

Abstract
The dorsal horn (DH) of the spinal cord is the integrative center that processes and transmits pain sensation. Abnormal changes in ion channel expression can enhance the excitability of pain-related DH neurons. Sodium-activated potassium (KNa) channels are highly expressed particularly in the central nervous system; however, information about whether rat DH neurons express the SLICK channel protein is lacking, and the direct effects on SLICK in response to inflammation and the potential signaling pathway mediating such effects are yet to be elucidated. Here, using cultured DH neurons, we have shown that tumor necrosis factor-α inhibits the total outward potassium current IK and the KNa current predominantly as well as induces a progressive loss of firing accommodation. However, we found that this change in channel activity is offset by the p38 inhibitor SB202190, thereby suggesting the modulation of SLICK channel activity via the p38 MAPK pathway. Furthermore, we have demonstrated that the tumor necrosis factor-α modulation of KNa channels does not occur at the level of SLICK channel gating but arises from possible posttranslational modification.
Keywords: p38 MAPK, SLICK channel, neuropathic pain, dorsal horn, TNF-α
Introduction
In recent years, sodium-activated potassium (KNa) channels have attracted considerable attention because of their role in maintaining the resting membrane potential and firing accommodation of the sensory neurons.1,2 KNa channels are encoded by the SLACK and SLICK genes. Although both channels may protect the neurons against noxious stimuli,3 SLICK may be more specialized for protective functions because of its higher sensitivity to intracellular Cl− and ATP. The dorsal horn (DH) of the spinal cord is the integrative center that processes and transmits pain sensation.4,5 Although KNa channels are particularly highly expressed in neurons,6 information about whether rat DH neurons express the SLICK channel protein was limited. In response to local inflammation, abnormal changes in ion channel expression can enhance the excitability of pain-related neurons;7 however, direct empirical evidence for a KNa channel contribution to neuronal excitability has been lacking.
Tumor necrosis factor-α (TNF-α), an important substance contributing to inflammation, is involved in the development of inflammatory pain, and the impairment of TNF signaling attenuates hypersensitivity in neuropathy.8–10 As previously reported, acute TNF-α stimulation at the dorsal root ganglion rapidly enhances TTX-R currents via the p38-dependent pathway,11 and TNF-α-induced p38 MAPK activation regulates TRPA1 and TRPV4 activity in odontoblast-like cells.12 Although TNF-α has been shown to modulate potassium channel expression and mediate thermal hyperalgesia,13 its direct effects on SLICK and the potential signaling pathway mediating such effects have yet to be elucidated.
We hypothesized that p38 MAPK pathway activation plays an important role in the modulation of the SLICK channel and then enhances the excitability of DH neurons. In this study, we addressed two questions using cultures of DH neurons. First, we explored whether the DH neurons express the SLICK channel protein. Second, we investigated whether p38 MAPK pathways are involved in the mechanism by which TNF-α acts on the SLICK channel.
Materials and methods
DH neuronal culture and treatments
All surgical procedures and experiments were approved by the Ethical Committee of Southeast University and were in accordance with the Guidelines for the Care and Treatment of Laboratory Animals of the US National Institutes of Health. The DHs from Sprague-Dawley female rats (1 day of age, Qinglongshan, Nanjing, China) were extracted for all experiments. The DHs were dissected and enzymatically digested with 0.25% trypsin at 37°C for 50 min. Mechanical dissociation and plating was then performed. The neurons were then placed in six-well plates with culture media (ThermoFisher, Shanghai, China) containing Neurobasal®-A media, B27, 2 mM l-glutamine, and the antibiotics penicillin and streptomycin. The cells were maintained in an incubator at 37°C and 5% CO2 throughout the experiments. All subsequent experiments were followed by cell culture for 1 week, and the number of cells in each well of the six-well plate reached 60,000–75,000. For TNF-α treatment experiments, neurons were grown in six-well plates before treatment with 10 ng/mL TNF-α in the presence or absence of 10 μM of p38 inhibitor SB202190 (Sigma, Gillingham, UK).
DH neuron electrophysiology
All data were acquired using Axon Clampex 10.6 (Molecular Devices). The high-pass filter setting was 2 kHz, and the sampling rate was 20 kHz. In the experimental process, we ensured that the quality of recording of DH neurons was determined by certain criteria, such as resting membrane potential, which is relatively stable; the series resistance was <30 MΩ throughout the recording process and the degree of variation was <20%; the input resistance was >400 MΩ and the degree of change was <35%. For current-clamp recordings, a Multiclamp 700B (Molecular Devices) was used, and data were analog-digital converted using an Axon 1440A instrument. Glass electrodes were pulled using a horizontal pipette puller (Sutter Instrument Company, Novato, CA, USA) and fire polished to have a resistance of 6–10 MΩ. The repetitive discharge of each cell was measured by the injection of 2.5× threshold stimuli for 1000 ms. The pipette solution contained the following (in mM): 124 K-gluconate, 2 MgCl2, 13.2 NaCl, 1 ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid [EGTA], 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 4 Mg-ATP, and 0.3 Na-GTP at pH 7.2. The bath solution contained (in mM) 140 NaCl, 5.4 KCl, 1 CaCl2, 1 MgCl2, 15.6 HEPES, and 10 glucose at pH 7.4. For the voltage-clamp, pipettes were filled with solution containing (in mM) 124 K-gluconate, 2 MgCl2, 13.2 NaCl, 1 EGTA, 10 HEPES, 4 Mg-ATP, and 0.3 Na-GTP at pH 7.2. The bath solution for all cells contained (in mM) 140 NaCl, 5.4 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose. When examining sodium-dependent potassium currents in isolation, NaCl was completely replaced with the impermeable cation N-methyl-d-glucamine (NMG-Cl) (Sigma). Identical bath and pipette solutions were used in both the voltage-clamp and current-clamp modes. In the voltage-clamp mode, the current was recorded at a holding potential of −70 mV, with 2000 ms pulses ranging between −80 and +50 mV.
Real-time polymerase chain reaction
The total RNA was extracted from the neuronal culture using the RNeasy Plus kit (Takara, RR047) according to the manufacturer’s recommendations. Before reverse transcription, the total RNA was treated with RQ1 RNase-Free DNase (Promega) to remove genomic DNA. The cDNA was synthesized using an Invitrogen SuperScript® III kit (ThermoFisher). The primers used for DH neuronal culture were as follows: for GAPDH, GTTACCAGGGCTGCCTTCTC (forward) and GATGGTGATGGGTTTCCCGT (reverse), and for SLICK, GTGTGTGTGCTTGTGTTGCAG (forward) and GAGCACCTCCCATACGTCTTC (reverse). The slopes of the primer efficiency equation for the primer pairs used in this study were between −3.1 and −3.6, giving reaction efficiencies between 90% and 110%, which are typically acceptable for a quantitative PCR assay. The running protocol comprised 40 cycles consisting of 95°C for 15 s and 60°C for 1 min using an Applied Biosystems 7500 Fast Real-time PCR system. Each sample was measured in triplicate.
Membrane protein biotinylation
DH neurons in six-well plates were used 30 min following incubation with 10 ng/mL TNF-α/10 μM of p38 inhibitor SB202190. Then, 160 μL of 10 mM Sulfo-NHS-SS-Biotin (ThermoFisher) was added to each well, and the plates were incubated at room temperature for 45 min. During this time, 100 μL of Pierce Streptavidin Magnetic Beads (ThermoFisher) were washed with phosphate-buffered saline (PBS) + 0.1% Tween. After 45 min, the cells were rinsed with PBS and lysed with a buffer containing PBS, 150 mM NaCl, 1% Nonidet P-40, and 0.1% sodium dodecyl sulfate, pH 8.0, and a protease inhibitor cocktail. The samples were then incubated with 50 μL of streptavidin beads (ThermoFisher) in a rotator overnight at 4°C. On the following day, the samples were washed twice in PBS. After removing the supernatant, the beads were resuspended in Laemmli sample buffer (Bio-Rad) and boiled for 5 min. The samples were then loaded onto a Ready Gel, and Western blot analysis was performed to detect membrane SLICK.
Cell viability assay
The cell cytotoxicity of TNF-α and the effect of p38 inhibitor SB202190 on the viability of DH neurons were evaluated using a 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay. Cells were incubated with 10 ng/mL TNF-α in the presence or absence of 10 μM SB202190. Afterward, 100 mL of a 0.5 mg/mL MTT solution (ThermoFisher) was added to the culture media. After incubation for 2 h at 37°C and the removal of culture medium, dimethyl sulfoxide was added to dissolve the formazan crystals. The absorbance at 570 nm was determined using an automatic microplate spectrophotometer. Data were analyzed using Prism GraphPad 6 software. Samples were measured in quintuplicate, and the experiment was performed in triplicate.
Statistical analysis
Statistical analyses were performed using SPSS statistical software (version 19.0, IBM). Data are presented as the mean ± standard error. Unpaired t-test analyses were used to determine statistical significance (p<0.05).
Results
Voltage-clamp whole-cell recordings were performed using these DH neurons. The average current density at 50 mV was 320.96±34.94 pA/pF (Figure 1A, n=5). When the sodium in the bath and pipette solution was replaced with the impermeant cation NMG, the IK of the DH neurons was greatly reduced, and the average current density at 50 mV was 197.07±20.98 pA/pF (Figure 1A, n=5), ~40% of its control value (Figure 1B, p<0.05, unpaired t-tests). Using whole-cell current-clamp recording on cultured DH neurons, we typically saw firing of <2 action potentials in 5 neurons, in response to 2.5× the stimulus threshold before TNF-α stimulation (Figure 2A). After the application of 10 ng/mL TNF-α, a progressive loss of firing accommodation was observed in 5 neurons (Figure 2B), and after 10 min, no firing accommodation was observed in 5 neurons (Figure 2C). However, in DH neurons incubated with 10 μM of p38 inhibitor SB202190 for 30 min before the short TNF-α treatment, no loss of firing accommodation was detected (Figure 2D, n=5).
Figure 1.

Voltage-clamp whole-cell recordings were conducted in DH neurons.
Notes: (A) Current was recorded at a holding potential of −70 mV, with 2000 ms pulses between −80 and +50 mV. Neuron was initially recorded in 140 mM external Na+ and then perfused with a solution in which NaCl was completely replaced with the impermeable cation N-methyl-d-glucamine (NMG-Cl). (B) Removal of extracellular Na+ reduces IK in DH neurons. *P<0.05.
Abbreviation: DH, dorsal horn.
Figure 2.
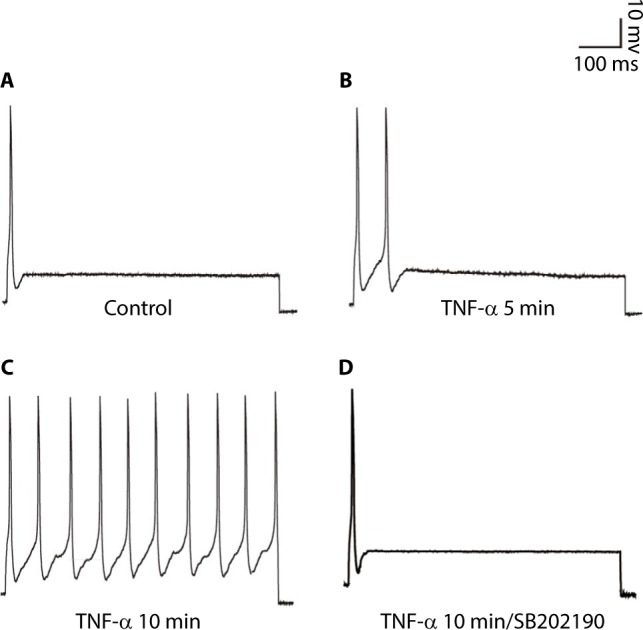
SB202190 restores the firing accommodation of DH neurons.
Notes: (A) Representative action potential recorded in untreated neurons. (B) 5 min application of 10 ng/mL TNF-α, neurons exhibited a progressive loss of firing accommodation. (C) 10 min application of 10 ng/mL TNF-α, neurons showed completed loss of firing accommodation. (D) DH neurons were incubated with 10 μM of p38 inhibitor SB202190 for 30 min before TNF-α treatment; no loss of firing accommodation was detected. Using whole-cell current-clamp on cultured DH neurons, stimulation was 2.5× threshold for 1000 ms.
Abbreviation: DH, dorsal horn.
Using whole-cell voltage-clamp recording, we assayed the effect of p38 MAPK activation on the IK in the DH neurons in the presence and absence of Na+ in the bath. In external solutions without Na+ (0 mM), the average current density at 5 mV was 152.23±21.44 pA/pF, and 10 min after the application of 10 ng/mL TNF-α, we observed a small but significant decrease in IK. The average current density at 50 mV was 140.46±20.19 pA/pF; only an 8% decrease in IK (Figure 3B, n=5) was observed when normalized to the control (Figure 3F, p<0.05, unpaired t-tests). In parallel experiments in which neurons were bathed in an extracellular bath containing physiological levels of Na+ (140 mM), the average current density at 50 mV was 250.19±34.85 pA/pF, and the application of 10 ng/mL TNF-α caused a much greater decrease in IK. The average current density at 50 mV was 166.92±20.92 pA/pF (Figure 3A, n=5). After the application of TNF-α, there was a 34% decrease in IK when normalized to the control (Figure 3E, p<0.01, unpaired t-tests), a significantly larger percentage decrease than in Na+-free conditions (p<0.05). These results strongly suggest that under physiological recording conditions, p38 MAPK strongly inhibits IK, particularly the KNa component of IK. The loss of firing accommodation caused by TNF-α could, therefore, likely be attributed to the inhibition of KNa. To test whether reduced p38 MAPK activity would produce the opposite effect, DH neurons were incubated with 10 μM of p38 inhibitor SB202190 for 30 min before TNF-α treatment, which produced a nonsignificant increase in total IK (Figure 3C). This effect was not seen upon replacing Na+ with NMG; instead, we observed a small decrease in the total outward current (Figure 3D), implying the presence of other unidentified potassium current(s) that are likely subject to p38 MAPK modulation.
Figure 3.

The KNa current is significantly decreased in DH neurons treated with TNF-α via p38 MAPK.
Notes: (A, B) Left, Representative traces of IK recorded before and after application of 10 ng/mL TNF-α in 140 and 0 mM Na+ external solution; Right, Current–voltage relationships before and after TNF-α treatment. (C, D) Left, Representative traces of IK recorded before and after application of 10 μM of p38 inhibitor SB202190 for 30 min and then 10 min TNF-α treatment in 140 mM and 0 mM Na+ external solution; Right, Current–voltage relationships before and after SB202190 and then TNF-α treatment. (E, F) Peak current after 10 ng/mL TNF-α and TNF-α combined SB202190 addition at 50 mV normalized to control for both 140 and 0 mM Na-containing extracellular solution. *P<0.05.
Abbreviations: DH, dorsal horn; TNF-α, tumor necrosis factor-alpha.
We determined whether TNF-α can decrease the expression of the channel protein to understand how it could decrease the current densities of SLICK channels. A surface-expression biotinylation assay showed that TNF-α decreased SLICK protein expression significantly (Figure 4A, n=5, p<0.05, unpaired t-tests). Meanwhile, we conducted parallel studies in which TNF-α was inhibited by pretreating neurons with 10 μM of SB202190 for 30 min followed by TNF-α treatment. In this case, surface SLICK channel expression was comparable to unstimulated neurons (Figure 4B). Furthermore, reverse transcription polymerase chain reaction analysis was performed on DH neurons incubated with TNF-α to determine whether the TNF-α-induced downregulation of SLICK proteins in DH neurons resulted from a reduction in transcription. We found no significant difference in SLICK mRNA expression compared with unstimulated neurons (Figure 4C). Our results showed that TNF-α directly decreased SLICK channel activity through possible posttranslational modification (PTM). We confirmed using the MTT assay that SB202190 in combination with TNF-α exerted no effect on the viability of DH neurons (Figure 5).
Figure 4.

TNF-α decreases membrane expression of SLICK channels via p38 MAPK through possible posttranslational modification in DH neurons.
Notes: (A) Representative blot of membrane SLICK biotinylation assay. Membrane biotinylation and precipitation by streptavidin followed by Western analysis using a SLICK-specific antibody were performed on untreated neurons, neurons treated with 10 ng/mL TNF-α for 10 min, or neurons pretreated with 10 μM of p38 inhibitor SB202190 for 30 min followed by 10 ng/mL TNF-α for 10 min. (B) Densitometric analysis of SLICK membrane expression when normalized to β-actin. (C) Normalized SLICK gene expression levels. *P<0.05.
Abbreviations: DH, dorsal horn; TNF-α, tumor necrosis factor-alpha.
Figure 5.
The cell cytotoxicity of TNF-α and the effect of p38 inhibitor SB202190 on the viability of DH neuron were evaluated using MTT assay.
Notes: MTT assay that SB202190 in combination with TNF-α exerted no effect on the viability of DH neurons.
Abbreviations: DH, dorsal horn; TNF-α, tumor necrosis factor-alpha.
Discussion
In this study, we provided the first evidence that the SLICK KNa channel is expressed in rat DH neurons and that TNF-α can reduce the expression of SLICK KNa channels at the protein level in DH neurons. Moreover, we showed that the inflammatory cytokine TNF-α decreases SLICK KNa expression at the membrane through a p38 MAPK-dependent pathway. Our study suggested that K+ exit decreased in DH neurons after TNF-α or p38 activation and reducing the number of KNa channels at the plasma membrane will result in loss of the firing accommodation.
Accumulating evidence has demonstrated that TNF-α is an effective activator of neuropathic pain.14 The MAPK family of kinases plays an important role in mammalian cell signaling. One kinase of the MAPK, p38, was reported to be increased in animal models of neuropathic pain.15 TNF-α activates multiple signaling pathways, including the p38 MAPK pathway,16 which is recognized as an important regulator of inflammatory pain,17 although TNF-α can also activate ERK or c-JUN of MAPK.18 Our results established the p38 MAPK-mediated downregulation of SLICK KNa expression in DH neurons and demonstrated, for the first time, a novel mechanism by which p38 MAPK could contribute to decreasing SLICK KNa channel responses via nontranscriptional mechanisms.
In this study, three types of firing patterns were identified in response to depolarizing current injections, namely, single-spiking, tonic-firing, and delayed-firing. We clamped only the single-spiking neurons and found that TNF-α can cause loss of firing accommodation, but this effect can be counteracted by p38 MAPK inhibitors. The findings were that in our culture preparation, the electrophysiological properties of DH neurons were similar to the properties of DH neurons recorded in young spinal cord slices.19 KNa channel expression decreases in vivo under pathological conditions accompanied by thermal hyperalgesia.20 Knockdown of KNa SLACK channels also decreases the firing accommodation in DRG neurons.21 We found that the SLICK channel mRNA remained unchanged, but the protein declined after exposure to TNF-α. Our results showed that these changes might be induced through the PTM of ion channels. Channel phosphorylation is the most common PTM. The action of kinases on SLICK channels is to decrease the current amplitude.22 Ubiquitylation is also a well-known PTM that negatively regulates the cell surface expression of many different plasma membrane proteins.23
We hypothesized that the mechanism underlying the neuropathic pain involved alterations in the SLICK channel, which could contribute to the abnormal excitability of DH neurons. These observations could explain our previous study, which showed that the TNF-α blood protein levels in the sciatica group were significantly higher than in healthy subjects.24 However, this sensitization could not be concluded to be a result of the direct phosphorylation or ubiquitylation of SLICK channels by p38 MAPK. In addition, to date, the onset of expression and detailed mapping of SLICK channel distribution in the DH during embryonic and postnatal development are missing, and therefore the effects of TNF-α may also change during this developmental period. Further work is needed to investigate this possibility.
Acknowledgments
The work was supported by the National Natural Science Foundation of China (Nos. 81572170 and 81572109). The authors thank Zi-Kai Zhou (Southeast University) for expert technical assistance. They also thank American Journal Expert for language polishing.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Tomasello DL, Gancarz-Kausch AM, Dietz DM, Bhattacharjee A. Transcriptional regulation of the sodium-activated potassium channel SLICK (KCNT2) promoter by nuclear factor-kappaB. J B Chem. 2015;290(30):18575–18583. doi: 10.1074/jbc.M115.643536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cervantes B, Vega R, Limon A, Soto E. Identity, expression and functional role of the sodium-activated potassium current in vestibular ganglion afferent neurons. Neuroscience. 2013;240:163–175. doi: 10.1016/j.neuroscience.2013.02.052. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharjee A, Joiner WJ, Wu M, Yang Y, Sigworth FJ, Kaczmarek LK. Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J Neurosci. 2003;27(37):11681–11691. doi: 10.1523/JNEUROSCI.23-37-11681.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Willis WD, Jr, Coggeshall RE. Sensory Mechanisms of the Spinal Cord. 3rd edition. Vol. 1. New York, NY: Kluwer Academic, Plenum Publishers; 2004. [Google Scholar]
- 5.Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139(2):267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhattacharjee A, Kaczmarek LK. For K+ channels, Na+ is the new Ca2+ Trends Neurosci. 2005;28(8):422–428. doi: 10.1016/j.tins.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Waxman SG, Zamponi GW. Regulating excitability of peripheral afferents: emerging ion channel targets. Nat Neurosci. 2014;17(2):153–163. doi: 10.1038/nn.3602. [DOI] [PubMed] [Google Scholar]
- 8.Ellis A, Bennett D. Neuroinflammation and the generation of neuropathic pain. Br J Anaesth. 2013;111(1):26–37. doi: 10.1093/bja/aet128. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Q, Yu J, Wang J, et al. The red nucleus TNF-α participates in the initiation and maintenance of neuropathic pain through different signaling pathways. Neurochem Res. 2015;40(7):1360–1371. doi: 10.1007/s11064-015-1599-9. [DOI] [PubMed] [Google Scholar]
- 10.Clark AK, Old EA, Malcangio M. Neuropathic pain and cytokines: current perspectives. J Pain Res. 2013;6:803–814. doi: 10.2147/JPR.S53660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin X. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci. 2006;26(1):246–255. doi: 10.1523/JNEUROSCI.3858-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Karim I, McCrudden MT, Linden GJ, et al. TNF-alpha-induced p38MAPK activation regulates TRPA1 and TRPV4 activity in odontoblast-like cells. Am J Pathol. 2015;185(11):2994–3002. doi: 10.1016/j.ajpath.2015.07.020. [DOI] [PubMed] [Google Scholar]
- 13.El Hachmane M-F, Rees KA, Veale EL, Sumbayev VV, Mathie A. Enhancement of TWIK-related Acid-sensitive potassium channel 3 (TASK3) two-pore domain potassium channel activity by tumor necrosis factor α. J Biol Chem. 2014;289(3):1388–1401. doi: 10.1074/jbc.M113.500033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andrade P, Hoogland G, Del Rosario JS, Steinbusch HW, Visser-Vandewalle V, Daemen MA. Tumor necrosis factor-α inhibitors alleviation of experimentally induced neuropathic pain is associated with modulation of TNF receptor expression. J Neurosci Res. 2014;92(11):1490–1498. doi: 10.1002/jnr.23432. [DOI] [PubMed] [Google Scholar]
- 15.Berta T, Qadri YJ, Chen G, Ji RR. Microglial signaling in chronic pain with a special focus on caspase 6, p38 MAP kinase, and sex dependence. J Dent Res. 2016;95(10):1124–1131. doi: 10.1177/0022034516653604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu H. Assembly of post-receptor signaling complexes for the tumor necrosis factor receptor superfamily. Adv Protein Chem. 2004;68:225–279. doi: 10.1016/S0065-3233(04)68007-7. [DOI] [PubMed] [Google Scholar]
- 17.Schäfers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-α induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. 2003;23(7):2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji RR, Gereau RWt, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60(1):135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruscheweyh R, Sandkühler J. Lamina-specific membrane and discharge properties of rat spinal dorsal horn neuronesin vitro. J Physiol. 2002;541(1):231–244. doi: 10.1113/jphysiol.2002.017756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang F, Wang X, Ostertag EM, et al. TMEM16C facilitates Na(+)-activated K+ currents in rat sensory neurons and regulates pain processing. Nat Neurosci. 2013;16(9):1284–1290. doi: 10.1038/nn.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nuwer MO, Picchione KE, Bhattacharjee A. PKA-induced internalization of slack KNa channels produces dorsal root ganglion neuron hyperexcitability. J Neurosci. 2010;30(42):14165–14172. doi: 10.1523/JNEUROSCI.3150-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santi CM, Ferreira G, Yang B, et al. Opposite regulation of Slick and Slack K+ channels by neuromodulators. J Neurosci. 2006;26(19):5059–5068. doi: 10.1523/JNEUROSCI.3372-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laedermann CJ, Abriel H, Decosterd I. Post-translational modifications of voltage-gated sodium channels in chronic pain syndromes. Front Pharmacol. 2015;6:263. doi: 10.3389/fphar.2015.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang K, Bao JP, Yang S, et al. A cohort study comparing the serum levels of pro-or anti-inflammatory cytokines in patients with lumbar radicular pain and healthy subjects. Eur Spine J. 2016;25(5):1428–1434. doi: 10.1007/s00586-015-4349-4. [DOI] [PubMed] [Google Scholar]