatase domain. Although a region of the Hsp70/90 peptide-binding site is accessible in the autoinhibited structure, high-affinity binding requires access to sites that are buried at the TPR–phosphatase domain interface. For Hsp70 or Hsp90 to engage Ppp5 with high affinity, the TPR domain would require to dissociate from the phosphatase domain, exposing the constitutively active catalytic site, stimulating phosphatase activity. Conversely, the phosphatase domain, by blocking a region of the Hsp90 peptide-binding site within the TPR domain, is predicted to antagonise Hsp90 binding, lowering the affinity of Hsp90 for full-length Ppp5 relative to the isolated TPR domain. To test these proposals, we performed a variety of kinetic and isothermal titration calorimetry (ITC) experiments. First we tested whether full-length Hsp90 and a peptide corresponding to the C-terminus of Hsp90 are capable of stimulating Ppp5 activity. Figure 6A and B shows the dose-dependent activation of Ppp5 by both Hsp90 protein and peptide. Full-length Hsp90 is a moderate activator of Ppp5, inducing ∼7-fold stimulation of the enzyme with a half-maximal activation (AC50) of ∼2 μM. The human Hsp90α peptide (TSRMEEVD) induces a similar fold activation but with a greatly reduced AC50 constant of 57 μM, indicating a significantly weaker association between Ppp5 and peptide. These results are in agreement with previous findings that a 12 kDa C-terminal domain of Hsp90 promoted a 10-fold stimulation of Ppp5 with an AC50 constant of 6 μM (Ramsey and Chinkers, 2002). Using ITC, we measured the affinity of Hsp90 peptide for both the isolated TPR domain and for Ppp5 (Figure 6C and D). Significantly, Hsp90 peptide binds the isolated TPR domain tightly, with a KD of 40 nM, some 500-fold higher than its affinity for the full-length protein (KD of 20 μM).
Figure 6.
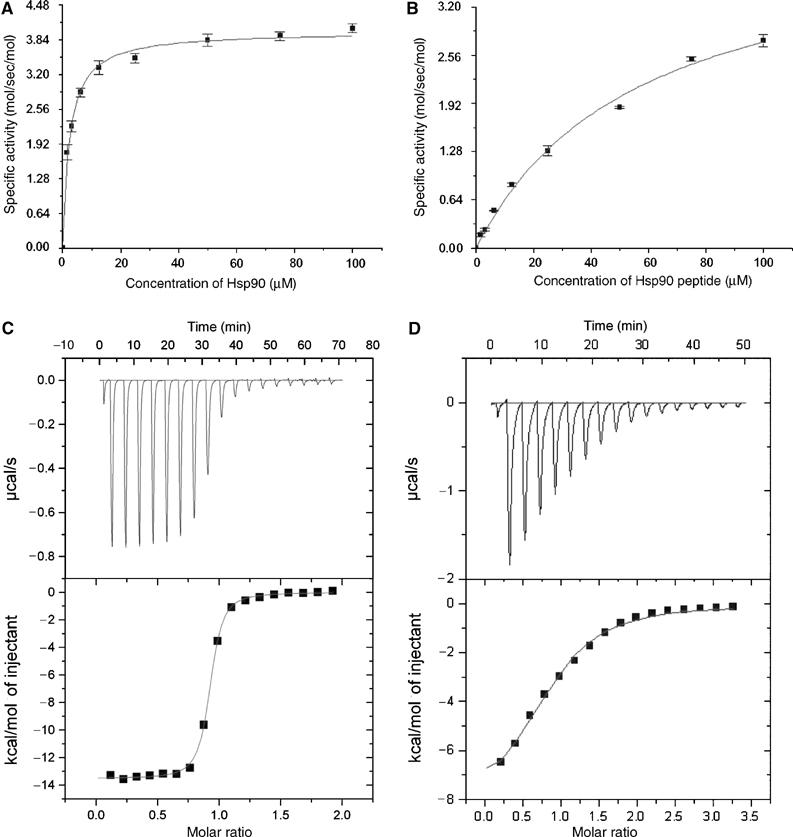
Hsp90 and Hsp90 peptides activate Ppp5 by binding to its TPR domain. (A) Dose-dependent activation of Ppp5 by full-length yeast Hsp90 and (B) Hsp90 C-terminal peptide. The data plotted are corrected for Ppp5 activity in the absence of activator. ITC plots for Hsp90 peptide interactions with (C) the isolated TPR domain (residues 19–146) and (D) full-length Ppp5.
These experiments suggest that the phosphatase domain partially blocks the Hsp90 peptide-binding site on the TPR domain in the context of the full-length protein, preventing optimal Hsp90–TPR domain interactions. Moreover, the relatively close correlation between the AC50 constant for the activation of Ppp5 by the Hsp90 peptide (57 μM) and the dissociation constant describing Hsp90–peptide–Ppp5 interactions (20 μM) suggests that binding of Hsp90 to Ppp5 is accompanied by activation of the phosphatase. Our findings that full-length Hsp90 is a more potent activator of Ppp5 than the C-terminal Hsp90 peptide indicate that residues N-terminal to the MEEVD motif, not present on our peptide, contribute to high-affinity Ppp5–Hsp90 interactions which could involve regions outside of the TPR domain. Consistent with the relative efficacies of Hsp90 and a short peptide to activate Ppp5, an acidic region (residues −9 to −13 relative to the Hsp90 C-terminus) is required for high-affinity Ppp5–Hsp90 interactions (Ramsey et al, 2000).
Arachidonoyl-CoA stimulates Ppp5 by inducing a structural change in the TPR domain
Soon after Ppp5's discovery, it was found to be stimulated by polyunsaturated fatty acids, with half-maximal activation at concentrations between 50 and 125 μM (Chen and Cohen, 1997; Skinner et al, 1997). That this activation involved the TPR domain was determined from the findings that only full-length Ppp5, and not the constitutively active catalytic domain, is activated by arachidonic acid (Chen and Cohen, 1997; Sinclair et al, 1999). Additionally, it has been shown that the TPR domain interacts with arachidonic acid, which protects against proteolytic cleavage of the TPR domain by subtilisin and trypsin (Sinclair et al, 1999). Moreover, Ppp5 and the isolated TPR domain, but not the isolated catalytic domain, interact directly with phosphatidylinositol lipids (Chen and Cohen, 1997). These original findings required concentrations of activator much greater than their physiological levels; however, recent data have extended these results revealing that long-chain fatty acyl-CoA esters activate Ppp5 at physiological concentrations of ∼1 μM (Ramsey and Chinkers, 2002). The CoA moiety functions to enhance the overall solubility of such long acyl chains in an aqueous environment. The most hydrophobic compounds, with chain lengths of 16 carbons or more, were found to be the most effective activators (Ramsey and Chinkers, 2002).
Some TPR domains have been found to be structurally unstable (Taylor et al, 2001; Jinek et al, 2004), and recent NMR data suggest that the isolated Ppp5 TPR domain, which is substantially unfolded at physiological relevant temperatures, becomes ordered in the presence of the Hsp90 MEEVD peptide (MJ Cliff et al, unpublished). Significantly, in the crystal structure of the isolated TPR domain, the molecule was stabilised by association of the C-terminus of a symmetry-related molecule into the peptide-binding groove (Das et al, 1998). Because of their extended conformation, TPR proteins differ from conventional globular proteins in having a high surface area to volume ratio and a relatively small hydrophobic core. These observations prompted us to test whether a possible mechanism of fatty acid-mediated activation of Ppp5 is due to fatty acid-induced disruption of the TPR structure, possibly by competing for the relatively weak hydrophobic interactions holding the fold together. Figure 7 shows the thermal-induced unfolding of the TPR domain as assayed by the α-helical content of the domain's circular dichroism signal. The addition of arachidonoyl-CoA promotes a significant reduction in α-helical content of the TPR domain below 300 K, and increases its melting temperature by ∼5°C. These results are consistent with the notion that arachidonoyl-CoA binds and stabilises an alternate conformation of the TPR domain with reduced helical content. Thus, one possible explanation for the ability of long-chain fatty acids to activate Ppp5 is that this induced conformational change results in the TPR domain being unable to form the specific contacts with the catalytic site channel of the Ppp5 phosphatase domain and αJ helix, required for autoinhibition.
Figure 7.
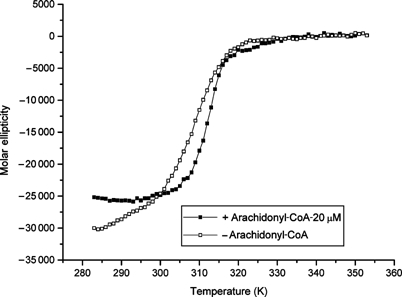
Arachidonoyl-CoA mediates TPR domain unfolding. Arachidonoyl-CoA affects the folding of the isolated TPR domain. The temperature dependence of the CD signal at 222 nm of amino acids 19–147 Ppp5 was recorded in the presence (closed symbols) and absence (open symbols) of 20 μM arachidonoyl-CoA. The melting temperature is increased from 34.6±0.3 to 39.9±0.2°C by the addition of arachidonoyl-CoA, although the apo-state loses helical content (∼15% of CD signal at 222 nm).
Concluding remarks
The structure of the autoinhibited state of Ppp5 reveals how the N-terminal TPR domain docks onto the phosphatase domain catalytic site thereby hindering access to substrate. Contacts between the phosphatase domain and TPR domain involve the intrarepeat turns of the TPR protein, a mode of TPR–protein interaction not observed previously. Interactions between the C-terminal αJ helix and the groove of the TPR domain stabilise the autoinhibited state, explaining biochemical data that the TPR domain and C-terminal 13 residues cooperate to suppress phosphatase activity. These interactions partially overlap the region of the TPR domain groove that is predicted to bind the C-terminus of Hsp90, indicating that sites within the TPR domain required for high-affinity Ppp5–Hsp90 interactions are blocked in the autoinhibited conformation. Hsp90 causes substantial activation of Ppp5 by competing for TPR–phosphatase domain contacts and allowing access to the catalytic site. The TPR domain of Ppp5 also interacts with other proteins, such as Ask1 (Morita et al, 2001), Gα12/Gα13 subunits (Yamaguchi et al, 2002) and DNA-PK (Wechsler et al, 2004), and it will be important to establish if these, and potentially other, TPR–protein interactions contribute to Ppp5 stimulation. The mechanism of Ppp5 autoinhibition by its N-terminal TPR domain, and activation by Hsp90, shares analogies to how SH2 domains regulate the activities of Src family kinases (Sicheri et al, 1997; Williams et al, 1997; Xu et al, 1997), and the protein tyrosine phosphatases SHP1/2 (Hof et al, 1998). In these proteins, the unliganded SH2 domains maintain their respective proteins in an autoinhibited state, by inducing an inactive catalytic domain, as in Src, or by blocking access to the catalytic site (SHP1/2). Engagement of phosphotyrosine proteins to their SH2 domains disrupts SH2–catalytic domain interactions, with concomitant stimulation of activity. Understanding how a TPR domain confers ligand-dependent regulation of enzyme activity provides new insights into the roles and mechanisms of TPR motifs, which occur in >150 human genes.
Materials and methods
Expression and purification of Ppp5, TPR domain and Hsp90
Human Ppp5 (residues 16–499) was amplified by PCR and cloned into the BamHI and XhoI sites of the pGEX 6P-2 vector with a His6 tag incorporated into the C-terminus of the protein. Expression of this protein was performed at 20°C overnight in Escherichia coli strain B834. Ppp5 was purified using a combination of glutathione–Sepharose affinity, S-Sepharose cation exchange (Amersham) and phenyl TSK hydrophobic interaction chromatography, prior to a desalting column, Ni-NTA agarose (Qiagen) and S200 gel filtration column (Amersham). The N-terminal His-tagged TPR domain (residues 19–146) was purified using Ni-NTA chromatography. His-tagged budding yeast Hsp90 was transformed into E. coli and purified (>90%) as described (Panaretou et al, 1998).
Crystallisation of Ppp5
Ppp5 was concentrated to 21 mg/ml. Crystals were grown using the under-oil batch method. Protein (1 μl) was mixed with an equal volume of crystallisation buffer (10% (w/v) polyethylene glycol 6000, 4% 2-methyl-2,4-pentanediol (MPD), 0.1 M NaHepes (pH 8.5) and 3 mM DTT) in individual wells of a 72-well polystyrene tray, immersed under silicon oil and incubated at 14°C.
Data collection and structure determination
Crystals were incubated in a cryoprotection buffer (13% (w/v) polyethylene glycol 6000, 18% MPD, 0.075 M NaHepes (pH 8.5)) for 20 s, prior to freezing in a nitrogen gas stream at 100 K. Data (low- and high-resolution passes) from a single crystal were collected at ID14-EH1, ESRF, Grenoble. Data were processed, scaled and merged using MOSFLM, TRUNCATE and SCALA (CCP4, 1991) (Table I).
Table 1.
Data collection and processing statistics
| Native Ppp5 data | |
|---|---|
| Data collection | ID14-EH1, ESRF |
| Wavelength (Å) | 0.979 |
| Space group | P21 |
| Unit cell (Å) | a=51.74, b=117.54, c=200.41, β=93.79° |
| Resolution (Å) | 30–2.9 (3.11–2.9) |
| Observations (N) | 369 163 (53 949) |
| Unique reflections (N) | 53 119 (10 083) |
| Redundancy | 6.9 (5.4) |
| Completeness (%) | 99.9 (99.9) |
| Rmerge | 0.118 (0.423) |
| I/σI | 3.4 (1.4) |
| Z | 4 |
| Refinement statistics | |
| Protein atoms (N) | 15 082 |
| Rcryst | 0.247 |
| Rfree | 0.291 |
| R.m.s.d. bond lengths (Å) | 0.0086 |
| R.m.s.d. bond angles (deg) |
1.37 |
| Highest shell values are in parentheses. |
Ppp5 structure was solved by molecular replacement methods with the initial phasing obtained by determining the position of the catalytic domain of the molecule using the structure of the catalytic subunit of PP1 as a search object with CNS (Brünger et al, 1998). Assuming a solvent content close to 50% predicts four molecules of Ppp5 per asymmetric unit; however, only two distinct orientations of PP1 could be obtained in cross-rotation searches. A strong self-vector peak at U=0.444, V=0.5, W=0.3644 on a native Patterson map indicated that two or more molecules per asymmetric unit shared identical orientations. We applied the translation vector to each of the two positioned PP1 catalytic subunits to generate the position of two other molecules. Attempts to position the TPR domain by molecular replacement methods failed; however, low-resolution refinement (8 Å) and atomic positional refinement at 3 Å, applying NCS restraints, and incorporation of the Ppp5 sequence improved phases sufficiently so that 2Fo−Fc maps revealed positions of three α-helices of the TPR domain. Iterative cycles of fitting TPR helices to electron density maps and refinement allowed the entire TPR domain to be located. The structure was refined using CNS (Brünger et al, 1998) and modelling building was performed using O (Jones et al, 1991).
Isothermal titration calorimetry
ITC was performed using a VP-ITC MicroCalorimeter (MicroCal). All protein and peptide samples were dialysed into a buffer comprising 25 mM Tris.HCl (pH 8.0), 50 mM NaCl, 1 mM MgCl2 and 3 mM DTT. Titrations were performed by injecting 19 consecutive aliquots (15 μl) of human Hsp90α peptide (sequence TSRMEEVD) into the ITC cell containing Ppp5 or the MEEVD peptide with the TPR domain (residues 19–146) at 25°C. ITC data were corrected for heats of dilution of the protein solution. Binding stoichiometry, enthalpy, entropy and binding constants were determined by fitting the corrected data to a bimolecular interaction model.
Activation of Ppp5 by HSP90 and peptide of HSP90
Cuvettes containing 50 mM pNPP, 0.5 mM MnCl2 and 50 mM NaCl in 100 mM Tris, pH 8.0, and activator (either full-length yeast Hsp90 or TSRMEEVD human Hsp90α peptide) were mixed at 22°C. The reaction was started by adding Ppp5 to a final amount of 0.5 μg. Production of pNP was followed by monitoring the increase in the absorbance at 410 nm. Standard error values were obtained from assays performed in triplicate. Activity was expressed as moles of pNP released per mole of Ppp5 per second, using a molar extinction coefficient of 15.1 × 103 mol pNP/L/cm (Ramsey and Chinkers, 2002).
Influence of arachidonoyl-CoA on the helical content and thermal stability of the TPR domain
The circular dichroism at 222 nm of 10 μM TPR domain of protein phosphatase 5 in 20 mM Tris, 50 mM NaCl and 5 mM dithioerythritol (DTE) was recorded for 1 min at 1°C intervals between 10 and 80°C with an equilibration time of 3 min between temperatures and a bandwidth of 2 nm, using an Aviv 202SF CD spectrometer with and without 20 μM arachidonoyl-CoA.
Acknowledgments
This work was funded by Cancer Research-UK (DB), the BBSRC (DB, JEL, MAW) and the Medical Research Council, UK (PTWC). JEL is a Wellcome Trust Senior Research Fellow. We are grateful for staff at ID14-EH1, ESRF for help with data collection and to L Pearl for discussions, and C Prodromou and C Vaughan for Hsp90 expression plasmids. Coordinates have been assigned ID codes 1wao.
References
- Ali A, Zhang J, Bao S, Liu I, Otterness D, Dean NM, Abraham RT, Wang XF (2004) Requirement of protein phosphatase 5 in DNA-damage-induced ATM activation. Genes Dev 18: 249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva AV, Kutuzov MA (1999) RdgC/PP5-related phosphatases: novel components in signal transduction. Cell Signal 11: 555–562 [DOI] [PubMed] [Google Scholar]
- Barton GJ, Cohen PTW, Barford D (1994) Conservation analysis and structure prediction of the protein serine/threonine phosphatases: sequence similarity with diadenosine tetraphosphatase from E. coli suggests homology to the protein phosphatases. Eur J Biochem 220: 225–237 [DOI] [PubMed] [Google Scholar]
- Borthwick EB, Zeke T, Prescott AR, Cohen PTW (2001) Nuclear localization of protein phosphatase 5 is dependent on the carboxy-terminal region. FEBS Lett 491: 279–284 [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- CCP4 (1991) The CCP4 Suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Chen M-S, Silverstein AM, Pratt WB, Chinkers M (1996) The tetratricopeptide repeat domain of protein phosphatase 5 mediates binding to glucocorticoid receptor heterocomplexes and acts as a dominant negative mutant. J Biol Chem 271: 32315–32320 [DOI] [PubMed] [Google Scholar]
- Chen MX, Cohen PTW (1997) Activation of protein phosphatase 5 by limited proteolysis or the binding of fatty acids to the TPR domain. FEBS Lett 400: 136–140 [DOI] [PubMed] [Google Scholar]
- Chen MX, McPartlin AE, Brown L, Chen YH, Barker HM, Cohen PTW (1994) A novel human protein serine/threonine phosphatase, which possesses four tetratricopeptide repeat motifs and localizes to the nucleus. EMBO J 13: 4278–4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinkers M (1994) Targeting of a distinctive protein-serine phosphatase to the protein kinase-like domain of the atrial natriuretic peptide receptor. Proc Natl Acad Sci USA 91: 11075–11079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinkers M (2001) Protein phosphatase 5 in signal transduction. Trends Endocrinol Metab 12: 28–32 [DOI] [PubMed] [Google Scholar]
- Cliff MJ, Williams MA, Brooke-Smith J, Barford D, Ladbury JE. Molecular recognition via coupled folding and binding in a TPR domain (submitted) [DOI] [PubMed]
- Cohen PTW (1997) Novel protein serine/threonine phosphatases: variety is the spice of life. Trends Biochem Sci 22: 245–251 [DOI] [PubMed] [Google Scholar]
- Das AK, Cohen PTW, Barford D (1998) The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein–protein interactions. EMBO J 15: 1192–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean DA, Urban G, Aragon IV, Swingle M, Miller B, Rusconi S, Bueno M, Dean NM, Honkanen RE (2001) Serine/threonine protein phosphatase 5 (PP5) participates in the regulation of glucocorticoid receptor nucleocytoplasmic shuttling. BMC Cell Biol 2: 6. Epub 2001 May 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff M-P, Cohen PTW, Reinemer P, Barford D (1995) Crystal structure of the catalytic subunit of human protein phosphatase 1 and its complex with tungstate. J Mol Biol 254: 942–959 [DOI] [PubMed] [Google Scholar]
- Goldberg J, Huang HB, Kwon YG, Greengard P, Nairn AC, Kuriyan J (1995) Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature 376: 745–753 [DOI] [PubMed] [Google Scholar]
- Griffith JP, Kim JL, Kim EE, Sintchak MD, Thomson JA, Fitzgibbon MJ, Fleming MA, Caron PR, Hsiao K, Navia MA (1995) X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12–FK506 complex. Cell 82: 507–522 [DOI] [PubMed] [Google Scholar]
- Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE (1998) Crystal structure of the tyrosine phosphatase SHP-2. Cell 92: 441–450 [DOI] [PubMed] [Google Scholar]
- Holmes CF, Maynes JT, Perreault KR, Dawson JF, James MN (2002) Molecular enzymology underlying regulation of protein phosphatase-1 by natural toxins. Curr Med Chem 9: 1981–1989 [DOI] [PubMed] [Google Scholar]
- Honkanen RE, Golden T (2002) Regulators of serine/threonine protein phosphatases at the dawn of a clinical era? Curr Med Chem 9: 2055–2075 [DOI] [PubMed] [Google Scholar]
- Huang S, Shu L, Easton J, Harwood FC, Germain GS, Ichijo H, Houghton PJ (2004) Inhibition of mammalian target of rapamycin activates apoptosis signal-regulating kinase 1 signalling by suppressing protein phosphatase 5 activity. J Biol Chem 279: 36490–36496 [DOI] [PubMed] [Google Scholar]
- Jinek M, Rehwinkel J, Lazarus BD, Izaurralde E, Hanover JA, Conti E (2004) The superhelical TPR-repeat domain of O-linked GlcNAc transferase exhibits structural similarities to importin alpha (2004). Nat Struct Mol Biol 11: 1001–1007 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kang H, Sayner SL, Gross KL, Russell LC, Chinkers M (2001) Identification of amino acids in the tetratricopeptide repeat and C-terminal domains of protein phosphatase 5 involved in autoinhibition and lipid activation. Biochemistry 40: 10485–10490 [DOI] [PubMed] [Google Scholar]
- Kissinger CR, Parge HE, Knighton DR, Lewis CT, Pelletier LA, Tempczyk A, Kalish VJ, Tucker KD, Showalter RE, Gastinel LN, Habuka N, Chen X, Maldonado F, Barker JE, Bacquet R, Villafranca JE (1995) Crystal structures of human calcineurin and the human FKBP12–FK506–calcineurin complex. Nature 378: 641–644 [DOI] [PubMed] [Google Scholar]
- Kita A, Matsunaga S, Takai A, Kataiwa H, Wakimoto T, Fusetani N, Isobe M, Miki K (2002) Crystal structure of the complex between calyculin A and the catalytic subunit of protein phosphatase 1. Structure 10: 715–724 [DOI] [PubMed] [Google Scholar]
- Lubert EJ, Hong Y, Sarge KD (2001) Interaction between protein phosphatase 5 and the A subunit of protein phosphatase 2A. J Biol Chem 276: 38582–38587 [DOI] [PubMed] [Google Scholar]
- Maynes JT, Bateman KS, Cherney MM, Das AK, Luu HA, Holmes CF, James MN (2001) Crystal structure of the tumor-promoter okadaic acid bound to protein phosphatase-1. J Biol Chem 276: 44078–44082 [DOI] [PubMed] [Google Scholar]
- Meek S, Morrice N, MacKintosh C (1999) Microcystin affinity purification of plant protein phosphatases: PP1C, PP5 and a regulatory A-subunit of PP2A. FEBS Lett 457: 494–498 [DOI] [PubMed] [Google Scholar]
- Morita K, Saitoh M, Tobiume K, Matsuura H, Enomoto S, Nishitoh H, Ichijo H (2001) Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J 20: 6028–6036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollendorf V, Donoghue DJ (1997) The serine/threonine phosphatase Ppp5 interacts with CDC16 and CDC27, two tetratricopeptide repeat-containing subunits of the anaphase-promoting complex. J Biol Chem 272: 32011–32018 [DOI] [PubMed] [Google Scholar]
- Panaretou B, Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH (1998) ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J 117: 4829–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey AJ, Chinkers M (2002) Identification of potential physiological activators of protein phosphatase 5. Biochemistry 41: 5625–5632 [DOI] [PubMed] [Google Scholar]
- Ramsey AJ, Russell LC, Whitt SR, Chinkers M (2000) Overlapping sites of tetratricopeptide repeat protein binding and chaperone activity in heat shock protein 90. J Biol Chem 275: 17857–17862 [DOI] [PubMed] [Google Scholar]
- Russell LC, Whitt SR, Chen M-S, Chinkers M (1999) Identification of conserved residues required for the binding of a tetratricopeptide repeat domain to heat shock protein 90. J Biol Chem 274: 20060–20063 [DOI] [PubMed] [Google Scholar]
- Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I (2000) Structure of TPR domain–peptide complexes: critical elements in the assembly of the Hsp70–Hsp90 multichaperone machine. Cell 101: 199–210 [DOI] [PubMed] [Google Scholar]
- Shao J, Hartson SD, Matts RL (2002) Evidence that protein phosphatase 5 functions to negatively modulate the maturation of the Hsp90-dependent heme-regulated eIF2α kinase. Biochemistry 41: 6770–6779 [DOI] [PubMed] [Google Scholar]
- Sicheri F, Moarefi I, Kuriyan J (1997) Crystal structure of the Src family tyrosine kinase Hck. Nature 385: 602–609 [DOI] [PubMed] [Google Scholar]
- Silverstein AM, Galigniana MD, Chen M-S, Owens-Grillo JK, Chinkers M, Pratt WB (1997) Protein phosphatase 5 is a major component of glucocorticoid receptor–hsp90 complexes with properties of an FK506-binding immunophilin. J Biol Chem 272: 16224–16230 [DOI] [PubMed] [Google Scholar]
- Sinclair C, Borchers C, Parker C, Tomer K, Charbonneau H, Rossie S (1999) The tetratricopeptide repeat domain and a C-terminal region control the activity of Ser/Thr protein phosphatase 5. J Biol Chem 274: 23666–23672 [DOI] [PubMed] [Google Scholar]
- Skinner J, Sinclair C, Romeo C, Armstrong D, Charbonneau H, Rossie S (1997) Purification of a fatty acid-stimulated protein-serine/threonine phosphatase from bovine brain and its identification as a homolog of protein phosphatase 5. J Biol Chem 272: 22464–22471 [DOI] [PubMed] [Google Scholar]
- Swingle MR, Honkanen RE, Ciszak EM (2004) Structural basis for the catalytic activity of human serine/threonine phosphatase 5. J Biol Chem 279: 33992–33999 [DOI] [PubMed] [Google Scholar]
- Taylor P, Dornan J, Carrello A, Minchin RF, Ratajczak T, Walkinshaw MD (2001) Two structures of cyclophilin 40: folding and fidelity in the TPR domains. Structure 9: 431–438 [DOI] [PubMed] [Google Scholar]
- Urban G, Golden T, Aragon IV, Cowsert L, Cooper SR, Dean NM, Honkanen RE (2003) Identification of a functional link for the p53 tumor suppressor protein in dexamethasone-induced growth suppression. J Biol Chem 278: 9747–9753 [DOI] [PubMed] [Google Scholar]
- Wechsler T, Chen BP, Harper R, Morotomi-Yano K, Huang BC, Meek K, Cleaver JE, Chen DJ, Wabl M (2004) DNA-PKcs function regulated specifically by protein phosphatase 5. Proc Natl Acad Sci USA 101: 1247–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JC, Weijland A, Gonfloni S, Thompson A, Courtneidge SA, Superti-Furga G, Wierenga RK (1997) The 2.35 Å crystal structure of the inactivated form of chicken Src: a dynamic molecule with multiple regulatory interactions. J Mol Biol 274: 757–775 [DOI] [PubMed] [Google Scholar]
- Xu W, Harrison SC, Eck MJ (1997) Three-dimensional structure of the tyrosine kinase c-Src. Nature 385: 595–602 [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Katoh H, Mori K, Negishi M (2002) Galpha(12) and Galpha(13) interact with Ser/Thr protein phosphatase type 5 and stimulate its phosphatase activity. Curr Biol 12: 1353–1358 [DOI] [PubMed] [Google Scholar]
- Zeke T, Morrice N, Vázquez-Martin C, Cohen PTW (2004) Human protein phosphatase 5 dissociates from heat shock proteins and is proteolytically activated in response to arachidonic acid and the microtubule depolymerising drug nocodazole. Biochem J Epub 21st September 2004 BJ20040690 [DOI] [PubMed] [Google Scholar]
- Zhao S, Sancar A (1997) Human blue-light photoreceptor hCRY2 specifically interacts with protein serine/threonine phosphatase 5 and modulates its activity. Photochem Photobiol 66: 727–731 [DOI] [PubMed] [Google Scholar]
- Zhou G, Golden T, Aragon IV, Honkanen RE (2004) Ser/thr protein phosphatase 5 (PP5) inactivates hypoxia-induced activation of an ASK-1/MKK-4/JNK-signalling cascade. J Biol Chem 279: 46595–46605 [DOI] [PubMed] [Google Scholar]
- Zuo Z, Dean NM, Honkanen RE (1998) Serine/threonine protein phosphatase type 5 acts upstream of p53 to regulate the induction of p21WAF1/Cip1 and mediate growth arrest. J Biol Chem 273: 12250–12258 [DOI] [PubMed] [Google Scholar]
- Zuo Z, Urban G, Scammell JG, Dean NM, McLean TK, Aragon I, Honkanen RE (1999) Ser/Thr protein phosphatase type 5 (PP5) is a negative regulator of glucocorticoid receptor-mediated growth arrest. Biochemistry 38: 8849–8857 [DOI] [PubMed] [Google Scholar]