

Abstract
Nuclear factor kappaB (NF-κB) plays an important role in the transcriptional regulation of genes involved in inflammation and cell survival. Here, we show that coactivator-associated arginine methyltransferase CARM1/PRMT4 is a novel transcriptional coactivator of NF-κB and functions as a promoter-specific regulator of NF-κB recruitment to chromatin. Carm1 knockout cells showed impaired expression of a subset of NF-κB-dependent genes upon TNFα or LPS stimulation. CARM1 forms a complex with p300 and NF-κB in vivo and interacts directly with the NF-κB subunit p65 in vitro. CARM1 seems to act in a gene-specific manner mainly by enhancing NF-κB recruitment to cognate sites. Moreover, CARM1 synergistically coactivates NF-κB-mediated transactivation, in concert with the transcriptional coactivators p300/CREB-binding protein and the p160 family of steroid receptor coactivators. For at least a subset of CARM1-dependent NF-κB target genes, the enzymatic activities of both CARM1 and p300 are necessary for the observed synergy between CARM1 and p300. Our results suggest that the cooperative action between protein arginine methyltransferases and protein lysine acetyltransferases regulates NF-κB-dependent gene activation in vivo.
Keywords: CARM1, ChIP, p300, p65/RelA, methylation
Introduction
Nuclear factor kappaB (NF-κB) is a widely expressed, inducible transcription factor, which plays a key role in the transcriptional regulation of a variety of genes involved in mammalian immune and inflammatory responses (Ghosh et al, 1998). NF-κB has additionally been implicated as an important regulator of cellular events such as apoptosis, cell proliferation and differentiation (Baldwin, 1996). NF-κB is composed of members of the Rel family, which in eukaryotes includes p50/p105 (NF-κB1), p52/p100 (NF-κB2), Rel (c-Rel), p65 (RelA) and RelB (Ghosh et al, 1998). These proteins share a conserved 300 amino-acid region within their amino-termini, known as the Rel-homology domain (RHD), which mediates dimerization, nuclear translocation, DNA-binding and interaction with heterologous transcription factors and NF-κB inhibitors (Ghosh et al, 1998). The specificity of NF-κB-dependent transcription is thought to be at least partially due to differential homo- and heterodimerization of its family members, leading to a range of DNA-binding and activation potential (Karin, 1998). The most abundant and best-studied form of NF-κB in cells is a ‘classical' heterodimer consisting of the two subunits p50 (NF-κB1) and p65 (RelA). Although all Rel family members bind to DNA, only p65 (RelA), Rel (c-Rel) and RelB contain a transactivation domain. In most differentiated unstimulated cells, NF-κB is sequestered in the cytoplasm as an inactive transcription factor complex by its physical association with one of the several inhibitors of NF-κB (IκB) (Baeuerle and Baltimore, 1988; Whiteside and Israel, 1997). Treatment of cells with extracellular stimuli such as cytokines, bacterial lipopolysaccharides (LPS) or potent oxidants leads to the rapid phosphorylation of the IκBs, which results in their ubiquitination and subsequent degradation by the 26S-proteasome pathway (Karin, 1998; Karin and Ben-Neriah, 2000). Consequently, NF-κB accumulates in the nucleus, binds to specific κB consensus sequences in the chromatin and activates specific subsets of genes.
The assembly of a higher order NF-κB transcription complex is an important stage in NF-κB-dependent transcription, involving multiple coactivator/cofactor–NF-κB–DNA interactions (Merika et al, 1998; Agalioti et al, 2000; Merika and Thanos, 2001). The two key coactivators of NF-κB, histone-acetyltransferases p300 and its homolog, the CREB-binding protein (CBP), directly associate with the NF-κB subunits p50 and p65 (Gerritsen et al, 1997; Perkins et al, 1997; Hassa et al, 2003). These coactivators are thought to promote the rapid formation of the pre-initiation and re-initiation complexes by bridging the sequence-specific activators (like NF-κB) to the basal transcriptional machinery, thereby facilitating multiple rounds of transcription (Goodman and Smolik, 2000). Additionally, the histone acetyltransferases p300 and CBP can modify the amino-terminal tails of nucleosomal histones, thereby altering the local chromatin structure (Schiltz et al, 1999; Kundu et al, 2000). It was proposed that the coactivator p300/CBP functions also as a signal integrator by coordinating diverse signal transduction events at the transcriptional level (Goodman and Smolik, 2000). Phosphorylation of p65 by protein kinase A (PKA) has been shown to stimulate NF-κB-dependent gene expression by enhancing p65 association with p300/CBP (Zhong et al, 1998). Previous reports have shown that NF-κB-dependent transcriptional complexes also require the p300/CBP-associated factor (P/CAF), the p160 family of steroid receptor coactivators and PC1/PARP-1 (Sheppard et al, 1999; Hassa et al, 2001, 2003). Thus, additional components might be required to stabilize the association of distinct NF-κB coactivator complexes.
Coactivator-associated arginine methyltransferase (CARM1/PRMT4) was identified as SRC-2/TIF2/GRIP1-binding protein and belongs to a family of arginine-specific protein methyltransferases, which includes at least seven members (PRMT1–7) (McBride and Silver, 2001; Miranda et al, 2004). CARM1 has been shown to synergistically stimulate transcription by nuclear receptors in combination with the p160 family of coactivators and forms a ternary complex with p300/CBP and SRC-2/TIF2/GRIP1 (Koh et al, 2001; Lee et al, 2002). After recruitment to the promoters of estrogen-responsive genes, CARM1 methylates specific arginine residues (Arg17 and Arg26) in the N-terminal tail of histone H3 as part of the transcriptional activation process (Bauer et al, 2002; Daujat et al, 2002). Recent studies broadened the targets of the transcriptional coactivator function of CARM1: CARM1 coactivates p53-dependent transcription and cooperates with β-catenin to enhance transcriptional activation by the lymphoid enhancer factor/T-cell factor (LEF1/TCF4) (Koh et al, 2002; An et al, 2004). Mice with a targeted disruption of Carm1 die during late embryonic development or immediately after birth, supporting the idea that CARM1 is a crucial coactivator for gene expression during late embryonic development (Yadav et al, 2003). Aberrant T-cell development in Carm1-deficient embryos was due to a partial developmental arrest in the earliest thymocyte progenitor subset, indicating that CARM1 plays a significant role in promoting the differentiation of early thymocyte progenitors (Kim et al, 2004).
Since the transcriptional coactivator CARM1 is capable of forming a complex with the known coactivators of NF-κB, p300/CBP and SRC-2, we tested whether CARM1 participates in NF-κB-dependent gene activation. Our results show that CARM1 directly binds to the NF-κB subunit p65 and synergistically coactivates NF-κB-mediated transactivation, in concert with the transcriptional coactivators p300/CBP and the p160 family of steroid receptor coactivators. CARM1 is responsible for H3(R17) methylation of NF-κB target genes in vivo and is required for NF-κB-regulated activation of a subset of genes. CARM1 functions by enhancing NF-κB recruitment to κB sites contained in H3(R17)-methylated promoters, although additional roles downstream of p65 recruitment mediated by its bridging factor/coactivator activity cannot be ruled out. These results suggest that the cooperative action between coactivators with histone arginine methyltransferase activities and at least two distinct classes of transcription coactivator molecules with histone acetyltransferase activities regulates NF-κB-dependent gene activation in vivo.
Results
NF-κB-dependent gene expression is impaired in Carm1(−/−) cells
To test whether CARM1 influences NF-κB-dependent gene expression, Carm1(+/+) or Carm1(–/–) MEFs were treated with LPS as indicated and the expression of NF-κB-dependent genes was assessed by RT–PCR (Figure 1A). The experiments revealed that LPS-induced levels of G-CSF, MIP-2, MCP-1, ICAM1 and IP-10 were impaired in Carm1(–/–) cells (Figure 1A). However, the expression of IκBα, IL-6, KC and COX-2 was not reduced (Figure 1A), indicating that only a subset of NF-κB-dependent genes requires CARM1 for induction. The expression of IκBα and IL-6 was even slightly upregulated in Carm1(–/–) cells, a result confirmed by quantitative real-time PCR (Figure 1A and data not shown). Similar results were obtained when cells were stimulated with TNFα (see below). To further investigate the relevance of CARM1 in NF-κB-dependent gene expression, Carm1(+/+) or Carm1(–/–) cells were transfected with the indicated NF-κB-dependent luciferase reporter constructs together with an expression vector for CARM1, and subsequently treated with the indicated stimuli (TNFα or LPS). Cell lysates were tested for luciferase activity as a measure of NF-κB activity (Figure 1B and C). Although TNFα and LPS were able to induce NF-κB-dependent transcriptional activation in Carm1(+/+) cells (Figure 1B), the NF-κB-dependent transcriptional activation was severely reduced in Carm1(–/–) cells upon stimulation with TNFα or LPS (Figure 1C). Re-expression of CARM1 in Carm1(–/–) cells synergistically enhanced NF-κB-dependent transcriptional activation five-fold in response to TNFα or LPS (Figure 1C). No synergistic activation was observed in Carm1(+/+) cells, indicating that endogenous CARM1 levels are sufficient to provide maximal coactivation (Figure 1B). The same transfection experiments with a reporter gene under the control of mutated κB sites revealed that the observed induction was NF-κB-specific. Together, these results indicate that CARM1 is required for NF-κB-dependent transactivation of extra-chromosomal templates upon stimulation with proinflammatory stimuli.
Figure 1.
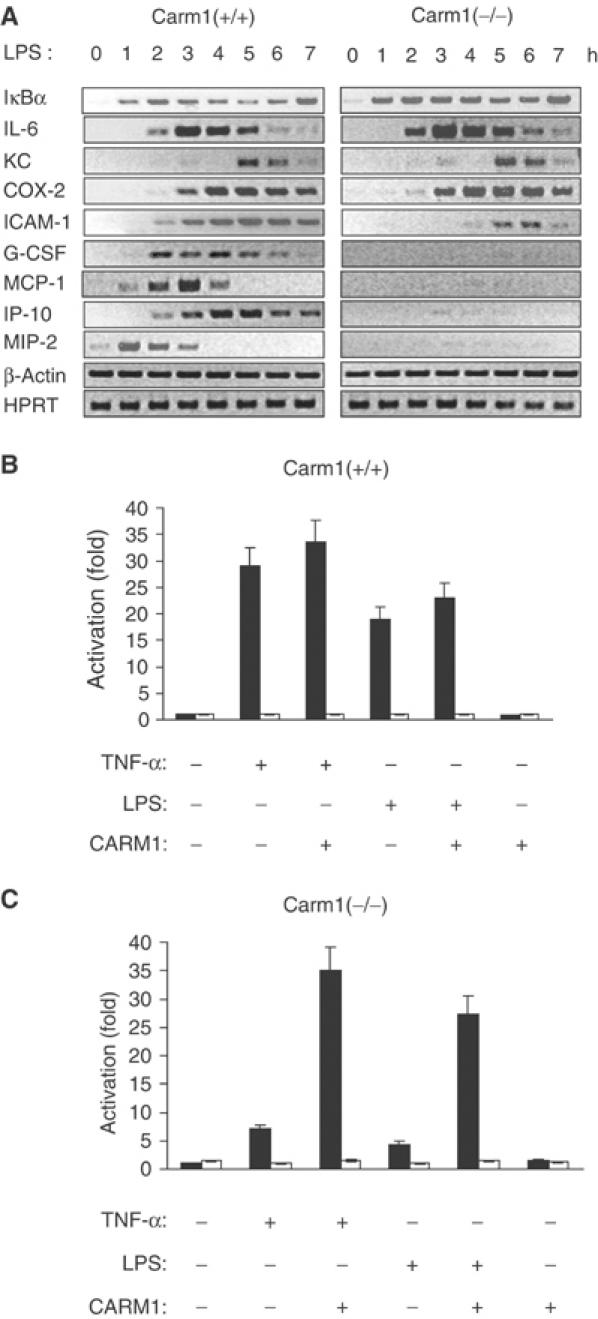
Impaired NF-κB-dependent gene expression in Carm1(−/−) cells in response to proinflammatory stimuli. (A) Impaired expression of MIP-2, MCP-1, G-CSF, ICAM1 and IP-10 in Carm1(−/−) MEF cells in response to LPS. Carm1(+/+) and Carm1(−/−) MEF cells were treated with LPS (10 μg/ml) and RNA isolated at the indicated time points, followed by RT–PCR determination of MIP-2, MCP-1, G-CSF, ICAM1, IP-10, IL-6, KC, COX-2, HPRT and β-actin mRNA. (B, C) Impaired NF-κB-dependent gene expression in Carm1(−/−) cells in response to TNFα and LPS. Carm1(+/+) MEF cells (B) and Carm1(−/−) MEF cells (C) were cotransfected with HIV-luc or HIVmut-κB-luc (2 μg) and pphRSVnt-β-gal (200 ng) together with CMV-CARM1 (500 ng) or CMV empty vector as indicated. Cells were subsequently stimulated with TNFα (10 ng/ml) or LPS (10 μg/ml) 24 h after transfection for 8 h. The indicated activation was determined by the ratio of the relative luciferase activity measured for the HIV-luc (black bars) or HIVmut-κB-luc (white bars) reporter gene after stimulation. The ratio obtained for untreated cells was arbitrarily set to 1. Error bars indicate s.e. of three independent experiments.
Normal NF-κB induction in Carm1(−/−) cells
To analyze whether the activation of the NF-κB-signaling pathway is affected in Carm1(−/−) cells upon stimulation with TNFα or LPS, we assessed the expression levels of important components of the NF-κB-signaling pathway by immunoblot and RT–PCR analysis (Figure 2A and B). The immunoblot analysis of nuclear and cytoplasmic extracts of Carm1(+/+) and Carm1(−/−) cells revealed that p65, c-Rel, RelB, p300 and PARP-1 are equally expressed in Carm1(+/+) and Carm1(−/−) cells (Figure 2A, left and right panels). Additionally, these experiments showed that there is equivalent nuclear translocation of NF-κB in Carm1(+/+) and Carm1(−/−) cells. Moreover, the expression levels of upstream signaling components, TLR-2, TLR-4 and IRAK4 mRNA, was not impaired in Carm1(−/−) cells (Figure 2B, right panel), compared to Carm1(+/+) cells (Figure 2B, left panel). We then investigated whether the degradation of IκBα might be affected due to the absence of CARM1. Carm1(+/+) and Carm1(−/−) cells were treated with TNFα or LPS for the indicated time points and the protein levels of IκBα subsequently tested by immunoblot analysis (Figure 2C). No differences in degradation and re-synthesis of IκBα could be observed between Carm1(+/+) and Carm1(−/−) cells upon stimulation with TNFα (Figure 2C) or LPS (data not shown). To further investigate whether the activation of the NF-κB-signaling pathway is affected in Carm1(−/−) cells upon stimulation, we analyzed the kinetics of nuclear translocation of p65 in Carm1(+/+) and Carm1(−/−) cells by immunofluorescence analysis. Kinetics of p65 nuclear entry in Carm1(−/−) cells was comparable to that observed in Carm1(+/+) cells (Figure 2D). Finally, we tested whether the DNA-binding activity of NF-κB on naked templates is affected in Carm1(−/−) cells. EMSA studies with DNA oligos corresponding to the κB sites in IL-6, IP-10 and MIP-2 promoter revealed no differences between Carm1(+/+) and Carm1(−/−) cells in DNA-binding activity of NF-κB (p50/p65) to nonchromatinized templates (data not shown).
Figure 2.
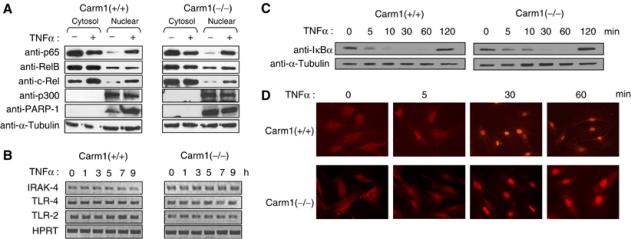
Expression levels of components of the NF-κB signaling pathway and nuclear–cytoplasmic shuttling of NF-κB is not affected in Carm1(−/−) MEFs. (A) Protein expression levels of p65, c-Rel, RelB, p300 and PARP-1 are not affected in Carm1(−/−) MEF cells. Carm1(+/+) and Carm1(−/−) MEF cells were treated with TNFα (10 ng/ml) for 20 min and cytoplasmic and nuclear extracts resolved by SDS–PAGE followed by subsequent immunoblot analysis for p65, c-Rel, RelB, p300 and PARP-1 (A, left and right panels). α-Tubulin was used as an internal standard. (B) mRNA expression levels of TLR4, TLR2 and IRAK4 are not impaired in Carm1(−/−) MEF cells. Carm1(+/+) and Carm1(−/−) MEF cells were treated with TNFα (10 ng/ml) (B, left and right panels) and RNA isolated at the indicated time points followed by RT–PCR analysis for TLR4, TLR2, IRAK4 and HPRT (B, left and right panels). (C) Degradation and re-synthesis of IκBα is not affected in Carm1(−/−) MEF cells. Carm1(+/+) and Carm1(−/−) MEF cells were treated with TNFα (10 ng/ml) (C, left and right panels) and whole-cell extracts isolated at the indicated time points and resolved by SDS–PAGE, followed by subsequent immunoblot analysis for IκBα. α-Tubulin was used as an internal standard. (D) Nuclear translocation of NF-κB is not delayed in Carm1(−/−) MEFs. Carm1(+/+) and Carm1(−/−) MEF cells were treated with TNFα (10 ng/ml) (D, upper and lower panels) and fixed with paraformaldehyde at the indicated time points followed by immunostaining for p65 and analysis by immunofluorescent microscopy.
CARM1 forms a complex with NF-κB in vivo and binds directly to p65 in vitro
To investigate whether CARM1 physically interacts with NF-κB in vivo, we immunoprecipitated CARM1 complexes from nuclear extracts upon stimulation of 293T cells with TNFα and tested the presence of p65 and p300 by immunoblot analysis using anti-p65, anti-CARM1 or anti-p300 antibodies. p65 and p300 formed a complex with CARM1 in the nucleus (Figure 3A). DNA did not mediate the association of p300 and p65 with CARM1 in the nucleus since the presence of ethidium bromide or DNAse1 did not affect p300/p65/CARM1 binding (data not shown). As these results strongly suggested that CARM1 would directly interact with at least one subunit of NF-κB, recombinant purified GST-p65 full length was bound to glutathione beads, followed by incubation with recombinant purified full-length His-CARM1 (Figure 3B). After extensive washes, bound proteins were resolved by SDS–PAGE, followed by immunoblot analysis for CARM1. CARM1 was able to bind directly to the NF-κB subunit p65 but not to the GST control (Figure 3B). To map the interaction domains within p65 and CARM1, GST and GST-fusion proteins expressing either the Rel-homology domain of p65 (RHD; aa 1–305), or the transactivation domain of p65 (aa 441–551), were used in GST pull-down experiments with in vitro translated full-length CARM1 or deletion mutants corresponding to the N-terminal domain (aa 1–148), catalytic domain (aa 140–480) or transactivation domain (aa 462–608), respectively. These experiments revealed that a region between aa 148 and 462 containing the catalytic domain but not the N- or C-terminal domain of CARM1 was able to selectively interact with the RHD of p65 (Figure 3C), confirming that the interactions described above were direct and not mediated by other proteins.
Figure 3.
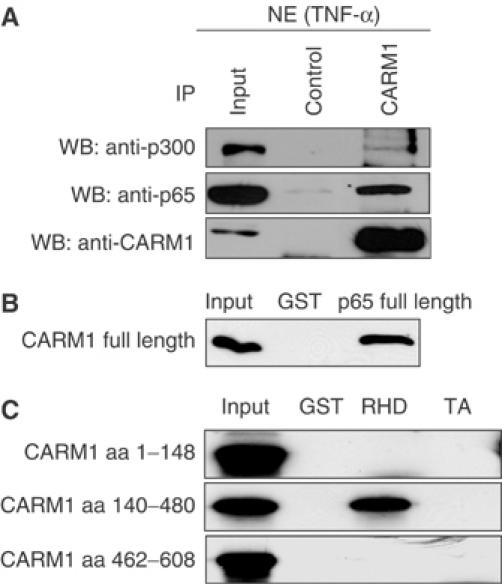
CARM1 forms a complex with NF-κB in vivo and directly binds to the NF-κB subunit p65 in vitro. (A) CARM1 and p65 form a complex in vivo. p65, CARM1 and p300 were coimmunoprecipitated (IP) in the presence of 120 mM NaCl from nuclear extract of TNFα-treated 293T cells (30 min, 10 ng/ml) using control IgGs and an anti-CARM1 antibody. Bound proteins were resolved by SDS–PAGE and subsequently detected by immunoblot (IB) analysis for p65, CARM1 and p300. Input lanes represent 10% of the input. (B) CARM1 directly binds to p65. Pull-down assays with purified GST and p65 full-length fused to GST (1 μg) and purified recombinant CARM1 full-length (0.1 μg) in the presence of 120 mM NaCl. Bound proteins were resolved by SDS–PAGE and detected by immunoblot analysis for CARM1. Input lanes represent 1% of the input. (C) The central domain of CARM1 binds to the RHD domain of p65. Pull-down assays with GST, the Rel-homology domain of p65 (RHD; aa 1–305) or the transactivation domain of p65 (TA; aa 441–551) fused to GST (1 μg) and different in vitro-translated CARM1 deletion mutants (N-terminal domain (aa 1–148), catalytic domain (aa 140–480) or transactivation domain (aa 462–608), respectively) in the presence of 120 mM NaCl. Bound proteins were resolved by SDS–PAGE visualized by autoradiography. Input lanes represent 1% of the input.
CARM1 coactivates NF-κB synergistically with p300/CBP
CARM1 and p300/CBP were recently shown to form a ternary complex and to function synergistically to enhance the activity of nuclear receptors (Lee et al, 2002). Thus, CARM1 in combination with p300 might also synergistically coactivate NF-κB-mediated transactivation. In order to directly test this possibility, we transfected Carm1(+/+) and Carm1(−/−) cells with expression vectors for CARM1 and p300 along with a NF-κB-dependent luciferase reporter; cells were subsequently treated with TNFα or LPS (Figure 4A and B). p300 and CARM1 synergistically stimulated NF-κB-mediated transcription in response to TNFα or LPS (Figure 4A and B). Expression of p300 synergistically enhanced reporter gene expression (up to three-fold) in Carm1(+/+) cells in response to TNFα or LPS (Figure 4A and B, left panel). However, in Carm1(−/−) cells, the synergy between p300 and TNFα or LPS was severely impaired (Figure 4A and B, right panel). Coexpression of CARM1 together with p300 caused a highly synergistic enhancement in Carm1(−/−) cells (Figure 4A and B, right panel), indicating that the presence of CARM1 itself is required for the synergistic transcriptional activation of NF-κB by p300 (Figure 4A and B).
Figure 4.
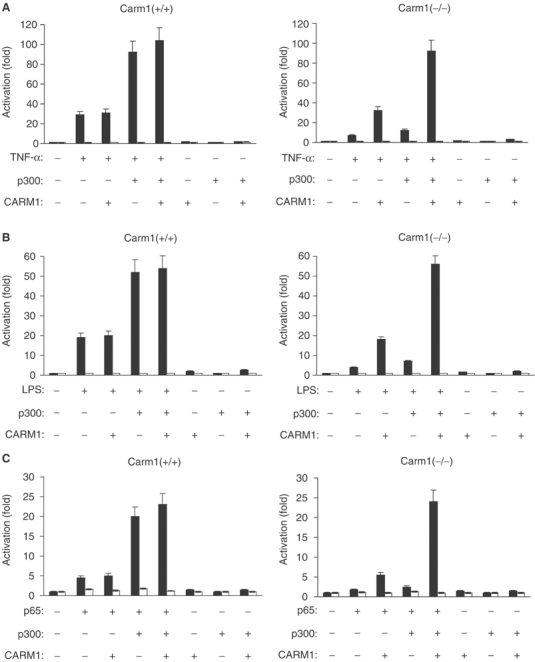
CARM1 synergistically coactivates the NF-κB-mediated transactivation, in concert with the transcriptional coactivator p300/CBP. (A, B) CARM1 synergistically activates together with p300 NF-κB-dependent gene expression in response to proinflammatory stimuli. Carm1(+/+) MEF cells (left panel) and Carm1(−/−) MEF cells (right panel) were cotransfected with HIV-luc or HIVmut-κB-luc (2 μg) and pphRSVnt-β-gal (200 ng), together with CMV-CARM1 (200 ng) and RSV-p300 (500 ng) or CMV and RSV empty vectors as indicated. Cells were subsequently stimulated with TNFα (10 ng/ml) (A) or LPS (10 μg/ml) (B) 24 h after transfection for 8 h. Cells were harvested 32 h after transfection and the indicated activation of NF-κB-dependent gene expression determined as described in Figure 1. (C) CARM1 synergistically activates together with p300 and p65-dependent gene expression. Carm1(+/+) MEF cells (left panel) and Carm1(−/−) MEF cells (right panel) were cotransfected with HIV-luc or HIVmut-κB-luc (2 μg) and pphRSVnt-β-gal (200 ng) together with CMV-p65 (80 ng) CMV-CARM1 (200 ng) and RSV-p300 (500 ng) or CMV and RSV empty vectors as indicated. The indicated activation of NF-κB-dependent gene expression was determined as described in Figure 1.
To confirm these results, experiments were repeated overexpressing p65 without exogenous stimulation. The amount of p65 expression vector strongly influenced the degree of cooperation among these coactivators; as previously shown for nuclear receptors (Lee et al, 2002), high synergistic enhancement of transcriptional coactivation was obtained only under conditions where limiting levels of p65 were expressed (data not shown). Therefore, we decided to proceed with low amounts of p65 expression vectors for all the following experiments. Carm1(+/+) and Carm1(−/−) cells were transfected with expression vectors for CARM1, p65 and p300 along with a NF-κB-dependent luciferase reporter containing wild-type or mutated κB sites. In the absence of cotransfected coactivator, the low levels of p65 used produced only a slight increase (4.5-fold) in luciferase activity in Carm1(+/+) cells (Figure 4C, left panel). No significant increase (1.5-fold) in luciferase activity could be observed in Carm1(−/−) cells (Figure 4C, right panel). Coexpression of CARM1 with p65 augmented NF-κB-dependent transcriptional activation in Carm1(−/−) cells to a similar extent as observed in Carm1(+/+) cells with p65 alone (Figure 4C, left panel). Coexpression of p300 with p65 synergistically enhanced reporter gene expression (four-fold) in Carm1(+/+) cells (Figure 4C, left and right panels), whereas no synergistic enhancement could be observed in Carm1(−/−) cells (Figure 4C, right panel). However, addition of CARM1 with p300 caused a highly synergistic enhancement in Carm1(−/−) cells (Figure 4C, right panel). No synergistic activation was observed in Carm1(+/+) cells, indicating that endogenous CARM1 is sufficient to provide maximal coactivation (Figure 4C, left panel). When p300 was substituted for CBP, a similar level of synergy was observed (data not shown).
CARM1 and SRC-2/TIF2/GRIP1 synergistically coactivate NF-κB-mediated transactivation
Next, we tested whether the combined coexpression of p65 together with CARM1, p300 and p160 family members might result in even stronger synergistic coactivation of NF-κB. Coexpression of SRC-2/TIF2/GRIP1 with p65 and p300 resulted in a additional (three-fold) synergistic increase in NF-κB activity in Carm1(+/+) cells (Figure 5A), compared to coexpression of p65 and p300. This could not be observed in Carm1(−/−) cells (Figure 5B). This synergy between p300 and p160 family members observed in Carm1(+/+) cells is in agreement with recent reports, showing that NF-κB-dependent transcriptional activity requires p300/CBP but also the p160 family of steroid receptor coactivators (Sheppard et al, 1999). Remarkably, strong synergistic coactivation of NF-κB by p300 and SRC-2/TIF2/GRIP1 could be observed in Carm1(−/−) cells only when CARM1 was present (Figure 5B), while additional expression of CARM1 did not enhance NF-κB-dependent transcriptional activation in Carm1(+/+) cells (Figure 5A). Combined coexpression of p65 together with CARM1, p300 and other p160 family members in Carm1(−/−) cells led to similar results, although the additional synergistic coactivation of NF-κB by SRC-1/NCoA-1 or SRC-3/p/CIP/RAC-3 was not as high as observed for SRC-2/TIF2/GRIP1 (data not shown). To further confirm the relevance of CARM1 for synergistic coactivation of NF-κB by p300 and p160 family members, we repeated these experiments in the monkey COS-1 cells (Figure 5C). The results of these experiments revealed that combined expression of p65 together with all three coactivators in COS-1 cells led to a similar level of coactivator synergy as observed in Carm1(−/−) cells (Figure 5C). Coexpression of CARM1 resulted in a strong synergistic coactivation, despite endogenous CARM1 expression in these cells. Thus, the endogenous levels of CARM1 seem to be limiting in these cells. Together, these results strongly suggest that CARM1 is required for synergistic coactivation of NF-κB-dependent gene activation by p300/CBP and p160 family members.
Figure 5.
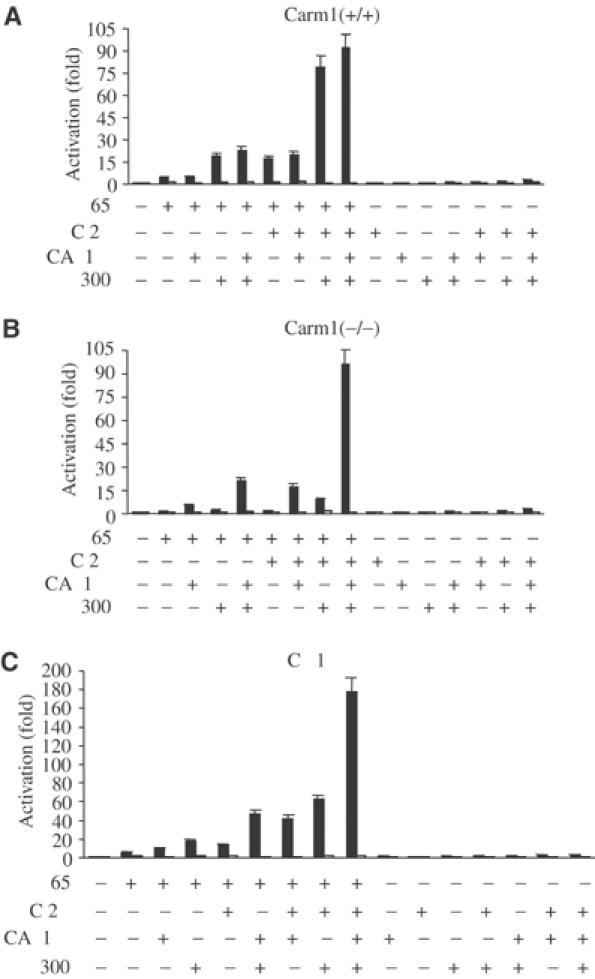
CARM1 synergistically coactivates NF-κB-dependent transcription in concert with the transcriptional coactivators p300/CBP and the p160 family of steroid receptor coactivators. (A, B) CARM1 synergistically activates together with p300 and SRC-2 p65-dependent gene expression. Carm1(+/+) MEF cells (A) and Carm1(−/−) MEF cells (B) were cotransfected with HIV-luc or HIVmut-κB-luc (2 μg) and pphRSVnt-β-gal (200 ng), together with CMV-p65 (80 ng), CMV-CARM1 (200 ng), CMV-SRC-2 (300 ng) and RSV-p300 (500 ng) or CMV and RSV empty vectors as indicated. Cells were harvested 32 h after transfection and the indicated activation of NF-κB-dependent gene expression determined as described in Figure 1. (C) CARM1 synergistically activates together with p300 and p65 NF-κB-dependent gene expression in COS-1 cells. COS-1 cells were cotransfected with HIV-luc or HIVmut-κB-luc (2 μg) and pphRSVnt-β-gal (200 ng) together with CMV-p65 (50 ng) CMV-CARM1 (200 ng), CMV-SRC-2 (300 ng) and RSV-p300 (500 ng) or CMV and RSV empty vectors as indicated. The indicated activation of NF-κB-dependent gene expression was determined as described in Figure 1.
CARM1 is required for H3(R17) methylation and recruitment of p65 to the MIP-2 and IP-10 promoters
We next tested both CARM1 and p65 recruitment as well as H3(R17) methylation at NF-κB-dependent genes using quantitative chromatin immunoprecipitation (ChIP) assays. First, we confirmed by real-time PCR the CARM1 dependency of NF-κB-dependent gene induction in TNFα-stimulated cells. While MIP-2, IP-10 and MCP-1 mRNA induction in response to TNFα was nearly completely abrogated in Carm1(−/−) cells, IL-6 mRNA expression was not affected or even increased in Carm1(−/−) cells, thus strengthening the concept that CARM1 requirement is strictly gene-specific (Figure 6A). An anti-CARM1 ChIP assay revealed that CARM1 is not only recruited to the CARM1 dependent MIP-2 and IP-10 promoters but also to the CARM1 independent IL-6 promoter in Carm1(+/+) cells, upon stimulation (Figure 6B). We next investigated H3(R17) methylation at all three genes using a ChIP assay with an antibody recognizing H3(R17) only when methylated. A strong increase in H3(R17) methylation was found to occur on MIP-2, IP-10 and IL-6 promoters in TNFα stimulated Carm1(+/+) cells (Figure 6C). No H3(R17) methylation could be detected on either gene in Carm1(−/−) cells, indicating that methylation of H3(R17) is entirely CARM1-dependent (Figure 6C). Surprisingly, an anti-p65 ChIP assay revealed a major defect in p65 recruitment to the MIP-2 and IP-10 promoters in Carm1(−/−) cells (Figure 6D), whereas no significant differences could be observed at IL-6 between Carm1(+/+) and Carm1(−/−) cells (Figure 6D), thus indicating that the mechanism underlying CARM1 requirement for transcriptional induction differs depending on the gene. While these results implicate that CARM1 functions in a gene-specific manner by enhancing NF-κB recruitment to regulatory sites, possibly through an increase in κB site accessibility, they do not rule out the possibility that further effects downstream of NF-κB recruitment also occur. Moreover, the results obtained with IL-6 indicate that H3(R17) methylation may not be required for the activity of all promoters at which it occurs.
Figure 6.
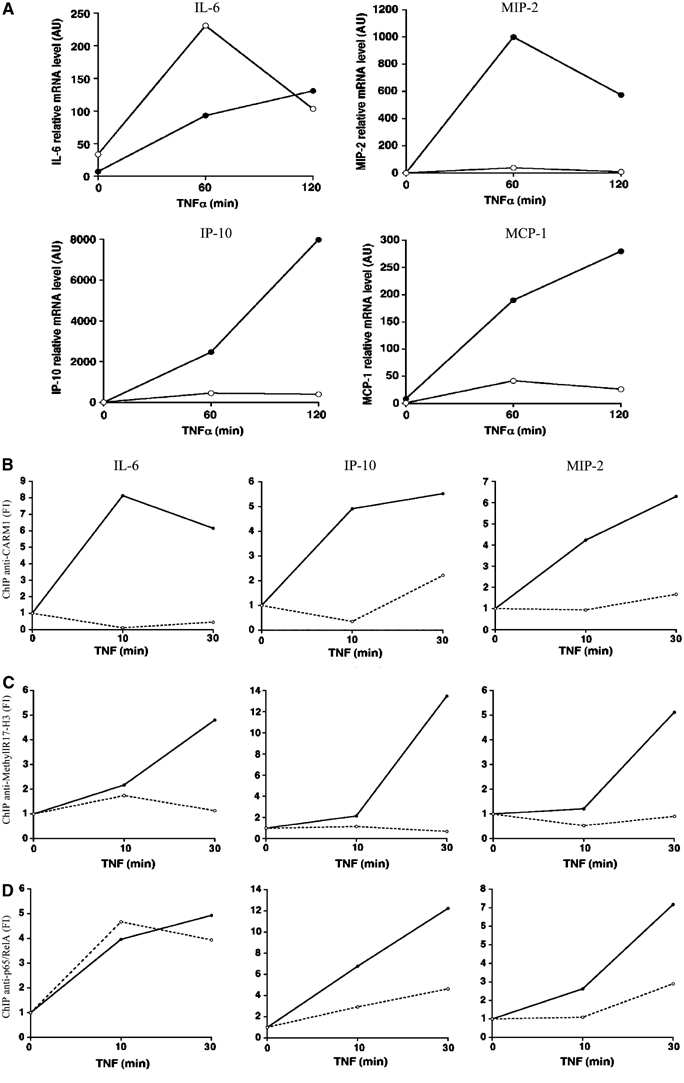
CARM1 functions in a gene-specific manner. (A) Quantification of TNFα-stimulated MIP-2, IP-10, MCP-1 and IL-6 transcription Carm1(+/+) MEF cells (filled circles) and Carm1(−/−) MEF cells (empty circles) by real-time PCR. (B) Quantitative ChIP assays with anti-CARM1 antibodies were performed for the indicated genes on TNFα-stimulated Carm1(+/+) (filled circles) and Carm1(−/−) MEF cells (empty circles). (C) Anti-methyl-H3(R17) ChIP assay for the indicated genes on TNFα-stimulated Carm1(+/+) (filled circles) and Carm1(−/−) MEF cells (empty circles). (D) ChIP assays with anti-p65 antibodies were performed for the indicated genes on TNFα-stimulated Carm1(+/+) (filled circles) and Carm1(−/−) MEF cells (empty circles).
The enzymatic activities of CARM1 and p300/CBP are required for full cooperativity and NF-κB-dependent transcription
Since our ChIP study revealed induced methylation of H3(R17) upon stimulation, the observed cooperativity between CARM1 and p300/CBP regarding NF-κB-dependent gene expression might require their enzymatic activities. We first tested whether in transient luciferase reporter assays the IL-6-, MIP-2 and IP-10 promoters are regulated in Carm1(+/+) and Carm1(−/−) cells, similarly as their endogenous counterparts (Figure 7A). These experiments revealed that all promoters requiring CARM1 for activity were similarly dependent on CARM1 when taken out of their natural context and tested in reporter assays. Re-expression of CARM1 in Carm1(−/−) cells fully rescued stimulus-induced activation of the MIP-2 and IP-10 promoter in Carm1(−/−) cells (Figure 7A). We next transfected Carm1(−/−) cells with expression vectors for p65, wild-type or enzymatic mutants of CARM1 and/or p300 along with a luciferase reporter under the control of the endogenous MIP-2 or IP-10 promoter (Figure 7B and C). Coexpression of wild-type CARM1 and p300 together with limiting levels of p65 in Carm1(−/−) cells caused a highly synergistic enhancement of transcription regulated from both MIP-2 and IP-10 promoters (Figure 7B and C). However, the cooperativity/synergy between p300 and CARM1 was severely impaired when an enzymatic mutant form of either CARM1 or p300 was coexpressed. Coexpression of an enzymatic mutant form of both CARM1 and p300 caused an even more severe reduction in p65-dependent activation of the IP-10 and MIP-2 promoter, indicating that the enzymatic activities of both CARM1 and p300/CBP are required for full synergistic enhancement of NF-κB-dependent transcription.
Figure 7.
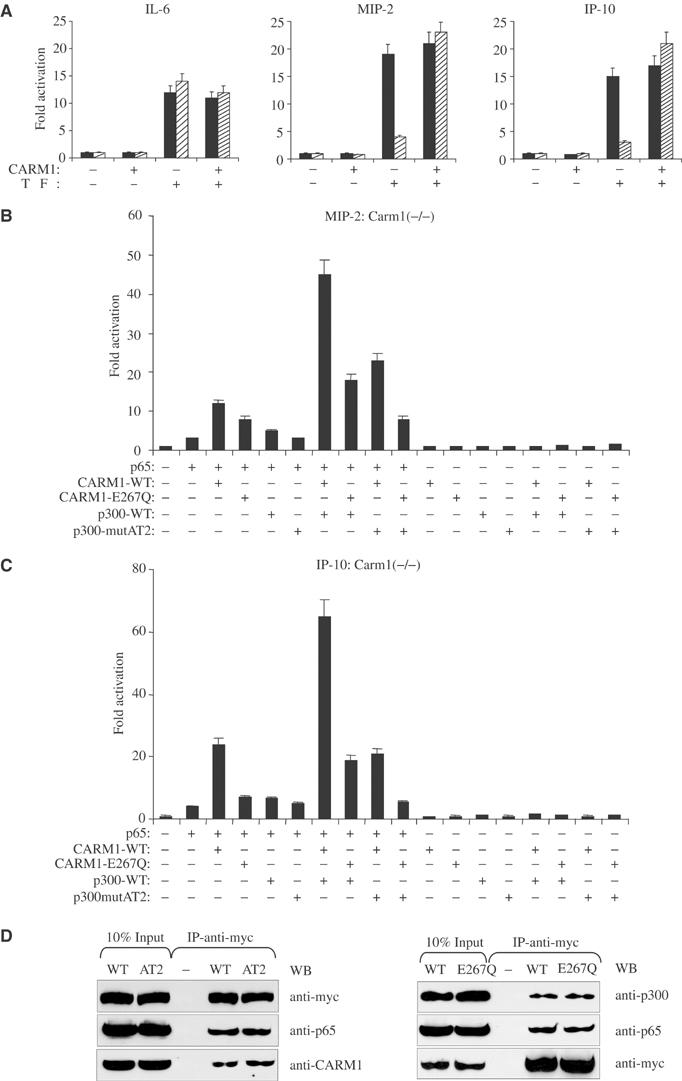
Enzymatic activities of CARM1 and p300/CBP are required for full cooperativity between CARM1 and p300. (A) Carm1(+/+) MEF cells (black bars) and Carm1(−/−) MEF cells (dashed bars) were cotransfected with pGL3-MIP-2(−770), pGL3-IP-10(−533) or pOLUC-IL-6(−303/+23) (5 μg) and pphRSVnt-β-gal (200 ng) alone or together with CMV-CARM1 (200 ng) or CMV empty vectors as indicated. Cells were subsequently stimulated with TNFα (10 ng/ml) for 6 h. Cells were harvested 32 h after transfection and the indicated activation of NF-κB-dependent gene expression determined as described in Figure 1. (B, C) Enzymatic activities of CARM1 and p300/CBP are required for full cooperativity between CARM1 and p300. Carm1(−/−) MEF cells were cotransfected with pGL3-MIP-2(−770) (B) or pGL3-IP-10(−533) (C) (5 μg) and pphRSVnt-β-gal (200 ng), together with CMV-p65 (80 ng) CMV-CARM1-WT or CARM1-E267Q (200 ng), CMV-p300-WT or p300mutAT2 (750 ng) and CMV empty vectors as indicated. Cells were harvested 32 h after transfection and the indicated activation of NF-κB-dependent gene expression determined as described in Figure 1. (D) Both wild type and mutant forms of CARM1 and p300 interact to the same extent with p65 in vivo. 293T cells were transfected with CMV expression vectors, either for wild type or enzymatic mutant forms of myc tagged p300 (left panel) or CARM1 (right panel) as well as pphCMV-kozak-myc empty vector. p65, CARM1 and p300 were coimmunoprecipitated (IP) from nuclear extract of TNFα-treated 293T cells (30 min, 10 ng/ml) using an anti-myc antibody. Bound proteins were resolved by SDS–PAGE and subsequently detected by immunoblot (IB) analysis for p65, CARM1 and p300. Input lanes represent 10% of the input.
To rule out the possibility that the observed effects were due to an impaired binding of p65 to the enzymatic mutants of CARM1 or p300, we performed co-immunoprecipitation assays with nuclear extracts isolated from TNFα stimulated 293T cells expressing either myc-tagged wild-type or enzymatic mutant forms of both, CARM1 or p300, respectively. Both wild-type and enzymatic mutant forms of CARM1 and p300 formed a comparable complex with p65 in the nucleus (Figure 7D left and right panels).
Discussion
The aim of this study was to investigate the role of CARM1, a coactivator for p53- and nuclear receptor-dependent transcription, in NF-κB-dependent gene (Koh et al, 2001; An et al, 2004). We provide both biochemical and genetic evidence that CARM1 is an essential NF-κB coactivator and a promoter-specific regulator of NF-κB recruitment to chromatin.
Impaired NF-κB-dependent transcription in Carm1-deficient cells
An impaired expression of ICAM-1, G-CSF, MCP-1, IP-10 and MIP-2 was found in TNFα or LPS stimulated Carm1(−/−) fibroblasts. Similarly, NF-κB-dependent reporter gene activity upon LPS or TNFα stimulation was four- to five-fold lower in Carm1(−/−) cells than in wt cells. Interestingly, the expression of IκBα, IL-6, KC and COX-2 was not reduced in Carm1(−/−) fibroblasts upon stimulation, indicating that only a subset of NF-κB-dependent genes is dependent on CARM1. It is unlikely that the canonical pathway of NF-κB transduction cascade is affected by lack of CARM1 since the upstream signaling transducers tested are equally expressed and IκBα degradation and re-synthesis as well as nuclear entry of p65/RelA was similar in Carm1(+/+) and Carm1(−/−) cells. Moreover, the DNA-binding activity of NF-κB(p65/p50) assayed in vitro on naked non-chromatinized templates was not impaired in nuclear extracts from Carm1(−/−) fibroblasts, irrespective of the κB site tested.
CARM1 forms a complex with p65 in vivo and in vitro
We could detect endogenous CARM1 in a nuclear complex with p65 and p300, upon TNFα stimulation. Moreover, CARM1 directly binds to the Rel homology domain p65 in vitro. The interaction is mediated through the central region of CARM1 containing the methyltransferase domain. Since this region is very conserved among members of the PRMT family (Zhang et al, 2000), p65 might also interact with other members such as PRMT1. The interaction between full-length p65 and CARM1 was weaker in vitro, when compared to the interaction in vivo. It is possible that the interaction between p65 and CARM1 might be modulated or stabilized by post-translational modifications of p65. Whether this modification could be phosphorylation of the Rel homology domain of p65 by protein kinase A (PKA) has yet to be investigated. Phosphorylation of p65 by PKA was shown to open the p65 structure and increase the efficiency of interaction with p300/CBP (Zhong et al, 1998). Whether p65, CARM1 and p300 exist in a single complex or different p65/CARM1 and p300/CARM1 complexes form during the transcriptional activation process in vivo remains to be elucidated.
Cooperation between CARM1, p300/CBP and p160 family of coactivators in NF-κB-mediated transcription
Depending on the stimulus and the cell type, multiple interactions and the combined actions of distinct transcriptional coactivators seem to be required for the assembly of NF-κB transcription complexes and transcriptional activity of NF-κB. The current study shows that CARM1 synergistically coactivates NF-κB-mediated transactivation, in concert with p300/CBP and the p160 family members. CARM1 was previously shown to function as a secondary coactivator for nuclear receptors (NRs) cooperating with p160 coactivators and p300/CBP under conditions where limiting levels of the NRs are expressed (Lee et al, 2002). Similarly, high synergistic enhancement of NF-κB coactivation by CARM1 and protein acetyltransferases was obtained only under conditions where limiting levels of p65 were expressed (data not shown).
Although NRs and NF-κB utilize similar sets of coactivators, there are qualitative differences in the assembly process of each coactivator component by NRs and NF-κB. p160 family members are shown to act as primary coactivators for nuclear receptors and recruit the secondary coactivators CBP/p300 and CARM1 (Lee et al, 2002). In contrast, p300/CBP serves as primary coactivator for NF-κB and provides a platform for the secondary coactivators p/CAF and p160 family members (Sheppard et al, 1999). Moreover, CARM1 directly interacts with the NF-κB subunit p65 in vitro and might act as a primary coactivator for NF-κB.
CARM1 coactivates NF-κB in a promoter-specific manner
At the estrogen-responsive promoter pS2 and p53-dependent promoter GADD45, CARM1 recruitment was shown to follow ER or p53 recruitment, respectively. Thus, CARM1 seems to be recruited by ER or p53 to these promoters (Daujat et al, 2002; Metivier et al, 2003; An et al, 2004). Surprisingly, our ChIP data revealed a major defect in p65 recruitment to the CARM1-dependent MIP-2 and IP-10 promoters in Carm1(−/−) fibroblast upon TNFα stimulation. In contrast, no significant differences could be observed at the IL-6 promoter between Carm1(+/+) and Carm1(−/−) fibroblasts. Since NF-κB dimers from Carm1(−/−) cells retained full DNA-binding activity in EMSA, it can be assumed that one critical role of CARM1 in vivo is to promote NF-κB recruitment to selected target genes that, due to their chromatin configuration, are unable to recruit NF-κB ‘by default'. This does not rule out the possibility that CARM1 exerts additional essential activities required for NF-κB to activate transcription after it is recruited to chromatin. While H3(R17) methylation at the IL-6 promoter is not important for p65 recruitment, in the case of the IP-10 and MIP-2 promoters the modification of H3 seems to be required. This might be due to a different histone positioning on the IL-6 promoter or due to an ‘open' and acessible chromatin conformation maintained by other transcription factors. In contrast, at the IP-10 and MIP-2 promoters, Arg methylation and most likely subsequent modifications of H3 and other histones might enhance κB site accessibility. The specific determinants dictating CARM1 dependency of NF-κB-regulated genes remain to be defined.
The importance of CARM1 enzymatic activity for NF-κB-dependent transcription is supported by complementation experiments in CARM1(−/−) cells using an enzymatic mutant of CARM1 and p300. Full synergistic enhancement of transcription regulated from the MIP-2 and IP-10 promoters was obtained only when wild-type forms of both CARM1 and p300 were coexpressed in CARM1(−/−) cells. Thus, for at least a subset of CARM1-dependent NF-κB target genes, the enzymatic activities of both CARM1 and p300 are necessary for NF-κB-dependent transcription. However, transiently transfected plasmids are thought not to be properly chromatinized, suggesting that nucleosomal histones are unlikely to represent the only relevant substrates whose Arg methylation is required for NF-κB-dependent transcription (Smith and Hager, 1997; Nan et al, 2004). Studies regarding the requirement of CARM1-dependent methylation of nonhistone proteins such as p300/CBP (Xu et al, 2001; Chevillard-Briet et al, 2002) in NF-κB-dependent transcription are ongoing. Since the enzymatic mutant of CARM1 could still partially contribute to NF-κB-dependent transcription, similar to p300/CBP, CARM1 might also act downstream of NF-κB recruitment as a bridging factor. In addition to central methyltransferase/p65-binding domain, the unique N- and C-terminal regions of CARM1 were recently shown to be also required for enhancement of transcriptional activation by nuclear receptors (Teyssier et al, 2002). The C-terminal part of CARM1 contains a transactivation domain, which might interact with not yet identified cofactors, thereby driving the equilibrium toward formation of the fully competent NF-κB-dependent coactivator-complex(es).
Interestingly, CARM1 was recruited in a stimulus-dependent manner to all tested promoters, and recruitment correlated with an increase in H3(R17) methylation. Given our results, it seems plausible that CARM1 recruitment to the MIP-2 and IP-10 promoters may be independent of p65 and precede p65 recruitment. It remains to be investigated whether other sequence-specific transcription factors known to cooperate with NF-κB in gene induction, such as STAT1, IRF3 or AP1, are required for CARM1 recruitment to those promoters. The IL-6 promoter reflects a complete different situation, where the TNFα-induced recruitment of CARM1 coincides with p65 recruitment in both Carm1(+/+) and Carm1(−/−) fibroblasts. This could imply that p65 itself might recruit CARM1 to the IL-6 promoter.
It is of great interest to elucidate the relationship of CARM1 to other NF-κB-associated coactivators regarding their relative contribution to NF-κB-dependent gene activation, with respect to different promoters and stimuli. A combination of microarray and genomewide Chip studies using conditional single, double or triple knockout mice or conditional knockin mice expressing enzymatic mutants of particular coactivators could verify their relative contribution and identify the sequential order of particular promoter-specific recruitment cascades in vivo.
Materials and methods
Plasmids
Expression vectors for p300 and p65 were described in Hassa et al (2003). pphRSV-nt-βgal and the NF-κB-dependent luciferase reporter constructs HIV-luc and HIV-mutκB-luc are described in Hassa et al (2001). pGL3-MIP-2(−770), pGL3-IP-10(−533) and pOLUC-IL-6(−303/+23) are described in Han et al (1999), Borgland et al (2000) and Walpen et al (2001). The different CMV-myc expression vectors for CARM1 were obtained by cloning the corresponding PCR products (CARM1 full-length, CARM1—aa 1–148, CARM1—aa 140–480 and CARM1—aa 462–608) in frame into pphCMV-T7-km-3 (Hassa et al, 2003). The enzymatic mutant form (E267Q) of CARM1 was created by site-directed mutagenesis as described in Lee et al (2002) and verified by sequencing. The CMV-kozak-myc-p300 wild type and p300mutAT2 (enzymatic mutant) were obtained by cloning the corresponding PCR products into pphCMV-T7-km-3. pBKS-p300mutAT2 is described in Kraus et al (1999). GST-fusion vectors for p65—aa 1–305 and p65—aa 441–551 were obtained by cloning the corresponding PCR products into pGex6P1.
Cell culture and transient transfection
Carm1(+/+) or Carm1(–/–) MEFs, 293T and COS-1 were grown in DMEM (Invitrogen). Cells were transfected as described previously, except that MEFs were grown for 12 h in DMEM containing 2% fetal calf serum before stimulation with TNFα or LPS (purchased from Sigma). The amount of DNA indicated in the figure legends was calculated for 10 ml of medium. Total amounts of DNA and equal molar ratios of promoters were kept constant in all setups by using empty vectors. For MEFs, only cell passages 2–5 were used for experiments. Owing to differences in transfection efficiencies, an expression plasmid of β-galactosidase (pph-RSV-nt-β-gal) was cotransfected as a transfection efficiency control, and luciferase activities were normalized based on β-galactosidase activity. Luciferase activity was measured as described in Hottiger et al (1998).
RNA isolation and RT–PCR analysis
RNA isolation and RT–PCR from Carm1(+/+) or Carm1(–/–) MEFs was performed according to the manufacturer's protocols (Invitrogen). Sequences of primers are available upon request. All PCR products were resolved by 1–2% agarose gel electrophoresis, and DNA bands were visualized by staining the gel with ethidium bromide.
Nuclear extracts, immunoprecipitation and immunoblotting
Nuclear, cytoplasmic and whole-cell extracts were prepared as described in Hottiger et al (1998) and Hassa et al (2003). All immunoprecipitation and immunoblotting analysis for CARM1, PARP-1, p65, c-Rel, RelB and p300 were performed as described previously (Hottiger et al, 1998; Hassa et al, 2003). Anti-RelA/p65 (C-20, sc-372), anti-p300 (sc-C260), mouse IgG (sc-2025), rabbit IgG (sc-2027), anti-c-Myc IgG (sc-2027) and anti-α-tubulin (sc-8035) antibodies were obtained from Santa Cruz Biotechnology; anti-PARP-1 antibody (clone C-2–10) was from Anawa trading SA (Switzerland) and anti-p300 antibody (14991A) was from BD PharMingen. The anti-CARM1 antibodies were either described in Kim et al (2004) or a generous gift from Dr S Richard (McGill University, Quebec, Canada) and Dr M Stallcup (University of Southern California, USA). The anti-dimethyl-H3-Arg17 antibody (07-214) was from Upstate. The anti-RelB and anti-c-Rel antibodies were kindly provided by Dr NR Rice (National Cancer Institute at Frederick, USA).
In vitro interaction and GST pull-down assay
Recombinant His-mCARM1 and GST-p65 full-length proteins were expressed in Sf21 cells. GST-p65—aa 1–305, -p65—aa 1–441 and -p65—aa 441–551 proteins were expressed in Escherichia coli and purified according to the manufacturer's protocols (Novagen, Amersham/Pharmacia Biosciences). All purified proteins were confirmed by Coomassie staining and Western blot analysis using the corresponding antibodies. Coupled in vitro transcription/translation reactions were carried out using the TNT T7-Quick system (Promega) according to the manufacturer's protocol. GST pull-down assays were performed in the presence of 120 mM NaCl as described previously (Hassa et al, 2003).
ChIP assay
ChIP was carried out as described previously (Saccani and Natoli, 2002). Sequences of promoter-specific primers and a detailed experimental protocol are available upon request.
Acknowledgments
We are grateful to WL Kraus (Cornell University, New York, USA), CK Glass (University of California at San Diego, USA), NR Rice (National Cancer Institute at Frederick, USA), J Pfeilschifter (University of Munster, Germany), AR Brasier (University of Texas Medical Branch, USA), S Richard (McGill University, Quebec, Canada), M Stallcup (University of Southern California, USA) and DA Muruve (University of Calgary, Canada) for the generous provision of reagents. We also thank A Hoffman (University of California at San Diego, USA) for his helpful advice and comments. This work was supported in part by the NIH grant DK62248 and, in part, by institutional grant ES07784 (to MTB), by Swiss National Science Foundation Grant 31-66720.01 (to GN), by Swiss National Science Foundation Grant 31-67771.02 and the Kanton of Zurich (to POH and MOH).
References
- Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D (2000) Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell 103: 667–678 [DOI] [PubMed] [Google Scholar]
- An W, Kim J, Roeder RG (2004) Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell 117: 735–748 [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Baltimore D (1988) I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science 242: 540–546 [DOI] [PubMed] [Google Scholar]
- Baldwin AS Jr (1996) The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol 14: 649–683 [DOI] [PubMed] [Google Scholar]
- Bauer UM, Daujat S, Nielsen SJ, Nightingale K, Kouzarides T (2002) Methylation at arginine 17 of histone H3 is linked to gene activation. EMBO Rep 3: 39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Bowen GP, Wong NC, Libermann TA, Muruve DA (2000) Adenovirus vector-induced expression of the C-X-C chemokine IP-10 is mediated through capsid-dependent activation of NF-kappaB. J Virol 74: 3941–3947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevillard-Briet M, Trouche D, Vandel L (2002) Control of CBP co-activating activity by arginine methylation. EMBO J 21: 5457–5466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daujat S, Bauer UM, Shah V, Turner B, Berger S, Kouzarides T (2002) Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr Biol 12: 2090–2097 [DOI] [PubMed] [Google Scholar]
- Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T (1997) CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci USA 94: 2927–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB (1998) NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 16: 225–260 [DOI] [PubMed] [Google Scholar]
- Goodman RH, Smolik S (2000) CBP/p300 in cell growth, transformation, and development. Genes Dev 14: 1553–1577 [PubMed] [Google Scholar]
- Han Y, Runge MS, Brasier AR (1999) Angiotensin II induces interleukin-6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear factor-kappa B transcription factors. Circ Res 84: 695–703 [DOI] [PubMed] [Google Scholar]
- Hassa PO, Buerki C, Lombardi C, Imhof R, Hottiger MO (2003) Transcriptional coactivation of nuclear factor-kappaB-dependent gene expression by p300 is regulated by poly(ADP)-ribose polymerase-1. J Biol Chem 278: 45145–45153 [DOI] [PubMed] [Google Scholar]
- Hassa PO, Covic M, Hasan S, Imhof R, Hottiger MO (2001) The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J Biol Chem 276: 45588–45597 [DOI] [PubMed] [Google Scholar]
- Hottiger MO, Felzien LK, Nabel GJ (1998) Modulation of cytokine-induced HIV gene expression by competitive binding of transcription factors to the coactivator p300. EMBO J 17: 3124–3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M (1998) The NF-kappa B activation pathway: its regulation and role in inflammation and cell survival. Cancer J Sci Am 4 (Suppl 1): S92–S99 [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y (2000) Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol 18: 621–663 [DOI] [PubMed] [Google Scholar]
- Kim J, Lee J, Yadav N, Wu Q, Carter C, Richard S, Richie E, Bedford MT (2004) Loss of CARM1 results in hypomethylation of thymocyte cyclic AMP-regulated phosphoprotein and deregulated early T cell development. J Biol Chem 279: 25339–25344 [DOI] [PubMed] [Google Scholar]
- Koh SS, Chen D, Lee YH, Stallcup MR (2001) Synergistic enhancement of nuclear receptor function by p160 coactivators and two coactivators with protein methyltransferase activities. J Biol Chem 276: 1089–1098 [DOI] [PubMed] [Google Scholar]
- Koh SS, Li H, Lee YH, Widelitz RB, Chuong CM, Stallcup MR (2002) Synergistic coactivator function by coactivator-associated arginine methyltransferase (CARM) 1 and beta-catenin with two different classes of DNA-binding transcriptional activators. J Biol Chem 277: 26031–26035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus WL, Manning ET, Kadonaga JT (1999) Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Mol Cell Biol 19: 8123–8135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu TK, Palhan VB, Wang Z, An W, Cole PA, Roeder RG (2000) Activator-dependent transcription from chromatin in vitro involving targeted histone acetylation by p300. Mol Cell 6: 551–561 [DOI] [PubMed] [Google Scholar]
- Lee YH, Koh SS, Zhang X, Cheng X, Stallcup MR (2002) Synergy among nuclear receptor coactivators: selective requirement for protein methyltransferase and acetyltransferase activities. Mol Cell Biol 22: 3621–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride AE, Silver PA (2001) State of the arg: protein methylation at arginine comes of age. Cell 106: 5–8 [DOI] [PubMed] [Google Scholar]
- Merika M, Thanos D (2001) Enhanceosomes. Curr Opin Genet Dev 11: 205–208 [DOI] [PubMed] [Google Scholar]
- Merika M, Williams AJ, Chen G, Collins T, Thanos D (1998) Recruitment of CBP/p300 by the IFN beta enhanceosome is required for synergistic activation of transcription. Mol Cell 1: 277–287 [DOI] [PubMed] [Google Scholar]
- Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F (2003) Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115: 751–763 [DOI] [PubMed] [Google Scholar]
- Miranda TB, Miranda M, Frankel A, Clarke S (2004) PRMT7 is a member of the protein arginine methyltransferase family with a distinct substrate specificity. J Biol Chem 279: 22902–22907 [DOI] [PubMed] [Google Scholar]
- Nan X, Hyndman L, Agbi N, Porteous DJ, Boyd AC (2004) Potent stimulation of gene expression by histone deacetylase inhibitors on transiently transfected DNA. Biochem Biophys Res Commun 324: 348–354 [DOI] [PubMed] [Google Scholar]
- Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ (1997) Regulation of NF-kappaB by cyclin-dependent kinases associated with the p300 coactivator. Science 275: 523–527 [DOI] [PubMed] [Google Scholar]
- Saccani S, Natoli G (2002) Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev 16: 2219–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiltz RL, Mizzen CA, Vassilev A, Cook RG, Allis CD, Nakatani Y (1999) Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. J Biol Chem 274: 1189–1192 [DOI] [PubMed] [Google Scholar]
- Sheppard KA, Rose DW, Haque ZK, Kurokawa R, McInerney E, Westin S, Thanos D, Rosenfeld MG, Glass CK, Collins T (1999) Transcriptional activation by NF-kappaB requires multiple coactivators. Mol Cell Biol 19: 6367–6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CL, Hager GL (1997) Transcriptional regulation of mammalian genes in vivo. A tale of two templates. J Biol Chem 272: 27493–27496 [DOI] [PubMed] [Google Scholar]
- Teyssier C, Chen D, Stallcup MR (2002) Requirement for multiple domains of the protein arginine methyltransferase CARM1 in its transcriptional coactivator function. J Biol Chem 277: 46066–46072 [DOI] [PubMed] [Google Scholar]
- Walpen S, Beck KF, Schaefer L, Raslik I, Eberhardt W, Schaefer RM, Pfeilschifter J (2001) Nitric oxide induces MIP-2 transcription in rat renal mesangial cells and in a rat model of glomerulonephritis. Faseb J 15: 571–573 [DOI] [PubMed] [Google Scholar]
- Whiteside ST, Israel A (1997) I kappa B proteins: structure, function and regulation. Semin Cancer Biol 8: 75–82 [DOI] [PubMed] [Google Scholar]
- Xu W, Chen H, Du K, Asahara H, Tini M, Emerson BM, Montminy M, Evans RM (2001) A transcriptional switch mediated by cofactor methylation. Science 294: 2507–2511 [DOI] [PubMed] [Google Scholar]
- Yadav N, Lee J, Kim J, Shen J, Hu MC, Aldaz CM, Bedford MT (2003) Specific protein methylation defects and gene expression perturbations in coactivator-associated arginine methyltransferase 1-deficient mice. Proc Natl Acad Sci USA 100: 6464–6468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhou L, Cheng X (2000) Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J 19: 3509–3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Voll RE, Ghosh S (1998) Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell 1: 661–671 [DOI] [PubMed] [Google Scholar]