

Abstract
Cellular quality control requires recognition of common features of misfolding, and so is not typically associated with the specific targeting of individual proteins. However, physiologically regulated degradation of yeast HMG-CoA reductase (Hmg2p) occurs by the HRD endoplasmic reticulum quality control pathway, implying that Hmg2p undergoes a regulated transition to a quality control substrate in response to a sterol pathway molecule. Using in vitro structural assays, we now show that the pathway derivative farnesol causes Hmg2p to undergo a change to a less folded structure. The effect is reversible, biologically relevant by numerous criteria, highly specific for farnesol structure, and requires an intact Hmg2p sterol-sensing domain. This represents a distinct lipid-sensing function for this highly conserved motif that suggests novel approaches to cholesterol management. More generally, our observation of reversible small-molecule-mediated misfolding may herald numerous examples of regulated quality control to be discovered in biology or applied in the clinic.
Keywords: ERAD, farnesol, HMG-CoA reductase, quality control, sterol-sensing domain
Introduction
Protein degradation fills two distinct roles in the cell: regulation and quality control. In regulation, a normal, functional protein is specifically recognized by degradation machinery to control a particular cellular process. In contrast, quality control requires recognition of structural hallmarks that distinguish the large set of abnormal proteins from their properly folded and assembled counterparts, independent of specific protein sequence and identity. Consequently, quality control and regulation are traditionally seen as unrelated processes. However, it is entirely reasonable that the high specificity of quality control could be harnessed by the cell for protein regulation. In studying the sterol pathway enzyme HMG-COA reductase (HMGR), we have found an example of this interface of regulation and quality control (Hampton, 2002a, 2002b), and show in the studies below that a reversible, small-molecule-mediated transition to a quality control substrate can be employed in the selective control of protein levels.
HMGR catalyzes a key step in sterol synthesis and is the primary drug target for treatment of high cholesterol. Both mammalian HMGR and the yeast isozyme Hmg2p undergo feedback-regulated, ubiquitin-mediated degradation in response to a specific signal from the sterol pathway (Ravid et al, 2000; Hampton, 2002b; Sever et al, 2003). Increases in the signal lead to increased degradation and a drop in enzyme steady-state level. Conversely, decreased signal leads to decreased degradation and a rise in steady-state level. This degradation leads to rapid control of Hmg2p level in response to changing demand for sterol pathway products.
In our genetic analysis of yeast Hmg2p-regulated degradation, we identified Hrd1p as the E3 ubiquitin ligase that mediates ubiquitination of Hmg2p (Hampton et al, 1996; Hampton and Bhakta, 1997; Gardner et al, 2000; Bays et al, 2001). Surprisingly, Hrd1p is a key quality control E3 of the endoplasmic reticulum (ER), required for degradation of numerous misfolded proteins, such as soluble CPY* and membrane-bound Pdr5p*, and for maintaining an acceptable level of normally produced misfolded proteins in the ER (Bordallo et al, 1998; Friedlander et al, 2000; Wilhovsky et al, 2000; Bays et al, 2001). Thus, the degradation of Hmg2p is an intersection between regulation and quality control. It is our goal to understand the underlying mechanism, both to allow comprehension of HMGR regulation, and as an example of harnessing quality control pathways for regulatory purposes in nature and the clinic.
Hmg2p catalyzes the synthesis of mevalonic acid, an early molecule in the sterol pathway. The signal for Hmg2p degradation is derived from the later pathway intermediate farnesyl pyrophosphate (FPP). When cellular FPP is abundant, the Hmg2p degradation rate is high; when the FPP level is low, the Hmg2p degradation rate is low. Mature Hmg2p is subject to this stability control and can undergo changes in degradative status rapidly and reversibly following changes in the FPP-derived signal (Hampton and Bhakta, 1997; Gardner and Hampton, 1999b). From our in vivo studies, we proposed that Hmg2p acquires structural features of a quality control substrate in response to the FPP-derived signal (Figure 1A) (Gardner et al, 2001b; Hampton, 2002a, 2002b). This regulated structural transition would occur in mature protein and be reversed rapidly when signal decreases. By this model, Hmg2p's status as a quality control substrate is regulated, so that when the FPP-derived signal is abundant Hmg2p acquires traits of a misfolded protein that allow Hrd1p-dependent degradation.
Figure 1.
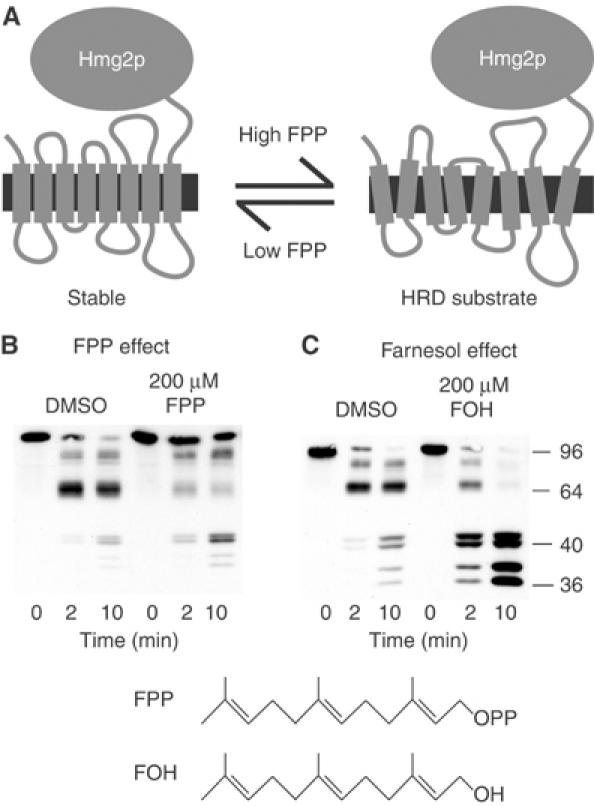
Farnesol but not FPP increased Hmg2p-GFP proteolysis rate. (A) Structural transition model of Hmg2p-regulated degradation. The large catalytic domain is on the cytosolic face of ER. (B) Effect of FPP. Microsomes bearing Hmg2p-GFP were resuspended in buffer with DMSO or 200 μM FPP, and then incubated with trypsin (15 μg/ml) at 30°C. Samples were removed at the indicated times and evaluated by 14% SDS–PAGE and immunoblotting of the lumenal Myc epitope. (C) Effect of farnesol (FOH). Microsomes bearing Hmg2p-GFP were resuspended in buffer with DMSO or 200 μM FOH, and then incubated with trypsin and evaluated as described above. Molecular weight markers (in kDa) are shown.
In vivo evidence for this structural transition model was obtained through subsequent experiments with chemical chaperones. These small molecules have been used to enhance protein folding in vivo and in vitro (Brown et al, 1996; Sato et al, 1996; Welch and Brown, 1996). Treatment of yeast with chemical chaperones such as glycerol strongly stabilizes Hmg2p, but has no effect on constitutively degraded ER degradation substrates (Gardner et al, 2001b; Shearer and Hampton, 2004). Hmg2p stabilization by chemical chaperones is rapidly reversible, implying that Hmg2p can undergo reversible changes in structure. In those studies, we devised a limited proteolysis assay to evaluate the structure of membrane-localized Hmg2p in response to chemical chaperones (Shearer and Hampton, 2004). As predicted from the in vivo stabilization, the presence of chemical chaperones dramatically reduces in vitro Hmg2p proteolysis, and this effect on Hmg2p structure is completely and rapidly reversible. Furthermore, altering the in vivo FPP-derived degradation signal with drugs has the expected effects on subsequently measured Hmg2p in vitro protease sensitivity: Hmg2p protease accessibility is higher in membranes prepared from cells with elevated FPP, and lower in membranes prepared from cells with diminished FPP. Thus, it seems that the in vivo degradation signal causes Hmg2p to be better recognized as an Hrd1p substrate.
The Hmg2p N-terminal transmembrane domain is both necessary and sufficient for proper regulated degradation (Hampton and Rine, 1994; Gardner and Hampton, 1999a). The simplest model for Hmg2p regulations is that an FPP-derived signal directly causes the Hmg2p transmembrane domain to adopt structural features of a quality control substrate, thus rendering the protein susceptible to Hrd1p-dependent degradation (Figure 1A). In this work, we demonstrate the validity of this structural transition model, and show that the FPP-derived molecule farnesol causes specific, reversible misfolding of Hmg2p in isolated ER microsomes. This in vitro conformational change is the same as that observed in vivo, biologically relevant by numerous criteria, and requires specific structural features in the signaling lipid. Thus, Hmg2p regulation provides an example of small molecule control of protein structure that allows programmed entry into a constitutive quality control pathway.
Results
In our previous work, we developed an in vitro limited proteolysis assay to study Hmg2p structure in isolated ER microsomes (Shearer and Hampton, 2004). We have used this assay in the studies below, revealing that a biological signal derived from the sterol pathway reversibly alters the folding state of Hmg2p. The assay was developed by constructing mycL-Hmg2p-GFP, a version of Hmg2p with the catalytic domain replaced with GFP, and a myc tag added to the first lumenal loop (Shearer and Hampton, 2004). The resulting noncatalytic protein, hereafter referred to as Hmg2p-GFP, is regulated normally in vivo. The use of GFP allows facile in vivo evaluation of protein levels by fluorescent techniques (Cronin and Hampton, 1999), and restricts the observed effects to the membrane domain, which is necessary and sufficient for FPP-mediated regulation of the entire Hmg2p molecule. Trypsin treatment of this protein in microsomes generates a characteristic time-dependent proteolytic pattern that is sensitive to chemical chaperones and alterations of the in vivo degradation signal. Specifically, these treatments cause changes in the rate of in vitro trypsinolysis without perturbing the pattern or the final products (Shearer and Hampton, 2004). For example, lowering in vivo FPP levels causes slower in vitro trypsinolysis of Hmg2p, whereas increasing in vivo FPP levels causes hastened in vitro trypsinolysis (Shearer and Hampton, 2004). We have now used this assay to investigate the nature and action of the degradation signal.
A central question in Hmg2p regulation is whether the FPP-derived signal works directly on the protein to effect structural change, or whether this occurs through another molecule or mechanism. We addressed this question by testing the effect of this pathway intermediate in the in vitro limited proteolysis assay (Figure 1B). FPP induced only a small increase in trypsinolysis rate (fold change of 1.80±0.08), so we turned our attention to the FPP-derived molecule farnesol.
When FPP is made more abundant in yeast by pharmacological or genetic means, the majority of the FPP is rapidly converted to the 15-carbon neutral lipid farnesol, making it a candidate for the in vivo degradation signal (Chambon et al, 1990; Gardner et al, 2001a). Indeed, treatment of ER microsomes with farnesol caused a dose-dependent increase in trypsinolysis rate of the lumenally tagged reporter that was maximal at 200 μM (fold change of 4.85±1.11) (Figure 1C). Note that this effect involved a change in rate but not proteolytic pattern. Longer incubation of untreated microsomes with trypsin results in the same characteristic quartet of bands seen after a much shorter incubation with farnesol (Shearer and Hampton, 2004). This increased accessibility was also observed using a separate, normally regulated Hmg2p-GFP variant tagged in a small portion of its fourth lumenal loop (unpublished data). Farnesol's effect on the lumenal reporter was saturable, reaching maximal effect near 200 μM, well below concentrations that alter membrane integrity—around 1 mM (unpublished data). This effect of farnesol was rapid, occurring with no need for preincubation (unpublished data).
We next evaluated the structural specificity of this effect. Farnesol is a 15-carbon hydrophobic alcohol that is potentially capable of affecting membrane dynamics independent of its own specific molecular structure. Thus, we examined the effects of several closely related lipids using the in vitro assay (see Figure 2D for structures). In striking contrast to farnesol, the linear 14- and 16-carbon alcohols tetradecanol and hexadecanol had no effect on Hmg2p trypsinolysis (Figure 2A), indicating that farnesol's effect could not be duplicated by simple lipids with similar properties.
Figure 2.
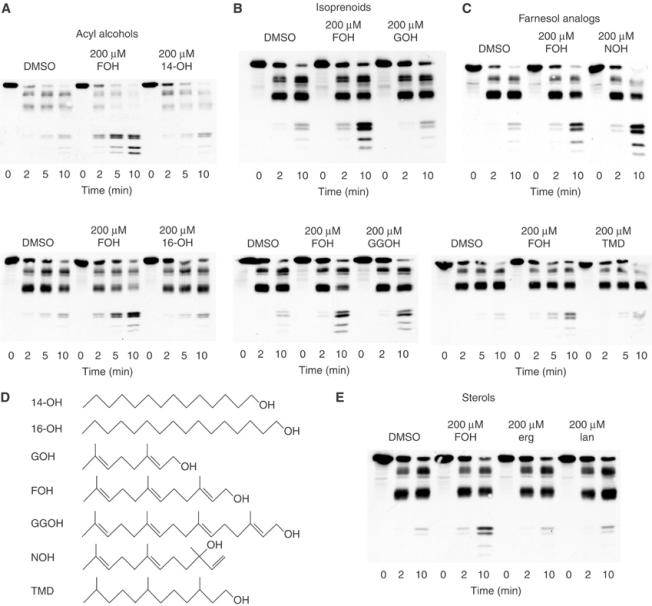
Lipid specificity of the farnesol effect. (A) Effects of the similarly sized alcohols tetradecanol (14-OH) and hexadecanol (16-OH). Microsomes bearing Hmg2p-GFP were resuspended in buffer with DMSO, 200 μM FOH, 200 μM 14-OH, or 200 μM 16-OH, and then incubated with trypsin (15 μg/ml) at 30°C. Samples were removed at the indicated times and evaluated by 14% SDS–PAGE and immunoblotting of the Myc epitope. (B) Effects of related isoprenoids geraniol (GOH) and geranylgeraniol (GGOH). Evaluation was as described in panel A. (C) Effects of the farnesol isomer nerolidol (NOH) and of saturated farnesol (TMD). Evaluation was as described in panel A. (D) Structures of the compounds tested. (E) Effects of the yeast sterols ergosterol (erg) and lanosterol (lan). Evaluation was as described in panel A.
We then tested several more structurally and biologically related compounds. The compound 10-carbon geraniol had no effect on the trypsinolysis rate of Hmg2p, whereas 20-carbon geranylgeraniol caused slightly more than a two-fold increase (2.48±0.04) (Figure 2B). Nerolidol, an isomer of farnesol, was as effective as farnesol in causing Hmg2p structural transition (Figure 2C). Fully hydrogenated farnesol, 3,7,11-trimethyldodecan-1-ol (TMD), had minimal effect (fold change of 1.46±0.31), demonstrating that farnesol's unique isoprene structure was necessary for promotion of the Hmg2p structural transition (Figure 2C). Given that Hmg2p is an integral membrane protein, it was important to consider the ability of the molecules tested to enter the membrane. According to published partition coefficients, all the small molecules evaluated except geraniol will partition almost completely (99% or more) into the lipid phase in our in vitro system, indicating equal accessibility to Hmg2p (Leo et al, 1971; Roullet et al, 1997). Even by our most conservative estimate, geraniol should partition at least 70% into the lipid phase. Testing with a higher concentration of geraniol to ensure amounts in the membrane equivalent to at least a 200 μM dose of the other molecules tested yielded no effect on Hmg2p trypsinolysis (unpublished data). Thus, the action of farnesol on Hmg2p-GFP structure was highly specific for a variety of features of this molecule.
Unlike Hmg2p, mammalian HMGR stability is regulated by sterols (Chin et al, 1985; Sever et al, 2003). Accordingly, we also tested the yeast sterols ergosterol and lanosterol in the limited proteolysis assay. As predicted from in vivo regulation of Hmg2p stability, neither sterol had any effect on Hmg2p-GFP conformation (Figure 2E), even when tested at concentrations 40-fold above sterol concentrations that affect mammalian SCAP (Brown et al, 2002).
The in vivo regulation of Hmg2p by the FPP-derived signal requires an intact COD1 gene and correct Hmg2p sequence. As a test of biological relevance, we evaluated the in vitro effect of farnesol on Hmg2p in these two circumstances where regulation is absent.
Cod1p is a P-type ATPase with unknown specificity that we discovered in a screen for genes required for proper regulation of Hmg2p (Cronin et al, 2000, 2002). In a cod1Δ mutant, Hmg2p undergoes constitutive HRD-dependent degradation that is largely unresponsive to alterations in FPP level. Using the limited proteolysis assay, we have recently shown that in the cod1Δ genetic background, Hmg2p has structural alterations that probably underlie its unresponsiveness to the degradation signal (Shearer and Hampton, 2004). Specifically, note that the quartet of bands in the 36–42 kDa range is replaced by a doublet, probably due to the increased N-terminal proteolysis that leads to appearance of the 16 kDa band at the bottom of the gel. This doublet was assayed for change in response to farnesol treatment, as it represents the same proteolysis sites as the quartet used in all other cases. As predicted from the in vivo lack of regulation, farnesol had minimal effect on in vitro proteolysis of Hmg2p in microsomes from a cod1Δ strain (fold change 1.60±0.03) (Figure 3A).
Figure 3.
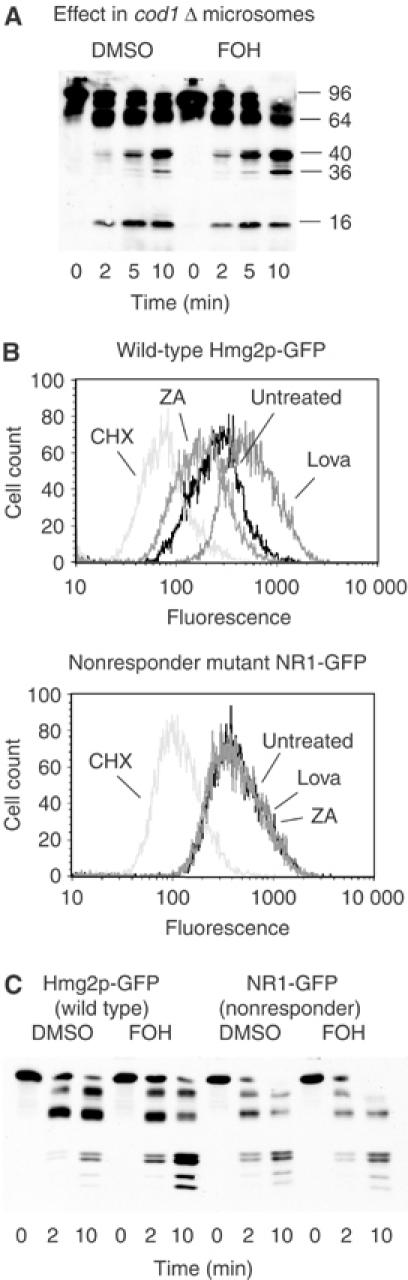
Unregulated mutants did not respond to farnesol. (A) Testing farnesol in a cod1Δ background. Microsomes from a cod1Δ strain expressing Hmg2p-GFP were resuspended in buffer with DMSO or 200 μM FOH, and then incubated with trypsin (15 μg/ml) at 30°C. Samples were removed at the indicated times and evaluated by 14% SDS–PAGE and myc immunoblotting. Molecular weight markers (in kDa) are shown. (B) Lack of regulation of nonresponder mutant NR1-GFP compared with wild-type Hmg2p-GFP. Cells expressing either Hmg2p-GFP or NR1-GFP were grown to early log phase and then incubated for 4 h with no drug, 25 μg/ml lovastatin (Lova), 10 μg/ml zaragozic acid (ZA), or 50 μg/ml cycloheximide (CHX) as indicated, and then assayed via flow cytometry for effects on Hmg2p stability. (C) Farnesol in vitro effect on wild-type Hmg2p-GFP and NR1-GFP. Microsomes bearing Hmg2p-GFP or farnesol nonresponder NR1-GFP were resuspended in buffer with DMSO or 200 μM FOH, and then incubated with trypsin (15 μg/ml) at 30°C. Samples were removed at the indicated times and evaluated by 14% SDS–PAGE and immunoblotting of the Myc epitope.
Our work on over 350 engineered mutants of Hmg2p revealed that regulated degradation requires numerous sequence elements in the transmembrane domain (Gardner and Hampton, 1999a). To further test the biological relevance of the in vitro effect of farnesol, we employed an Hmg2p variant that is degraded in an Hrd1-dependent manner, yet is totally unresponsive to the in vivo FPP-derived degradation signal. In this variant, residues 348–352 at the end of the sixth transmembrane span are altered from TFYSA to ILQAS. A mutant version of the lumenally tagged Hmg2p-GFP that incorporated this change, termed NR1-GFP, was expressed in yeast to evaluate the effects of this mutation on the in vitro assay. We first confirmed that NR1-GFP was degraded by the HRD pathway in vivo, using flow cytometry as in many previous studies (Gardner and Hampton, 1999a; Cronin et al, 2000). Addition of cycloheximide to halt protein synthesis caused the expected time-dependent loss of the fluorescent protein (Figure 3B, ‘CHX'), which was entirely dependent on the HRD pathway (unpublished data). We next confirmed that degradation of NR1-GFP was unregulated. Cells expressing NR1-GFP or Hmg2p-GFP were treated with drugs that alter FPP levels, and hence Hmg2p degradation, and the steady-state cellular fluorescence measured by flow cytometry. Wild-type reporter responded in the expected manner to changes in FPP: lovastatin, which reduces FPP levels, caused increased Hmg2p-GFP levels; zaragozic acid, which increases FPP abundance, decreased Hmg2p-GFP levels (Figure 3B). In contrast, NR1-GFP steady-state levels remained unchanged when cells expressing this mutant were subjected to those same treatments (Figure 3B). Thus, NR1-GFP underwent normal, HRD-dependent degradation that was completely unresponsive to the in vivo regulatory signal.
Microsomes from strains expressing NR1-GFP were tested for the in vitro effect of farnesol in the limited proteolysis assay. While a wild-type Hmg2p-GFP control showed the expected increase in trypsinolysis, the unregulated NR1-GFP mutant was not responsive to farnesol (Figure 3C). Importantly, the cleavage pattern of NR1-GFP was identical to that of the wild-type protein, indicating no obvious conformational defect. Nevertheless, NR1-GFP was completely refractory to farnesol in vitro just as it was unresponsive to the degradation signal in vivo. Several other Hmg2p regulatory mutants from our collection were similarly tested, and all showed the same correlation between in vivo regulation and in vitro response to farnesol (unpublished data).
The in vivo degradation rate of Hmg2p changes rapidly in response to alterations in regulatory signal. A sudden increase in FPP level, as when cells are treated with zaragozic acid, causes accelerated Hmg2p degradation (Hampton and Bhakta, 1997). A sudden decrease in FPP, as when cells are treated with lovastatin, causes immediate stabilization of the entire pool of Hmg2p (Hampton and Rine, 1994; Gardner and Hampton, 1999b). Thus, it appears that the effects of the in vivo signal on Hmg2p are readily reversible. We next tested whether this was the case for the in vitro effect of farnesol as well.
To test for reversibility, we needed to remove farnesol from the microsomes following incubation. We initially tried simple resuspension of farnesol-treated microsomes in fresh buffer, but this did not reverse farnesol's effect. We reasoned that the highly hydrophobic farnesol molecule might preferentially partition into the microsomal membrane. Thus, to remove farnesol from treated membranes, we provided a ‘lipid sink' in the form of additional microsomal membranes from a strain not expressing Hmg2p-GFP. Microsomes from a strain expressing Hmg2p-GFP were treated with DMSO or farnesol. These microsomes were then split into two samples and mixed with additional microsomes lacking tagged protein (the sink membranes). The additional microsomes were also treated with either DMSO or farnesol, providing four conditions as shown in a diagram of the experiment (Figure 4A). We used a two-fold excess of sink membranes added to the microsomes containing Hmg2p-GFP. Each sample was then resuspended in new buffer and assayed via limited proteolysis. Farnesol-enhanced proteolysis depended entirely on the presence of this molecule at the time of trypsinization. The effects of farnesol treatment on Hmg2p-GFP were abrogated by addition of sink membranes without farnesol (Figure 4B, first and fourth groups), whereas inclusion of farnesol with the sink membranes preserved the in vitro effect (Figure 4B, third group). Finally, addition of farnesol only at the time of the addition of sink membranes also caused the in vitro effect on Hmg2p-GFP proteolysis (Figure 4B, second group). Thus, pretreatment with farnesol could be readily reversed if the properties of the lipid were considered in the procedure.
Figure 4.
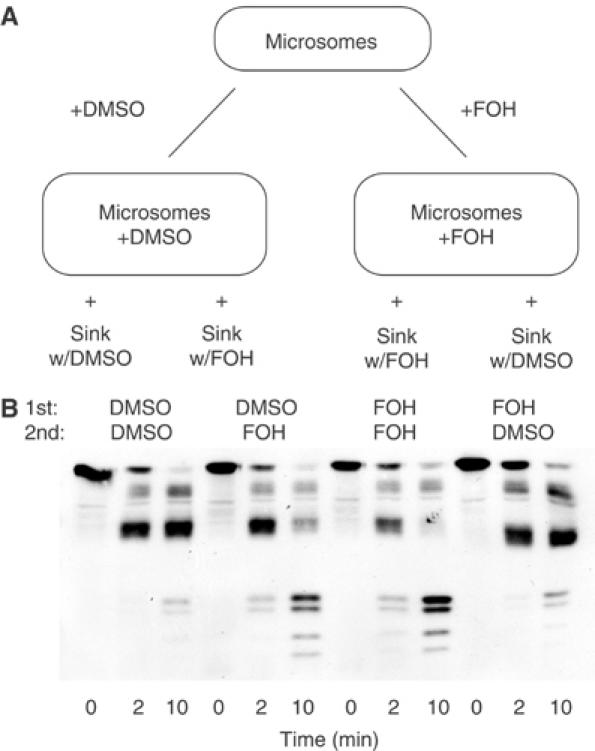
The farnesol effect is reversible. (A) Diagram of the reversibility experiment. Microsomes bearing Hmg2p-GFP were resuspended in buffer with DMSO or 200 μM FOH. Each sample was then split in two and combined with additional sink membranes (lacking Hmg2p-GFP) treated with DMSO or 200 μM FOH. (B) Outcome of reversibility treatments. Mixes resulting from the procedure described above were pelleted, resuspended in buffer, and then incubated with trypsin (15 μg/ml) at 30°C. Samples were removed at the indicated times and evaluated by 14% SDS–PAGE and immunoblotting of the Myc epitope. The row labeled ‘1st' indicates the initial treatment, while the row labeled ‘2nd' indicates whether sink membranes with or without farnesol were added.
Taken together, the above observations indicated that farnesol specifically caused Hmg2p to undergo a reversible structural change to a more protease-accessible state. Our model of Hrd1p-mediated Hmg2p-regulated degradation predicts that farnesol should induce a structural change to a more misfolded state. We addressed this using chemical chaperones and thermal denaturation assays.
Chemical chaperones such as glycerol aid in the proper folding of misfolded proteins. If farnesol were inducing Hmg2p to acquire traits of a misfolded protein in vitro, then it follows that chemical chaperones would antagonize this effect, promoting correct folding (Brown et al, 1996; Sato et al, 1996). Accordingly, we tested the effect of glycerol on the in vitro action of farnesol in Hmg2p. Addition of 20% glycerol to farnesol-treated membranes antagonized the effect of farnesol, resulting in a substantially slower trypsinolysis rate in the case of farnesol and glycerol together versus farnesol alone (Figure 5). Importantly, we have shown previously that the presence of 20% glycerol has no effect on the activity of trypsin in this assay (Shearer and Hampton, 2004). Thus, the farnesol-induced alteration in Hmg2p structure was sensitive to chemical chaperones, implying that the protein in this state was less fully folded.
Figure 5.
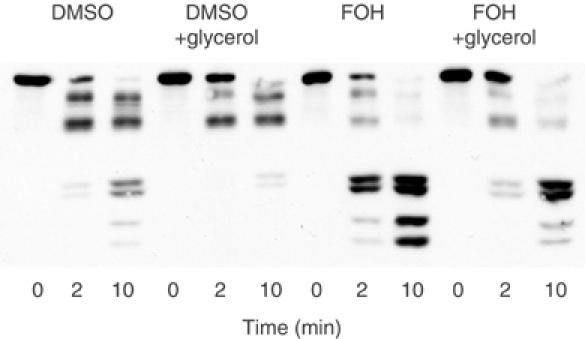
The chemical chaperone glycerol antagonizes the farnesol effect. Microsomes bearing Hmg2p-GFP were resuspended in buffer with DMSO or 200 μM FOH, with or without 20% glycerol, and then incubated with trypsin (15 μg/ml) at 30°C. Samples were removed at the indicated times and evaluated by 14% SDS–PAGE and immunoblotting of the Myc epitope.
We tested if Hmg2p-GFP in the presence of farnesol was more misfolded by employing a thermal denaturation assay, in which the rate and extent of protein aggregation at elevated temperature under various conditions is determined. Upon heat treatment, poorly folded proteins are more prone to aggregation than those that are fully folded. Thus, if Hmg2p were adopting traits of a misfolded protein in response to farnesol, we would expect to see an increased tendency to aggregate in the presence of farnesol when incubated at high temperatures. Microsomes treated with DMSO or farnesol were incubated at 70°C for the indicated times and subjected to SDS–PAGE gel electrophoresis and immunoblotting for the Myc tag in Hmg2p-GFP. In this assay, the stacking gel is included in the analysis to allow detection of low-mobility aggregates that can form after heat denaturation. As shown, incubation of microsomes at 70°C caused the time-dependent appearance of such aggregates, indicated first as immunoreactivity at the stack/running gel boundary (Figure 6A, Stack) and later as low-mobility immunoreactivity near the top of the stack (Figure 6A, LMI). In the presence of farnesol, Hmg2p-GFP showed a greater proclivity for aggregation, with similar low-mobility aggregates appearing approximately four-fold faster than in the untreated samples (Figure 6A). Importantly, farnesol did not cause aggregation of Hmg2p-GFP in the absence of heating, as indicated by the lack of aggregates at time zero, but rather enhanced the rate of heat denaturation. Thus, farnesol treatment caused membrane-localized Hmg2p-GFP to behave more like a misfolded protein. This unfolding effect of farnesol had the same lipid specificity observed in our in vitro proteolysis experiments: increased aggregation tendency was most strongly caused by farnesol, and to a lesser extent by geranylgeraniol, while geraniol and tetradecanol were without effect (unpublished data). Because this independent assay does not require the lumenal epitope tag of Hmg2p-GFP, we also tested native, catalytically active Hmg2p and observed the same effects of farnesol on thermal denaturation. Although this is not surprising considering that Hmg2p-GFP regulation matches that of the native protein in vivo, it was important to confirm that the effects observed with the reporter generalize to the authentic protein.
Figure 6.
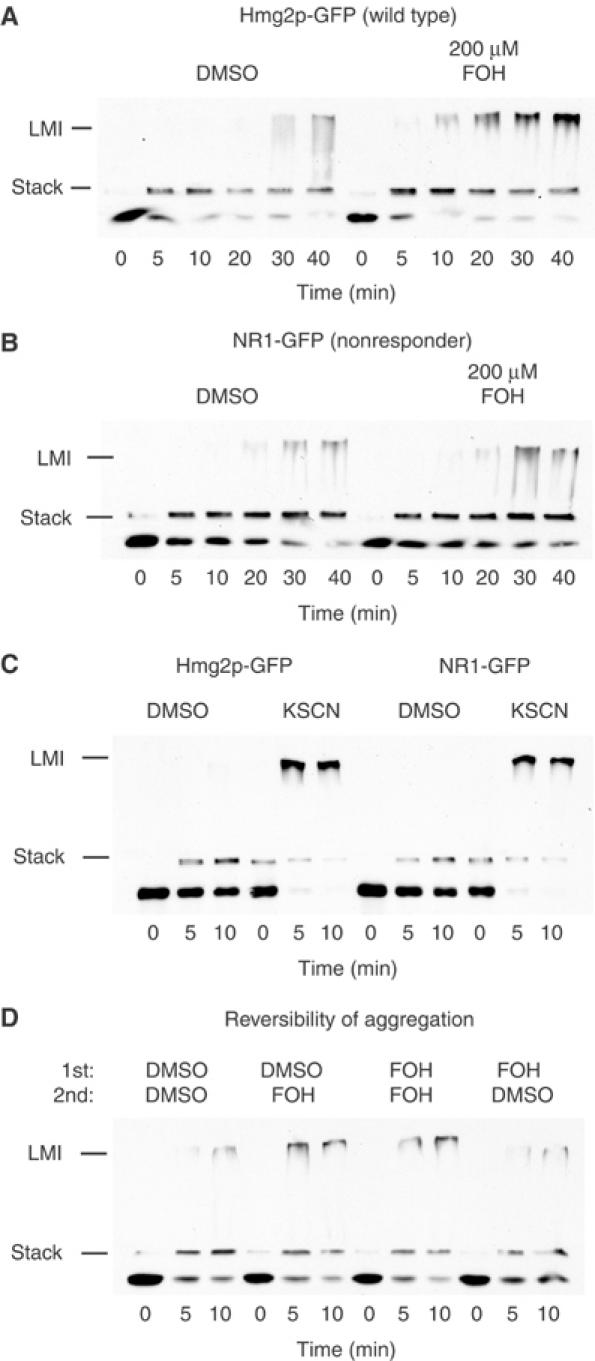
Farnesol specifically and reversibly enhances thermal denaturation of Hmg2p-GFP. (A) Effect of farnesol on thermal denaturation of Hmg2p-GFP. Microsomes bearing Hmg2p-GFP were resuspended in buffer with DMSO or 200 μM FOH, and then incubated at 70°C. Samples were removed at the indicated times and evaluated by SDS–PAGE and immunoblotting of the Myc epitope, including both the 2% stack and the 14% gel. Lines indicate the boundary between the gel and the stack (Stack) and LMI indicates the low-mobility immunoreactivity, or Hmg2p-GFP aggregates. (B) Effect of farnesol on thermal denaturation of NR1-GFP. Microsomes bearing NR1-GFP were evaluated for the effect of farnesol as described above. (C) Effect of the chaotrope potassium thiocyanate (KSCN) on thermal denaturation of Hmg2-GFP and NR1-GFP. Microsomes with Hmg2p-GFP or NR1-GFP were resuspended in buffer with or without 125 mM KSCN, and then incubated at 70°C and evaluated as described above. (D) Testing the reversibility of the farnesol effect in the thermal denaturation assay. Microsomes containing Hmg2p-GFP were prepared as described in Figure 4A, then pelleted, resuspended, and incubated at 70°C for the indicated times, and then evaluated as described above.
We used the farnesol-unresponsive NR1-GFP mutant to evaluate the biological relevance of farnesol-enhanced thermal denaturation. Microsomes bearing NR1-GFP were subjected to the thermal denaturation assay in the presence and absence of farnesol. In contrast to wild-type Hmg2p-GFP, farnesol had no enhancing effect on the thermal denaturation of the unresponsive mutant (Figure 6B). Importantly, NR1-GFP was equally responsive to the nonspecific denaturant potassium thiocyanate (KSCN), which caused identical increases in the rate of thermal denaturation in both mutant and wild type (Figure 6C). Thus, the unresponsiveness of NR1-GFP to farnesol was specific for that molecule, and not due to a general inability of the mutant protein to undergo thermal denaturation.
The effects of farnesol on Hmg2p in the limited proteolysis assay were reversible, when appropriate measures were taken to remove the farnesol from treated microsomes (Figure 4B). The in vitro effects of farnesol in the thermal denaturation assay were similarly reversible. Microsomes were prepared and treated with DMSO or farnesol followed by DMSO or farnesol-treated sink membranes as described above for the limited proteolysis reversibility experiment (as diagrammed in Figure 4A). The resulting samples were then subjected to the thermal denaturation assay. Conditions that reversed the effect of farnesol in the Hmg2p limited proteolysis assay—addition of sink membranes lacking farnesol similarly reversed its effect in the aggregation assay (Figure 6D). Thus, the farnesol-promoted increase in thermal denaturation was reversible, just as we observed with the farnesol-induced conformational change.
Taken together, these results indicated that the enhanced thermal sensitivity of Hmg2p caused by farnesol had the same biologically relevant properties as its action in the limited proteolysis assay: reversibility, lipid specificity, and a requirement for correct Hmg2p sequence.
Discussion
We believe Hmg2p is the first example of a normal protein that undergoes small-molecule-regulated quality control degradation. Since proposing this model (Gardner et al, 2001b; Hampton, 2002a, 2002b), we have directed our studies toward discovering how this is achieved. We have previously suggested that Hmg2p undergoes a structural transition in response to an FPP-derived molecular degradation signal, acquiring traits of a misfolded protein. In this state it would be recognizable by the HRD quality control pathway, and thus ubiquitinated and degraded.
Consistent with this idea, we previously showed that Hmg2p undergoes a structural transition when the in vivo FPP-derived signal for degradation is abundant (Shearer and Hampton, 2004). Using two independent in vitro assays, we have now demonstrated that the FPP-derived molecule farnesol caused a structural transition that was quite specific for the farnesol structure, met all criteria for in vivo relevance, was reversible, and did indeed involve a transition between a more and less folded form of Hmg2p.
We first evaluated the possibility that degradation signal directly affected Hmg2p structure with the in vitro limited proteolysis assay. The FPP derivative farnesol had a dramatic effect on the limited proteolysis of Hmg2p. Ours and other studies have shown that farnesol is produced in abundance when FPP levels are elevated by drugs or genetic manipulation (Chambon et al, 1990; Gardner et al, 2001a), making it a reasonable candidate for the FPP-derived signal that regulates Hmg2p stability in vivo. As we have observed with the in vivo signal, farnesol acted rapidly and reversibly in this in vitro assay. The concentrations of farnesol that affect Hmg2p structure (50–200 μM) are in the same range as those required for another in vivo function of farnesol—quorum sensing in the yeast Candida albicans (30–300 μM) (Hornby et al, 2001; Ramage et al, 2002). It is quite possible, however, that the concentrations of farnesol required for in vivo regulation are less than those needed for the in vitro effect. While the in vitro assay demands changes in the whole pool of Hmg2p, in vivo regulation of Hmg2p levels may occur by continuous regulated degradation of a small fraction of the Hmg2p pool at a given time.
The biological relevance of the farnesol-induced structural transition was confirmed by analysis of pertinent cis and trans regulatory mutants. In a cod1Δ mutant, Hmg2p is mostly unresponsive to the in vivo degradation signal (Cronin et al, 2000), due to alterations in its structure that likely abolish regulation (Shearer and Hampton, 2004). In the same manner, limited proteolysis of Hmg2p in microsomes derived from a cod1Δ strain was largely unaffected by farnesol. An independent test of relevance was provided by the unregulated in cis NR1-GFP mutant of Hmg2p. This variant of Hmg2p was completely unregulated in vivo, and was completely unresponsive to farnesol in the limited proteolysis assay. These results with unregulated mutants demonstrated that the in vitro effect of farnesol adhered closely to the in vivo properties of Hmg2p regulation. Furthermore, these experiments showed that the actions of farnesol were not due to general effects on membranes or membrane proteins, since its effects were highly dependent on the correct structure and sequence of Hmg2p.
Our model of Hmg2p regulation predicts that farnesol promotes acquisition of quality control traits in Hmg2p, a prediction borne out by these in vitro studies. The chemical chaperone glycerol antagonized farnesol's effect in vitro, which implied that farnesol was promoting a less folded state. Farnesol also promoted enhanced thermal aggregation of Hmg2p-GFP, indicating that the protein was more misfolded in the presence of farnesol. Enhancement of Hmg2p-GFP thermal denaturation was farnesol specific, did not occur with the NR1-GFP unresponsive mutant, and was fully reversible. Thus, farnesol had precisely the same features in both in vitro assays of its effect on Hmg2p.
Farnesol has previously been implicated in the degradation of mammalian HMGR, where it has been shown to increase degradation rate when added to intact or permeabilized cells (Correll et al, 1994; Meigs et al, 1996). However, in some circumstances, farnesol appears to cause nonspecific aggregation of membrane proteins in permeabilized cells (Meigs et al, 1996). These concerns do not pertain to our studies. In both the limited proteolysis and thermal denaturation assays, the effects of farnesol were reversible. Furthermore, farnesol did not itself cause Hmg2p aggregation, but only enhanced denaturation of Hmg2p at high temperature as a specific readout of tendency to misfold. Finally, farnesol had no effect on the unresponsive NR1-GFP mutant and a limited effect on Hmg2p in cod1Δ membranes, ruling out a generalized influence of farnesol on ER membrane proteins in these studies.
Taken together, the work presented above showed that Hmg2p underwent a rapid, reversible, farnesol-dependent transition to a more misfolded state, one that presumably is better recognized by the Hrd1p quality control pathway. It will be important to next determine if this altered state is indeed recognized by the Hrd1p ubiquitination machinery, a goal that we are actively pursuing.
The effect of farnesol on Hmg2p structure was quite specific for this lipid. A variety of other lipids, including linear alcohols of similar size, other isoprene compounds, fully hydrogenated farnesol, and the natural sterols ergosterol and lanosterol, were largely ineffective at altering Hmg2p structure. Both FPP and GGOH had small effects, perhaps as a function of their similarity to farnesol. Each compound includes the farnesol backbone as part of its structure. It is therefore reasonable to imagine that these molecules might have some ability to act like farnesol. Nerolidol, the only other compound tested that matched farnesol's efficacy, is a very close, albeit nonbiological, isomer (see Figure 2D). Taken together, the results from these lipids pointed to a fairly strict structural requirement for farnesol, presenting the possibility that the signal is recognized by Hmg2p in a specific manner.
The transmembrane domain of Hmg2p mediates regulated degradation (Gardner and Hampton, 1999a), and this portion of the molecule, as found in Hmg2p-GFP, is sufficient for the in vitro effects of farnesol. This portion of the Hmg2p protein has a conserved sequence motif known as the sterol-sensing domain (SSD), which is found in a number of proteins involved in lipid regulation, including mammalian HMGR, the sterol regulator SCAP, the developmental proteins Patched and Dispatched, and the lipid storage-related protein NPC1 (Hua et al, 1996; Carstea et al, 1997; Kuwabara and Labouesse, 2002). Based on the functions of these proteins, the SSD was hypothesized to sense cholesterol or related sterols. However, later work with Drosophila SCAP indicated that the SSD in that protein specifically responds to a lipid signal unrelated to sterols (Dobrosotskaya et al, 2002), implying that the SSD has a broader role in recognition of diverse lipid signals. Adding to this idea, our studies indicate that the Hmg2p SSD is involved in farnesol sensing. Supporting this, we have observed numerous mutations in the conserved SSD that alter the in vivo response of Hmg2p to the degradation signal (Gardner and Hampton, 1999a). In particular, the dramatic, completely unresponsive NR1-GFP mutation is a conservative five-amino-acid replacement (from TFYSA to ILQAS) that alters a phenylalanine found in nearly all SSDs, and one that is required for sterol regulation of HMGR in mammals (Xu and Simoni, 2003). When comparison is restricted to only HMGR from various organisms, several additional identities appear in this region (consensus TFxxAxxS), perhaps indicating a particular role in HMGR regulation for this conserved SSD segment. It has been shown that both NPC1 and SCAP bind directly to cholesterol in their SSDs (Ohgami et al, 2004; Radhakrishnan et al, 2004). Further, SCAP undergoes an SSD-dependent conformational change in the presence of sterols (Brown et al, 2002). The SCAP structural change is qualitatively different from that seen in Hmg2p, involving an altered proteolytic pattern rather than a global change in accessibility. Furthermore, sterols do not cause SCAP degradation. Regardless, structural response to lipid signals may prove to be a general trait of SSD proteins. Thus, the SSD is most likely an ancestral lipid sensor that has evolved to discriminate a variety of lipid signals. In this sense, SSDs have a broad conservation of purpose, rather than a conservation of specific effector lipid. The slight variation of SSD to mean ‘signal-sensing domain,' or perhaps ‘state-sensing domain' might thus be appropriate for this ancient membrane motif. It will be important to more fully examine the role of the SSD in Hmg2p regulation, with particular focus on the model that farnesol binds directly to this domain.
How related are regulation of Hmg2p and mammalian HMGR? Both are subject to regulated degradation via the ubiquitin–proteasome machinery, driven by specific pathway lipids, and requiring an intact SSD. Hmg2p degradation depends primarily on the FPP-derived signal, with little or no dependence on sterols (however see Gardner et al, 2001a). Mammalian HMGR degradation is regulated primarily by sterols, but it too shows a separate requirement for an early isoprene, which has been proposed to be farnesol (Correll et al, 1994; Meigs et al, 1996) or more recently, geranylgeraniol (Sever et al, 2003). The Insig proteins play a key role in HMGR-regulated degradation (Sever et al, 2003), and the distant yeast homologs NSG1 and NSG2 play a distinct but related role in Hmg2p degradation (Flury et al, manuscript in preparation). Given these similarities, it is conceivable that quality control machinery could be involved in the regulated destruction of HMGR as well. If mammalian HMGR is recognized by a quality control pathway, this would be another example of what is likely to be a common regulatory strategy: small-molecule-promoted entry into constitutive quality control degradation pathways.
Regulated entry into a constitutive quality control pathway represents a novel yet reasonable combination of two common elements of cellular physiology. Many proteins undergo reversible structural change in response to small molecules, often allosteric alterations that regulate their activities (Monod et al, 1963; Gerhart and Schachman, 1968; Pardee and Reddy, 2003). Quality control is similarly ubiquitous, being found across all kingdoms of life (Dougan et al, 2002; Hampton, 2002a; Volker and Lupas, 2002). Regulated degradation of Hmg2p could be considered in a sense an allosteric transition to a misfolded state. Given the large number of examples of small molecule regulation of protein structure and the ubiquity of quality control pathways, we expect many examples of small-molecule-regulated quality control will be discovered.
Beyond this probable natural role, small molecule regulation of protein quality control also holds promise for drug development. From our data, it is conceivable that compounds could be developed to modulate the degradation rate of a single target protein by specifically causing the appropriate structural changes. The most immediate application would be in designing molecules that stimulate the degradation of HMGR, providing a novel mechanism to lower cholesterol and achieve the many other benefits currently associated only with the inhibitors of HMGR enzymatic activity, the statins (Marz and Koenig, 2003; Stuve et al, 2003; Gresser and Gathof, 2004). The ubiquity of quality control pathways, and the specificity with which they operate implies that this strategy of small molecule protein targeting could have many uses in the laboratory and the clinic. Pharmaceutical adaptation of this approach could include targeting of proteins that normally are not degraded by such pathways, or the enhancement of quality control degradation for cases, such as numerous neurodegenerative diseases, in which natural quality control of key proteins is not sufficiently robust. The required knowledge of how quality control can be harnessed will come as we identify more proteins that, like Hmg2p, selectively enter these pathways in a regulated manner.
Materials and methods
Restriction enzymes and T4 DNA ligase were obtained from New England Biolabs. Lovastatin and zaragozic acid were generously provided by Dr Samuel Wright (Merck, Rahway, NJ). 9E10 cell culture supernatant was produced in our lab from cells (CRL 1729, American Type Culture Collection) grown in RPMI1640 culture medium (GIBCO BRL) with 10% fetal calf serum and supplements. HRP-conjugated goat anti-mouse antibodies were purchased from Jackson Immuno Research. 3,7,11-Trimethyldodecan-1-ol was synthesized in the lab of Seiichi Matsuda (Rice University). Chemiluminescence immunodetection reagents were obtained from Perkin Elmer. All other chemical reagents were obtained from Sigma or Fisher.
Plasmid construction and DNA manipulation
Plasmid pRH1692 (integrating) expressed the NR1-GFP unregulated variant of lumenally myc-tagged Hmg2-GFP with amino acids 348–352 switched from TFYSA to ILQAS, and was constructed by replacing the SphI–BglII segment of pRH1581, the plasmid expressing MycL-Hmg2p-GFP (Shearer and Hampton, 2004), with the SphI–BglII fragment from pRH902 (Gardner and Hampton, 1999a).
Yeast culture and strains
Yeast strains were grown in minimal media (Difco Yeast Nitrogen Base) with glucose and the appropriate supplements as described previously, or in YPD, if so noted (Hampton and Rine, 1994). Experiments were performed at 30°C unless otherwise noted. Yeast was transformed with plasmid DNA by the LiOAc method.
All strains used were isogenic (Table I). RHY2842 was made by targeted integration of the plasmid pRH1692 at the StuI site of the ura3-52 locus in RHY519 (Gardner et al, 1998), then selection for Ura+ prototrophy.
Table 1.
Yeast strains
| Strain | Genotype |
|---|---|
| RHY2723 | MATa ade2-101 met2 lys2-801 his3Δ200 hmg1∷LYS2 hmg2∷HIS3 ura3-52∷URA3∷HMG2cd∷mycL-Hmg2p-GFP |
| RHY2803 | MATa ade2-101 met2 lys2-801 his3Δ200 hmg1∷LYS2 hmg2∷HIS3 ura3-52∷URA3∷HMG2cd∷mycL-Hmg2p-GFP hrd1Δ∷KanMX |
| RHY2853 | MATalpha ade2-101 met2 lys2-801 his3200 ura3-52∷URA3∷mycL-Hmg2p-GFP cod1Δ∷KanMX |
| RHY2842 | MATa ade2-101 met2 lys2-801 his3Δ200 hmg1∷LYS2 hmg2∷HIS3 ura3-52∷URA3∷HMG2cd∷NR1-GFP |
| RHY2866 | MATa ade2-101 met2 lys2-801 his3Δ200 hmg1∷LYS2 hmg2∷HIS3 ura3-52∷URA3∷HMG2cd∷NR1-GFP hrd1Δ∷KanMX |
RHY2866 was derived from RHY2842 by transformation with the hrd1Δ∷KanMX deletion cassette excised from pRH1122 (Gardner and Hampton, 1999a), then selection on YPD with G418.
Flow cytometry of yeast cells
Flow cytometry was carried out as described previously (Cronin et al, 2000). Briefly, cells were grown to early log phase (OD600 0.1) in minimal medium, and then incubated with drugs for the times indicated and analyzed for individual cell fluorescence with a BD Biosciences FACSCalibur flow cytometer. All histograms in this paper represent results from 10 000 cells.
Microsome preparation
Microsomes were prepared as described previously (Shearer and Hampton, 2004). Briefly, cells were grown to mid-log phase in YPD, pelleted, and subjected to lysis. The resulting lysates were cleared of debris, and then centrifuged to pellet microsomes, which were resuspended in physiological buffer at 6 μl buffer per OD of starting cells.
Limited proteolysis assay on microsomes
The assay was performed on microsomes as described (Shearer and Hampton, 2004). Briefly, resuspended microsomes were treated with indicated lipids (dissolved in DMSO) or with a DMSO-only vehicle control, and then an aliquot was removed as a no trypsin control. Trypsin was then added and the remaining microsomes were incubated at 30°C. Aliquots were removed at the indicated times and the reaction halted by addition of SDS–urea sample buffer. DMSO added alone had no additional effect when compared to buffer only (unpublished data).
Samples were resolved on 14% SDS–PAGE, then transferred with 15% methanol to nitrocellulose and immunoblotted for myc epitope, all as described (Hampton and Rine, 1994; Gardner et al, 1998). Fold differences in trypsinolysis rate were calculated by measuring the total intensity of the characteristic quartet of bands at the 10 min time point under the compared conditions using NIH Image 1.63, using at least three experiments per calculation.
Thermal denaturation assay
Microsomes were prepared as described above, and then treated with lipids as indicated. Following treatment, 30 μl sample aliquots were transferred to ultra-thin-wall PCR tubes, one per desired time point, and placed on ice for 20 min. Tubes were then placed in a Perkin Elmer GeneAmp PCR System 2400 thermocycler holding at 70°C and incubated for the indicated times. At the end of the incubation, samples were placed on ice for 2 min and then prepared for electrophoresis by addition of 30 μl 2 × USB (75 mM MOPS, pH 6.8; 4% SDS; 200 mM DTT; 0.2 mg/ml bromophenol blue; 8 M urea) followed by solubilization for 10 min at 55°C.
Samples were resolved on 14% SDS–PAGE with a 2% stacking gel, and then transferred from both gel and stack with 15% methanol to nitrocellulose and immunoblotted for myc epitope as described (Hampton and Rine, 1994).
Acknowledgments
We thank members of the Hampton lab for continuous intellectual and inspirational support. We also thank Seiichi Matsuda and Hui Shan for synthesis of fully hydrogenated farnesol. RYH wishes further to acknowledge Douglass Golenbock for scientific drive and lifelong friendship. AGS wishes to thank Omar Bazirgan for continuing good suggestions on experimental design, and Christine Rhee for good advice generally. This work was supported by NIH (NIDDK) grant #GM51996-06 and an AHA Established Investigator Award. AGS was supported in part by CMG NIH Training grant #GM07240.
References
- Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY (2001) Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol 3: 24–29 [DOI] [PubMed] [Google Scholar]
- Bordallo J, Plemper RK, Finger A, Wolf DH (1998) Der3p–Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol Biol Cell 9: 209–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AJ, Sun L, Feramisco JD, Brown MS, Goldstein JL (2002) Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol Cell 10: 237–245 [DOI] [PubMed] [Google Scholar]
- Brown CR, Hong-Brown LQ, Biwersi J, Verkman AS, Welch WJ (1996) Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones 1: 117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstea ED, Morris JA, Coleman KG, Loftus SK, Cummings DZC, Gu J, Rosenfeld M, Pavan WJ, Krizman DB, Nagle J, Polymeropoulos MH, Sturley SL, Ioannou YA, Higgins ME, Comly M, Cooney A, Brown A, Kaneski CR, Blanchette-Mackie EJ, Dwyer NK, Neufeld EB, Chang TY, Liscum L, Strauss JF III, Ohno K, Zeigler M, Carmi R, Sokol J, Markie D, O'Neil RR, Van Viggelen OP, Elleder M, Patterson MC, Brady RO, Vanier MT, Pentchev PG, Tagle DA (1997) Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 277: 228–231 [DOI] [PubMed] [Google Scholar]
- Chambon C, Ladeveze V, Oulmouden A, Servouse M, Karst F (1990) Isolation and properties of yeast mutants affected in farnesyl diphosphate synthetase. Curr Genet 18: 41–46 [DOI] [PubMed] [Google Scholar]
- Chin DJ, Gil G, Faust JR, Goldstein JL, Brown MS, Luskey KL (1985) Sterols accelerate degradation of hamster 3-hydroxy-3-methylglutaryl coenzyme A reductase encoded by a constitutively expressed cDNA. Mol Cell Biol 5: 634–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correll CC, Ng L, Edwards PA (1994) Identification of farnesol as the non-sterol derivative of mevalonic acid required for the accelerated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem 269: 17390–17393 [PubMed] [Google Scholar]
- Cronin SR, Hampton RY (1999) Measuring protein degradation with green fluorescent protein. In Methods in Enzymology, Green Fluorescent Protein, Conn PM (ed) Vol. 302, pp 58–73. London, UK; San Diego, CA: Academic Press Inc. [DOI] [PubMed] [Google Scholar]
- Cronin SR, Khoury A, Ferry DK, Hampton RY (2000) Regulation of HMG-CoA reductase degradation requires the P-type ATPase Cod1p/Spf1p. J Cell Biol 148: 915–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin SR, Rao R, Hampton RY (2002) Cod1p/Spf1p is a P-type ATPase involved in ER function and Ca2+ homeostasis. J Cell Biol 157: 1017–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrosotskaya IY, Seegmiller AC, Brown MS, Goldstein JL, Rawson RB (2002) Regulation of SREBP processing and membrane lipid production by phospholipids in Drosophila. Science 296: 879–883 [DOI] [PubMed] [Google Scholar]
- Dougan DA, Mogk A, Bukau B (2002) Protein folding and degradation in bacteria: to degrade or not to degrade? That is the question. Cell Mol Life Sci 59: 1607–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T (2000) A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol 2: 379–384 [DOI] [PubMed] [Google Scholar]
- Gardner R, Cronin S, Leader B, Rine J, Hampton R (1998) Sequence determinants for regulated degradation of yeast 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell 9: 2611–2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Hampton RY (1999a) A ‘distributed degron' allows regulated entry into the ER degradation pathway. EMBO J 18: 5994–6004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Hampton RY (1999b) A highly conserved signal controls degradation of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase in eukaryotes. J Biol Chem 274: 31671–31678 [DOI] [PubMed] [Google Scholar]
- Gardner RG, Shan H, Matsuda SP, Hampton RY (2001a) An oxysterol-derived positive signal for 3-hydroxy-3-methylglutaryl-CoA reductase degradation in yeast. J Biol Chem 276: 8681–8694 [DOI] [PubMed] [Google Scholar]
- Gardner RG, Shearer AG, Hampton RY (2001b) In vivo action of the hrd ubiquitin ligase complex: mechanisms of endoplasmic reticulum quality control and sterol regulation. Mol Cell Biol 21: 4276–4291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, Hampton RY (2000) Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol 151: 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart JC, Schachman HK (1968) Allosteric interactions in aspartate transcarbamylase. II. Evidence for different conformational states of the protein in the presence and absence of specific ligands. Biochemistry 7: 538–552 [DOI] [PubMed] [Google Scholar]
- Gresser U, Gathof BS (2004) Atorvastatin: gold standard for prophylaxis of myocardial ischemia and stroke—comparison of the clinical benefit of statins on the basis of randomized controlled endpoint studies. Eur J Med Res 9: 1–17 [PubMed] [Google Scholar]
- Hampton RY (2002a) ER-associated degradation in protein quality control and cellular regulation. Curr Opin Cell Biol 14: 476–482 [DOI] [PubMed] [Google Scholar]
- Hampton RY (2002b) Proteolysis and sterol regulation. Annu Rev Cell Dev Biol 18: 345–378 [DOI] [PubMed] [Google Scholar]
- Hampton RY, Bhakta H (1997) Ubiquitin-mediated regulation of 3-hydroxy-3-methylglutaryl-CoA reductase. Proc Natl Acad Sci USA 94: 12944–12948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Gardner RG, Rine J (1996) Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell 7: 2029–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Rine J (1994) Regulated degradation of HMG-CoA reductase, an integral membrane protein of the endoplasmic reticulum, in yeast. J Cell Biol 125: 299–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornby JM, Jensen EC, Lisec AD, Tasto JJ, Jahnke B, Shoemaker R, Dussault P, Nickerson KW (2001) Quorum sensing in the dimorphic fungus Candida albicans is mediated by farnesol. Appl Environ Microbiol 67: 2982–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Nohturfft A, Goldstein JL, Brown MS (1996) Sterol resistance in CHO cells traced to point mutation in SREBP cleavage-activating protein. Cell 87: 415–426 [DOI] [PubMed] [Google Scholar]
- Kuwabara PE, Labouesse M (2002) The sterol-sensing domain: multiple families, a unique role? Trends Genet 18: 193–201 [DOI] [PubMed] [Google Scholar]
- Leo A, Hansch C, Elkins D (1971) Partition coefficients and their uses. Chem Rev 71: 525–616 [Google Scholar]
- Marz W, Koenig W (2003) HMG-CoA reductase inhibition: anti-inflammatory effects beyond lipid lowering? J Cardiovasc Risk 10: 169–179 [DOI] [PubMed] [Google Scholar]
- Meigs TE, Roseman DS, Simoni RD (1996) Regulation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase degradation by the nonsterol mevalonate metabolite farnesol in vivo. J Biol Chem 271: 7916–7922 [DOI] [PubMed] [Google Scholar]
- Monod J, Changeux JP, Jacob F (1963) Allosteric proteins and cellular control systems. J Mol Biol 6: 306–329 [DOI] [PubMed] [Google Scholar]
- Ohgami N, Ko DC, Thomas M, Scott MP, Chang CC, Chang TY (2004) Binding between the Niemann-Pick C1 protein and a photoactivatable cholesterol analog requires a functional sterol-sensing domain. Proc Natl Acad Sci USA 101: 12473–12478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee AB, Reddy GP (2003) Beginnings of feedback inhibition, allostery, and multi-protein complexes. Gene 321: 17–23 [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A, Sun LP, Kwon HJ, Brown MS, Goldstein JL (2004) Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol Cell 15: 259–268 [DOI] [PubMed] [Google Scholar]
- Ramage G, Saville SP, Wickes BL, Lopez-Ribot JL (2002) Inhibition of Candida albicans biofilm formation by farnesol, a quorum-sensing molecule. Appl Environ Microbiol 68: 5459–5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravid T, Doolman R, Avner R, Harats D, Roitelman J (2000) The ubiquitin–proteasome pathway mediates the regulated degradation of mammalian 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem 275: 35840–35847 [DOI] [PubMed] [Google Scholar]
- Roullet J, Luft UC, Xue H, Chapman J, Bychkov R, Roullet CM, Luft FC, Haller H, McCarron DA (1997) Farnesol inhibits L-type Ca2+ channels in vascular smooth muscle cells. J Biol Chem 272: 32240–32246 [DOI] [PubMed] [Google Scholar]
- Sato S, Ward CL, Krouse ME, Wine JJ, Kopito RR (1996) Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem 271: 635–638 [DOI] [PubMed] [Google Scholar]
- Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA (2003) Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J Biol Chem 278: 52479–52490 [DOI] [PubMed] [Google Scholar]
- Shearer AG, Hampton RY (2004) Structural control of endoplasmic reticulum-associated degradation: effect of chemical chaperones on 3-hydroxy-3-methylglutaryl-CoA reductase. J Biol Chem 279: 188–196 [DOI] [PubMed] [Google Scholar]
- Stuve O, Youssef S, Steinman L, Zamvil SS (2003) Statins as potential therapeutic agents in neuroinflammatory disorders. Curr Opin Neurol 16: 393–401 [DOI] [PubMed] [Google Scholar]
- Volker C, Lupas AN (2002) Molecular evolution of proteasomes. Curr Top Microbiol Immunol 268: 1–22 [DOI] [PubMed] [Google Scholar]
- Welch WJ, Brown CR (1996) Influence of molecular and chemical chaperones on protein folding. Cell Stress Chaperones 1: 109–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhovsky S, Gardner R, Hampton R (2000) HRD gene dependence of endoplasmic reticulum-associated degradation. Mol Biol Cell 11: 1697–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Simoni RD (2003) The inhibition of degradation of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase by sterol regulatory element binding protein cleavage-activating protein requires four phenylalanine residues in span 6 of HMG-CoA reductase transmembrane domain. Arch Biochem Biophys 414: 232–243 [DOI] [PubMed] [Google Scholar]