

Abstract
Chronic sun exposure can lead to severe skin disorders such as carcinogenesis. The cell death process triggered by ultraviolet B (UVB) irradiation is crucial because it protects the surrounding tissue from the emergence and the accumulation of cells that bear the risk of becoming transformed. Here, we show that repression of NF-κB and Egr-1 expression drastically inhibits UVB-mediated cell death. Furthermore, we demonstrate that Egr-1 is induced upon UVB irradiation through NF-κB activation and the binding of p65/RelA within the Egr-1 promoter. We show that Egr-1 contributes to the regulation of the Gadd45a and Gadd45b genes, which are involved in the control of cell cycle, DNA repair and apoptosis, by direct binding to their promoter. Our study demonstrates for the first time a signaling cascade involving sequential activation of NF-κB, Egr-1 and Gadd45 to induce UVB-mediated cell death. Failure in the induction of each protagonist of this pathway alters the UVB-mediated cell death process. Therefore, impairment of the cascade could be at the onset of skin carcinogenesis mediated by genotoxic stress.
Keywords: apoptosis, Egr-1, Gadd45, NF-κB, UVB
Introduction
Ultraviolet (UV) light is a major environmental damaging agent that can cause several disorders such as suppression of immune functions, premature skin aging, keratosis and cutaneous malignancy. Among the two types of UV (UVA, UVB) that reach the surface of the earth, UVB radiation (280–320 nm) consists of the most energetic and DNA-damaging rays (Cleaver and Crowley, 2002) and accounts for a large part of gene mutations due to chronic solar exposure. Skin represents the first line of defense against UV injury, and is the site of a large variety of malignant neoplasms such as melanomas and also basal cell (BCC) or squamous cell carcinoma (SCC), both derived from keratinocytes. Although BCC and SCC are not as critical for human health as melanomas, they are the most frequent cutaneous malignancies (Cleaver and Crowley, 2002). Incidence of UV light on skin carcinogenesis is now well documented. Indeed, chronic sun exposure promotes the precancerous stage of actinic keratosis, leading then to the induction and development of skin cancers (Soehnge et al, 1997; Armstrong and Kricker, 2001). This event has been modeled as a multistage process in which UV triggers somatic mutations followed by clonal expansion of damaged cells, progression into a precancerous state and likely to uncontrolled proliferation (Brash, 1997). However, the cells are able to repair the damages that occur following such stress by activating gene clusters involved in the control of DNA repair, cell cycle checkpoints and proliferation (Li et al, 2001; Dazard et al, 2003). The cells that are severely UV damaged and unable to repair are eliminated toward apoptosis. In that particular context, this process appears to be a protective mechanism preventing the emergence and the accumulation of cells that bear the risk of becoming transformed (Kulms and Schwarz, 2002a). Therefore, failure of such a mechanism is likely to contribute to the progression into the cancerous state.
The tumor suppressor gene Egr-1 (Sukhatme et al, 1988) was shown to be upregulated at a transcriptional level in several cell types such as fibroblasts, mammary epithelia and glioblastoma, following UV irradiations (Huang et al, 1999). Egr-1 belongs to a group of early response genes transiently induced by several environmental signals such as growth factors, hormones, UV, stretch and hypoxia. Many important biological functions like neurite outgrowth (Harada et al, 2001), wound repair (Khachigian et al, 1996), growth control (Perez-Castillo et al, 1993; Hofer et al, 1996) and apoptosis (Muthukkumar et al, 1995; Ahmed et al, 1996; Virolle et al, 2001) are under control of Egr-1. In addition, the functional importance of this transcription factor in cancer has been emphasized by its description as a tumor suppressor gene in mammary epithelia and as an important progression factor in prostate cancer cells (Abdulkadir et al, 2001; Baron et al, 2003; Virolle et al, 2003). Egr-1 has been implicated in tumor progression in mouse epidermis (Riggs et al, 2000). However, further investigations are required to determine the precise role of Egr-1 in epidermal cells undergoing genotoxic stress.
Although several gene profiling studies have been performed in UV-irradiated keratinocytes, only little information is available to characterize gene activation cascades and their functional capabilities during the epidermal response. In the present study, our aim is to understand the molecular basis of Egr-1 induction and its function related to UVB-mediated genotoxic stress in epidermal cells. We show that NF-κB activation is necessary for UVB-mediated transcriptional Egr-1 induction through the binding of p65/RelA within the Egr-1 promoter. Moreover, blockade of Egr-1 and NF-κB expression drastically reduces UVB-mediated cell death. Furthermore, we show that Egr-1 contributes to the regulation of expression of the Gadd45 gene family by binding within their regulatory sequences. Induction of Gadd45 genes following genotoxic stress is very important because many reports argue in favor of their contribution to promoting apoptosis and thus preventing skin carcinogenesis (Hildesheim et al, 2002; Yoo et al, 2003).
Results
UVB stress mediates Egr-1 induction in human skin
To determine whether the transcription factor Egr-1 is upregulated in human skin following UVB stress, we performed immunostaining analysis on human foreskin sections 2 h after irradiation with a single dose of 60 mJ/cm2 UVB. The experiment revealed a strong fluorescent signal corresponding to Egr-1 protein expression distributed to each epidermal layer, while it remained undetectable in control epidermis (Figure 1A). In order to demonstrate the transcriptional induction of Egr-1, expression of Egr-1 transcripts was analyzed by quantitative real-time RT–PCR from three epidermis tissue samples nonirradiated or separately irradiated at 60 or 100 mJ/cm2. A five-fold and up to nine-fold induction was detected respectively at 60 and 100 mJ/cm2 as compared to the nonirradiated control condition (Figure 1B). These results showed that, in human skin, Egr-1 protein is kept at a very low expression level but becomes strongly expressed following UVB stress. The time course of expression of Egr-1 protein was assessed after UVB irradiation using an immunoblotting procedure. Nuclear proteins were extracted from human keratinocytes every 30 min, from 1 to 5 h after UVB irradiation. As seen in Figure 1C, significant Egr-1 expression peaked at 1.5 h after irradiation and remained stable at least for 5 h. To determine if UVB-mediated Egr-1 induction was related to an increase in gene transcription, we tested expression of Egr-1 mRNA by real-time RT–PCR analysis after a single pulse irradiation (60 mJ/cm2) according to a time course from 10 min to 3 h (Figure 1D). The results showed a three-fold stimulation in Egr-1 mRNA production 1 and 2 h after irradiation and a slight decrease thereafter. To confirm this transcriptional activation, we have isolated and cloned 1.4 kb of genomic DNA located within Egr-1 5′ regulatory sequences (pEgr-1-luc) into a luciferase reporter gene. This genomic DNA corresponds to the core promoter, plus the 5′ noncoding region of the Egr-1 mRNA. This construct was then transfected into human keratinocytes that were irradiated or not with increasing doses of UVB (40–200 mJ/cm2) (Figure 1E). Under these conditions, Egr-1 promoter activity was stimulated four-fold whatever the magnitude of UVB doses tested. These results demonstrate that UVB stress-mediated Egr-1 induction occurred through increased transcription events.
Figure 1.

UVB treatment induces Egr-1 expression. (A) Three tissue sections of human foreskin separately irradiated (UVB 60 mJ/cm2) or not (Ctl) were subjected to immunohistochemical staining with an anti-Egr-1 antibody as described in Materials and methods (e: epidermis; d: dermis). (B) Total RNAs from control (Ctl) or irradiated epidermis tissue were analyzed for Egr-1 induction by quantitative real-time RT–PCR as described in Materials and methods. (C) UVB irradiation induces Egr-1 protein expression. Nuclear extracts were prepared from HaCaT cells according to a time course following UVB irradiation (60 mJ/cm2). Western blot analysis was then performed with antibodies to Egr-1 and ERK as loading control. (D) UVB irradiation induces Egr-1 mRNA expression. Total RNAs were prepared from HaCaT cells according to a time course following UVB irradiation (60 mJ/cm2). Egr-1 mRNA expression was then assessed by one-step real-time RT–PCR as described in Materials and methods. The fold inductions correspond to the ratio between control untreated and irradiated cells. (E) UVB irradiation induces Egr-1 promoter activity. Egr-1 promoter was cloned in a luciferase reporter construct and transfected into immortalized human keratinocytes (KHSV) irradiated or not by increasing dose of UVB.
Functional analysis of UVB-mediated Egr-1 promoter induction
To determine the pathway involved in UVB-mediated Egr-1 expression, 334 and 570 bp were deleted from the 5′ end of pEgr-1-luc to create F4 and F3 promoter fragments, respectively. These constructs were then transiently transfected into human keratinocytes irradiated or not 24 h later by a single UVB dose of 60 mJ/cm2. The results showed a significant increase in the transcription activity of pEgr-1-luc and F4 constructs, while F3 remained unresponsive (Figure 2). This result delineates a critical sequence of 200 bp located at position −448/−248 from the transcription initiation start point. Further serial deletions within this 200 bp, located precisely at −447/−397, −447/−347, −447/−297 and −447/−247, were made to create respectively Δ1 (Figure 2), Δ2, Δ3 and Δ4 constructs (not shown). As seen in Figure 2, the deletion of the first 50 bp (Δ1) was sufficient to decrease the promoter induction drastically. This 50 bp nucleotide sequence was divided into two regions of 23 and 27 bp that were alternately deleted to create the Δ5 and Δ6 constructs, respectively (Figure 2). The Δ6 construct (deletion of 27 bp) abolished the Egr-1 promoter response to UVB stimulation (Figure 2). Computational sequence analysis revealed a canonical NF-κB binding motif (CGGAAATGCC) within this 27-nucleotide-long sequence. The mutation of this putative Kappa B binding site (mutNF-κB construct) into an SpeI restriction site was sufficient to suppress the promoter induction (Figure 2). These results demonstrate that the KB binding site located at −425/−417 is necessary to promote UVB-mediated Egr-1 transcriptional induction.
Figure 2.
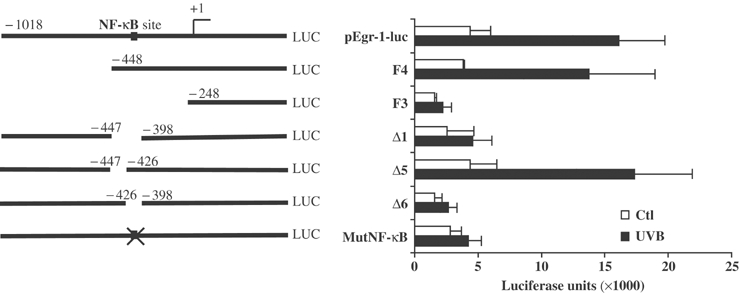
A KB binding site is required for the UVB-mediated Egr-1 promoter induction. Full-length, deleted and mutated Egr-1 regulatory sequences are represented on the left. The numbers indicate the position of the deleted fragments relative to the transcription start site (+1, arrow). The cross represents the mutation of the NF-κB binding site. (Right) pEgr-1-luc and its deleted/mutated fragments were transiently transfected into KHSV stimulated (black) or not (white) with UVB irradiation. The values are expressed as luciferase units.
The transcription factor NF-κB binds and regulates Egr-1 promoter during UVB stress
In order to test NF-κB activation in keratinocytes, a luciferase reporter construct containing six canonical KB binding sites was transfected into epidermal cells. The results showed a significant increase in the luciferase activity (about five-fold) upon UVB treatment (Figure 3A). In order to assess the time course of NF-κB activation, we performed an electrophoretic mobility shift assay (EMSA) experiment at 40 min and 1 h following UV treatment using nuclear extracts from irradiated normal human keratinocytes (NHKs) and a radiolabeled probe corresponding to the NF-κB site located on the Egr-1 promoter. Our experiment detected a significant binding of NF-κB mostly composed of RelA NF-κB family member as soon as 40 min after treatment (Figure 3B). This suggests that NF-κB is activated by UVB stress at early stage and is correlated to the induction of Egr-1 transcripts that occurs within 1 h after irradiation. To demonstrate the direct binding of NF-κB onto the Egr-1 promoter in the chromatin context of epidermal cells, we used in vivo cells crosslinking and chromatin immunoprecipitation experiment (ChIP). UVB-irradiated HaCaT cells were crosslinked and NF-κB targets were recovered by immunoprecipitation with a specific antibody raised against p65/RelA. The detection of Egr-1 promoter in the captured fragments of genomic DNA was performed by PCR amplification with primers designed to specifically recognize Egr-1 promoter regions flanking the KB binding site (Figure 3C). As expected, efficient promoter amplification could be observed using control DNA genomic input (lanes 1) or nonimmunoprecipitated chromatin fragment (lanes 2 and 5). The Egr-1 promoter was detected only in the UVB-irradiated samples (lanes 3), while it remained undetectable in nonimmunoprecipitation (lanes 4 and 7) and nonirradiated control conditions (lanes 6). These results demonstrate the direct binding of NF-κB onto the Egr-1 promoter after UVB irradiation in the chromatin context of living cells. To determine NF-κB involvement in the UVB-mediated Egr-1 induction, Egr-1 mRNA expression was tested by real-time RT–PCR at 1 h after stress in wild-type (WT) and RelA−/− mouse embryonic fibroblasts (MEFs) (Figure 3D). The expected Egr-1 induction observed in WT MEFs was significantly reduced in RelA−/− MEFs. Therefore, the lack of RelA gene impairs Egr-1 induction during the cellular response to UVB irradiations. The sum of our experiments clearly demonstrates that the UVB-induced Egr-1 expression is directly mediated by the activation and the binding of NF-κB onto Egr-1 promoter.
Figure 3.
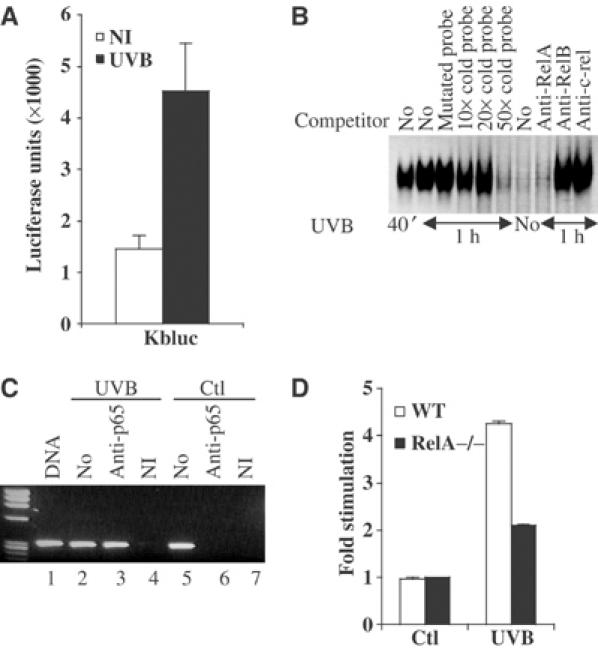
NF-κB binds and regulates Egr-1 promoter expression. (A) Six canonical NF-κB binding sites were cloned within a luciferase reporter construct (Kbluc). This construct was then transiently transfected into KHSV cells UVB irradiated (black) or not (white). (B) At 40 min and 1 h following irradiation, NHKs were harvested and nuclear lysate was prepared for EMSA. When indicated, the binding reaction was preincubated with NF-κB mutated cold probe and increasing amount of homologous cold probe, or RelA-, RelB- and cRel-specific antibodies. (C) ChIP experiment. HaCaT cells were UVB irradiated (lanes 2–4) or not (lanes 5–7). Chromatin extracts were immunoprecipitated with specific antibodies to p65(RelA) (lanes 3 and 6) or a nonimmune control serum (lanes 4 and 7). Lanes 1 correspond to the control genomic DNA input. Lanes 2 and 5 correspond to the control nonimmunoprecipitated chromatin. The detection of captured Egr-1 promoter was performed by PCR as described in Materials and methods. (D) Blockade of RelA expression alters Egr-1 induction. Total mRNAs were extracted from RelA−/− (black) and WT (white) MEFs UVB (60 mJ/cm2) irradiated or not. Egr-1 mRNA expression was then assessed by real-time one-step RT–PCR as described in Materials and methods. The fold inductions correspond to the ratio between untreated control and irradiated cells.
Inhibition of Egr-1 expression promotes epidermal cell survival
To assess the role of the Egr-1 protein in the control of cell survival following UVB-mediated genotoxic stress, we rendered keratinocytes deficient for Egr-1 expression using an siRNA approach. To design the siRNA, we used an oligonucleotide sequence that we previously described to be very efficient and highly specific to Egr-1 (Baron et al, 2003; Virolle et al, 2003). This sequence has been cloned into an adenovirus to create pAD-siRNA-Egr-1 construct so as to lead to a small hairpin RNA (shRNA) structure under the control of the H1 promoter (Brummelkamp et al, 2002). In parallel, the adenovirus control construct pAD-siRNA-Ctl, which corresponds to an irrelevant siRNA, has been constructed. To test the efficiency of this strategy, UVB-irradiated HaCaT cells were infected with decreasing amounts of pAD-siRNA-Egr-1. As seen in Figure 4A, UV and epidermal growth factor (EGF) stimulation increased Egr-1 expression. Infection with pAD-siRNA-Egr-1 drastically inhibited Egr-1 expression in a dose-dependent fashion, but infection with pAD-siRNA-Ctl had no effect. No change in ERK expression could be observed throughout this experiment.
Figure 4.

Egr-1 and NF-κB activations are required for UVB-mediated cell death. (A) Inhibition of Egr-1 expression by siRNA targeted to Egr-1. HaCaT cells were infected with pAD-siRNA-Egr-1 and pAD-siRNA-Ctl adenovirus. After 24 h, the cells were UVB irradiated and lysed. The samples were analyzed by Western blotting with antibodies to Egr-1. The membrane was reprobed with antibodies to ERK as internal control. (B) HaCaT and NHKs were infected with pAD-siRNA-Egr-1 and pAD-siRNA-Ctl adenovirus. The cells were then UVB irradiated (60 mJ/cm2) and the remaining cells were observed in the dishes 24 h later. The percentage of dead cells was assessed by MTT staining (C) and induction of the cleavage of caspase 3/7 is represented in (D). Egr-1−/− (black) and RelA−/− (gray) MEFs are less sensitive to death than WT MEFs (white). Cells were UVB irradiated (60 mJ/cm2) and cell death and apoptosis were assessed 24 h later by MTT staining (E) and caspase assay (F).
Next we studied whether significant inhibition of Egr-1 expression could affect keratinocytes survival during UVB irradiation. NHKs and HaCaT were irradiated with a single dose of 60 mJ/cm2 UVB. Interestingly, siRNA inhibition of Egr-1 expression rendered the keratinocytes more resistant to UVB-mediated cell death (Figure 4B). Cell death was estimated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) staining as described in Materials and methods (Figure 4C). After a single dose of UVB irradiation, nearly 60% of keratinocytes (NHKs and HaCaT) were sensitive to cell death. Inhibition of Egr-1 expression with pAD-siRNA-Egr-1 drastically decreased the amount of dead cells to 20% (Figure 4C) with a significant inhibition of caspase 3/7 activation (Figure 4D) while pAD-siRNA-Ctl did not alter the process. Furthermore, siRNA expression had no effect on cell viability in nonirradiated keratinocytes. To confirm the requirement of Egr-1 expression in UVB-mediated death, we used WT, Egr-1−/− and RelA−/− MEFs in the same experimental settings. While an average of 40% of WT MEFs died upon UVB treatment, only 15% of Egr-1−/− cells were induced to death (Figure 4E). Interestingly, RelA−/− cells were also more resistant, displaying an average of 15% of dead cells. Activation of caspase 3/7 was also significantly reduced in Egr-1−/− and RelA−/− cells (Figure 4F). These results therefore demonstrate that cells lacking either Egr-1 or RelA expression are more resistant to cell death triggered by UV-mediated genotoxic stress.
Egr-1 contributes to Gadd45a and Gadd45b expression during UVB stress
Gadd45a and Gadd45b genes are rapidly induced upon DNA damage, and the corresponding proteins are involved in DNA repair, cell cycle checkpoint and apoptosis. The transcriptional induction of Gadd45a and Gadd45b genes, measured by real-time PCR, occurred at 3 h after irradiation (Figure 5A). This was in perfect correlation with Egr-1 protein induction (Figure 1B). Moreover, mRNA expression level of Gadd45a and Gadd45b upon UVB irradiation was strongly reduced in Egr-1−/− and RelA−/− MEFs compared to WT MEFs (Figure 5B). In order to assess whether or not Egr-1 was capable of contributing to the regulation of Gadd45a and Gadd45b genes following UVB irradiation, we used HaCaT cells rendered deficient for Egr-1 expression after infection with pAD-siRNA-Egr-1 virus. Total RNAs were extracted 3 h after UVB treatment and were tested for Gadd45a and Gadd45b expression by real-time RT–PCR. The results showed that inhibition of Egr-1 expression altered significantly both basal (Figure 6A) and stimulated Gadd45a expression (Figure 6B). Cumulatively, these defaults led to a severe impairment in Gadd45a expression following UVB stress as compared to Egr-1-expressing cells (Figure 6C). Inhibition of Egr-1 expression also altered the UVB-mediated Gadd45b stimulation (Figure 6D). However, no significant changes were observed in the basal expression (Figure 6E). This event remained unaffected when the cells were infected with siRNA-Ctl virus. Our results suggest that Egr-1 is required to allow total Gadd45 gene expression upon UVB exposure. To gain further insights into the mechanism of Gadd45a and Gadd45b gene stimulation, their promoters were cloned into luciferase reporter vectors. These constructs were then transiently cotransfected into keratinocytes with an Egr-1 eukaryotic expression vector. The results showed that exogenous Egr-1 expression was sufficient to stimulate the Gadd45a and Gadd45b promoters (Figure 7A). In order to demonstrate the binding of Egr-1 protein onto these promoters in the context of living keratinocytes, we performed a ChIP experiment on HaCaT cells irradiated or not by a single dose of UVB (Figure 7B). The detection of the human Gadd45a and Gadd45b promoters was performed by PCR analysis with specific primer pairs located in the core promoter and the 5′ noncoding region of each gene. Efficiency of the primers was tested on genomic DNA input (lanes 1 and 6). The results showed a significant amplification of Gadd45b and Gadd45a promoters in chromatin, from irradiated conditions, immunoprecipitated with anti-Egr-1 (lanes 5 and 10). No amplification occurred either in control condition (lanes 4 and 9) or in nonimmune serum immunoprecipitations (lanes 2, 3, 7 and 8). In order to confirm the binding of Egr-1 on the Gadd45a promoter, we performed an EMSA using as probe the genomic DNA fragment amplified in the ChIP experiment. The results showed a DNA/protein complex efficiently competed by an antibody specific to Egr-1 or a canonical Egr-1 cold probe but not by a nonimmune serum or a mutated Egr-1 cold probe (Figure 7C). Our results clearly demonstrate that Egr-1 regulates Gadd45a and Gadd45b promoter activities by direct binding to their endogenous regulatory sequences contributing thus to the stimulation of the expression of these genes. In order to assess the contribution of Gadd45a and Gadd45b expression in the apoptosis process, we have transiently transfected these two genes in keratinocytes. At 30 h after transfection, dead cells and activation of caspase 3/7 were measured (Figure 7D). The experiment showed that expression of Gadd45a induced a nine-fold activation of caspase 3/7 and resulted in 45% of dead cells. The effect of Gadd45b expression, although reduced as compared to Gadd45a, displayed a 2.5-fold activation of caspases and 11% of dead cells. Our results indicate that expression of Gadd45 genes contributes to the apoptosis process in keratinocytes.
Figure 5.
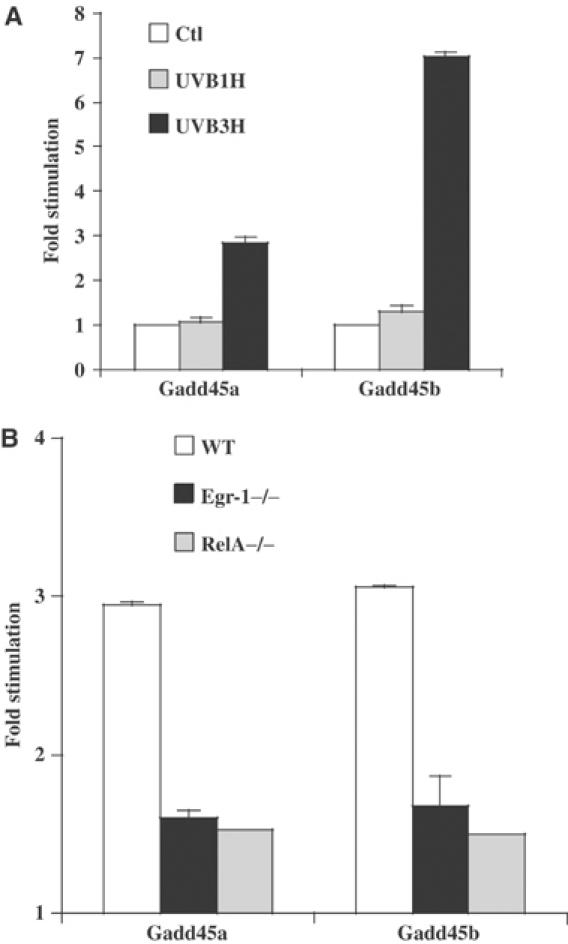
Gadd45a and Gadd45b mRNAs are expressed upon UVB irradiation. (A) HaCaT cells were UVB stimulated (60 mJ/cm2) and total RNAs were extracted at 1 and 3 h after irradiation. The results are expressed as the ratio between unstimulated and stimulated cells. Gadd45 mRNA expression was assessed by real-time one-step RT–PCR as described in Materials and methods. (B) Egr-1−/− (black), RelA−/− (gray) and WT (white) MEFs are irradiated or not by a single dose of 60 mJ/cm2 UVB. After 3 h, total RNAs are extracted and Gadd45a and Gadd45b mRNA expression level is assessed by real-time RT–PCR and compared to the basal expression in the WT MEFs. The results are the mean of the ratio between each MEF cell line irradiated by a single dose of UVB (60 mJ/cm2) and the nonirradiated WT MEFs considered as calibrator for the basal Gadd45 gene expression.
Figure 6.

Egr-1 and NF-κB contribute to the regulation of Gadd45a and Gadd45b genes. HaCaT cells irradiated or not with a single dose of UVB (60 mJ/cm2) were infected with pAD-siRNA-Egr-1 and pAD-siRNA-Ctl adenovirus as described in Materials and methods. Total RNAs were extracted and Gadd45 expression was assessed by real-time one-step RT–PCR. (A) Gadd45a basal expression in the presence or absence of Egr-1. (B) UVB-mediated induction of Gadd45a in the presence or absence of Egr-1. The fold induction represents the ratio between the UVB-stimulated condition and the nonirradiated condition. (C) Relative expression of Gadd45a. The fold stimulation corresponds to the UVB-irradiated conditions versus the nonirradiated, Egr-1-expressing condition. (D) UVB-mediated induction of Gadd45b in the presence or absence of Egr-1. The fold induction represents the ratio between the UVB-stimulated condition and the nonirradiated condition. (E) Gadd45b basal expression in the presence or absence of Egr-1.
Figure 7.
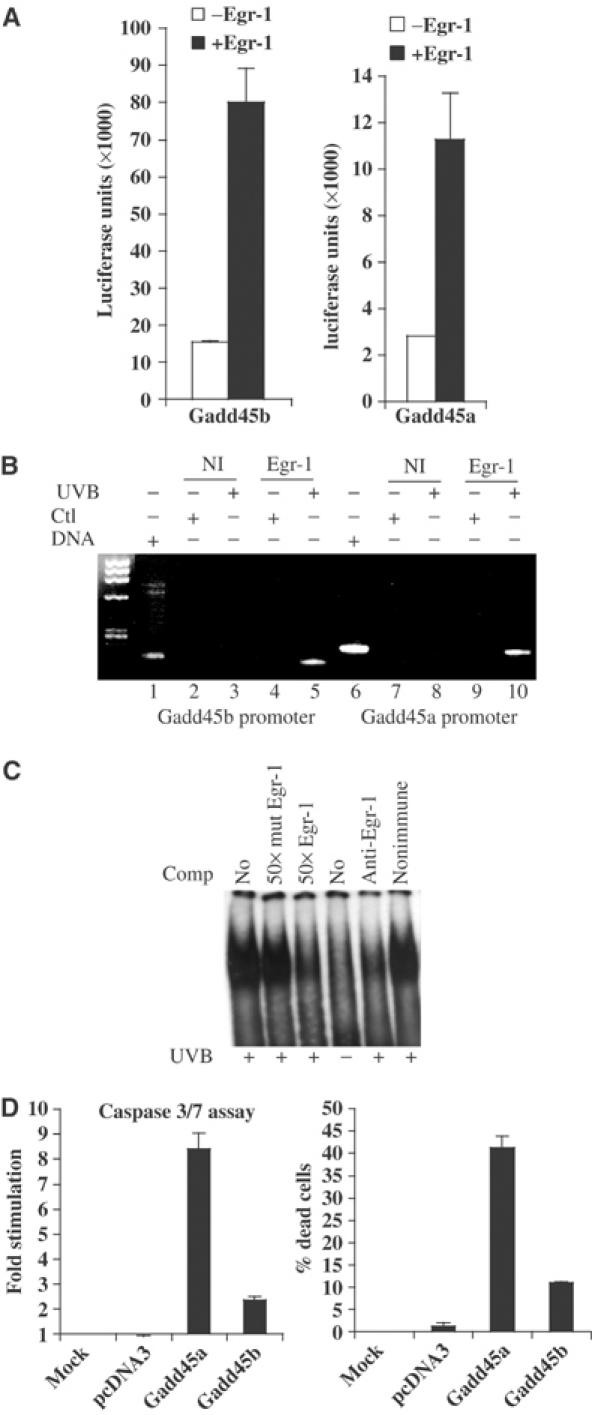
Egr-1 binds and regulates Gadd45a and Gadd45b promoters. (A) Gadd45b and Gadd45a promoters were isolated and cloned into a luciferase reporter gene as described in Materials and methods. These constructs were then cotransfected with an Egr-1-expressing vector in KHSV cells. The values are expressed as luciferase units. (B) ChIP experiment on UVB-irradiated HaCaT cells (lanes 3, 5, 8 and 10) or nonirradiated cells (lanes 2, 4, 7 and 9). Chromatin extracts were immunoprecipitated with specific antibodies to Egr-1 (lanes 4, 5, 9 and 10) or a nonimmune control serum (lanes 2, 3, 7 and 8). Lanes 1 and 6 correspond to the control genomic DNA input. The detection of Gadd45b (lanes 1–5) and Gadd45a (lanes 6–10) promoters was performed by PCR with the appropriate pair of primers as described in Materials and methods. (C) At 2.5 h following irradiation, NHKs were harvested and nuclear lysate was prepared for EMSA. When indicated, the binding reaction was preincubated with canonical Egr-1 cold probe, Egr-1 mutated cold probe, antibody specific to Egr-1 and nonimmune serum. (D) Empty vector (pcDNA3), Gadd45a and Gadd45b were expressed in KHSV by transient transfection. After 30 h, caspase 3/7 activation (left) and the percentage of dead cells (right) were assessed.
Discussion
We describe a cascade of gene stimulation required to mediate epidermal cell death upon UVB irradiation. We provide evidence that activation and binding of the NF-κB member p65(RelA) to a canonical KB site located in the Egr-1 promoter is required for Egr-1 induction upon UVB irradiation. Egr-1, in turn, directly binds onto the Gadd45a and Gadd45b promoters to stimulate transcription directly, thus contributing to the inducible expression of these genes. A previous study has reported that Oct-1 and NF-YA proteins play a central role in activation of the Gadd45a cloned promoter (Jin et al, 2001). Nevertheless, our results show that invalidation of Egr-1 alters both basal and inducible endogenous Gadd45a expression, indicating that another mechanism also contributes to the regulation of this gene. Oct-1, NF-YA and Egr-1 are nuclear proteins induced after exposure to multiple DNA-damaging agents. Therefore, these proteins are putatively able to participate in the cellular response to genotoxic stress. Based on these observations, it is likely that optimal Gadd45a expression in response to such stress would occur through the coordination and cooperation of these transcription factors. Gadd45 genes belong to growth arrest and DNA damage-inducible genes, which play a prominent role in the control of growth, cell cycle checkpoint and nucleotide excision repair through their association with p21WAF/CIP1, Cdc2/CyclinB and PCNA (Smith et al, 1994; Kearsey et al, 1995; Zhan et al, 1999; Vairapandi et al, 2002). Several lines of evidence directly implicate Gadd45a and Gadd45b in the apoptotic process via activation of the JNK and p38 MAPK signaling pathway (Hildesheim et al, 2002; Yoo et al, 2003). Knockout studies demonstrate that mice lacking the Gadd45a gene significantly impair apoptosis and are susceptible to ionizing radiation, UVB and 3,12-dimethylbenzaanthracene induced tumors (Hollander et al, 2001; Hildesheim et al, 2002). Upon genotoxic stress, Gadd45 genes behave as a DNA damage gatekeeper by killing the cells that fail to repair. This process is important to avoid accumulation of mutations that could likely be at the onset of carcinogenesis. Gadd45 genes are therefore important for the regulation of cell fate upon genotoxic stress to promote tumor suppression. According to this, it has been demonstrated that UVB-mediated Gadd45 genes induction is strongly altered in tumor epidermal cells (Dazard et al, 2003). As a result, alteration of the pathways responsible for the induction of these genes may have serious consequences on the physiology of the cell.
Nothing is known about Egr-1 function during stress response or carcinogenesis in human skin context. The siRNA strategy we used allowed a physiological analysis of Egr-1 functions during UVB stress in epidermal cells. In accordance with the fact that Gadd45 gene expression is altered in Egr-1-deficient cells, we found that inhibition of Egr-1 expression in epidermal cells strongly reduces the rate of death mediated by UVB. The strong resistance to UVB-mediated cell death also observed in Egr-1−/− MEFs supports this observation. The fact that the sole inhibition of Egr-1 expression severely impairs the cell death process suggests this transcription factor as an important early regulator for epidermal homeostasis during the genotoxic stress response. Apoptosis during UVB stress injury in skin is particularly important because it is a protective response to eliminate potential transformed cells from the surrounding tissue (Kulms and Schwarz, 2002a, 2002b). Egr-1 can therefore be considered as a potent tumor suppressor in skin.
We have demonstrated that NF-κB activation is required for Egr-1 induction after UVB irradiation. Accordingly, RelA−/− MEFs displayed both a strong resistance to UVB-induced cell death and reduced Gadd45 gene expression. These results confirm that Egr-1 acts in the same pathway as NF-κB. In addition, our results point to NF-κB as the trigger responsible for a part of the cellular death response to UVB, through activation of a cascade that sequentially induces Egr-1 and Gadd45 gene expression. The finding that NF-κB, and especially p65(RelA), can promote cell death contrasts with its largely documented antiapoptotic action in a variety of experimental systems (Barkett and Gilmore, 1999; Dixit and Mak, 2002). NF-κB prevents cell death through expression of several target genes coding for proteins that interfere with caspase activation (Bcl-xl, X-IAP, IAP1 and 2, IEX-1L and Bfl1) (Barkett and Gilmore, 1999). Nevertheless, NF-κB is also able to promote apoptosis, for instance following DNA damage, through the induction of proapoptotic genes that code for the death receptor Fas and its ligand (Kasibhatla et al, 1998; Barkett and Gilmore, 1999; Pahl, 1999). p65(RelA) was shown to associate with histone deacetylases to repress antiapoptotic genes in particular settings such as UVC or daunorubicin stimulation (Campbell et al, 2004). Furthermore, it is now well accepted that inhibition of NF-κB activation, especially RelA containing complex, promotes growth and carcinogenesis in skin (Dajee et al, 2003; Takao et al, 2003). NF-κB is constitutively present and sequestered in the cytoplasm and can be rapidly activated upon genotoxic stress. In consequence, this transcription factor could be considered as an early mediator of the genotoxic stress response. Therefore, identification of other target transcription factors that, like Egr-1, may orchestrate the onset of the cellular genotoxic stress response would be of great interest for our understanding of mechanisms involved in DNA repair and tumor suppression.
In conclusion, our study demonstrates for the first time a signaling cascade involving sequential activation of NF-κB, Egr-1 and Gadd45 genes to induce UVB-mediated cell death in epidermal cells. Failure in the induction of each protagonist of this pathway alters the UVB-mediated cell death process. This process is crucial to eradicate (the cells that bear the risk of becoming tumorigenic; therefore, impairment of this cascade could be at the onset of skin carcinogenesis mediated by genotoxic stress.
Materials and methods
Cell culture
Cells were cultivated at 37°C in 5% CO2. NHKs were isolated as described elsewhere (Vailly et al, 1998). NHKs were cultured in keratinocytes-SFM medium (GIBCO) supplemented with bovine pituitary extract and EGF. Immortalized rela−/− MEFs and matching WT cells were provided by Professor Ron Hay (University of St Andrews). MEFs derived from egr-1-null mice were obtained from Eileen Adamson (The Burnham Institute, Cancer Research Center, La Jolla, CA). Immortalized HaCaT cells and MEFs were maintained in Dulbecco's modified Eagle's medium supplemented with 10% bovine fetal serum. KHSV cell line (derived from normal human primary keratinocytes immortalized by SV-40 infection) was maintained as described (Virolle et al, 2002). When indicated, the cells received a single dose of 60 mJ/cm2 UVB radiations (4 UVB lamp T20M; Bio-Link BLX-312, Vilbert Lourmat, France).
Immunofluorescence
Samples of human skin irradiated or not with a single dose of UVB (60 mJ/cm2) were included in optimal cutting temperature (OCT; Tissue Tek) 3 h after irradiation. Cryo-cuts (5 μm) were fixed for 20 min at room temperature in paraformaldehyde. The paraformaldehyde was neutralized with phosphate buffered saline (PBS) and nonspecific binding was blocked by incubation for 10 min in a solution of 10 mM NH4Cl and 5% BSA. The cells were then incubated with antibodies to Egr-1 diluted in 1% BSA (1:50) at room temperature. Cells were washed and exposed to secondary antibody (1:1000). Fluorescence was imaged using a Zeiss Axiophot microscope.
Transfection and adenovirus infections
The day before transfection, KHSV cells were seeded in 24-well plates at a density of 80 000 cells per well. Cells were transfected with FuGen-6 reagent (Roche) as described elsewhere (Virolle et al, 2002). The day after transfection, cells were irradiated and after 3 h luciferase activity was measured using a microlumat LB96P, EG&G Berthold. The results are the average of three separate experiments in quadruplicate.
HaCaT cells were plated 1 day before infection in 35 mm wells at a density of 180 000 cells per dish. Infections were made at a multiplicity of infection (m.o.i.) of 12. At 24 h after infection, cells were extensively rinsed with PBS and irradiated.
Cell death measurement was performed 24 h later in a 96-well plate (20 000 cells per well) by MTT assay (Sigma, M-2128). Briefly, the medium was aspirated from the wells, cells were rinsed with PBS and 100 μl MTT (Sigma-Aldrich) solution (final concentration, 0.5 mg/ml) was added. Cells were incubated for 5 h, after which the medium was again aspirated and the precipitated formazan dissolved by adding 100 μl DMSO and placing the resulting mixture on a shaker for 10 min. Samples were read at 560 nm.
The detection of caspase 3/7 activity was performed using the Caspase-GloTM3/7 Assay according to the manufacturer's protocol (Promega).
Plasmids and recombinant viruses
The 1.4 kb DNA fragment upstream of Egr-1 ATG was PCR amplified on genomic DNA using specific primers (forward 5′-CACCCAGGCCTCTCTTGGGGCAATCA-3′, reverse 5′-TGTCCATGGTGGGCGAGTGA-3′). This fragment was cloned into pENTR shuttle vector (Gateway Technology Invitrogen) to obtain pENTR-pEgr-1. The pEgr-1-luc construct was obtained by recombination between pENTR-pEgr-1 and the gateway adapted pGl3 basic luciferase vector (Clontech). F2, F3 and F4 fragments were PCR amplified using pEgr-1-luc as template with the following specific primers: forward F2 5′-CACCCGCTCTCACGGTCCCTGAGGT-3′, F3 5′-CACCGCGGCGGCTAGAGCTCTAGG-3′, F4 5′-CACCCGAGGGAGCA ACCAGCTGCGA-3′ and reverse 5′-TGTCCATGGTGGGCGAGTGA-3′. PCR fragments were then cloned in the destination pGl3 basic luciferase vector. The deleted constructs Δ1, Δ2, Δ3, Δ4, Δ5 and Δ6 and the mutated mutNF-κB construct were created according to the Quickchange protocol (Stratagene) using the following primers:
Δ15′-TTGGAACCAGGGAGGAGGGAGGGAGGATCC CCCGCCGGAACAACCCTTAT-3′,
Δ25′-TTGGAACCAGGGAGGAGGGAGGGAGCCCGA TATGGCCCGGCCGCTTCCGG-3′,
Δ35′-TTGGAACCAGGGAGGAGGGAGGGAGGGGAG GGGCAACGCG GGAACTCCGG-3′,
Δ45′-TTGGAACCAGGGAGGAGGGAGGGAGGCGGC GGCTAGAGCT CTAGGCTTCC-3′,
Δ55′-TTGGAACCAGGGAGGAGGGAGGGAGCGGAA ATGCCATATAA GGAGCAGGA-3′,
Δ65′-AGCGAGGGAGCAACCAGCTGCGACCGATCCC CCGCCGGAAC AACCCTTAT-3′, mutNF-κB 5′-GGAGCAACCAGCTGCGACCCGACTAGTCCATA TAAGGAGCAGGAAGGATC-3′.
The Egr-1-specific antisense oligonucleotide E5, described elsewhere (Baron et al, 2003), was cloned into the expression vector psiRNA-H1neo (Invitrogen) according to an shRNA design described elsewhere (Brummelkamp et al, 2002). The cassette containing the H1 promoter, E5shRNA and the polymerase termination stop was isolated by PCR amplification from the psiRNA-H1neo construct (forward primer 5′-CGATACTAGTAATATTTGCATGT-3′, reverse primer 5′-CCCTAACTGACACACATTCC-3′), subcloned in an entry gateway plasmid (pENTR vector) and then introduced by recombination into pAD/PL promoter-less adenoviral vector (Gateway Invitrogen) to create the pAd-siRNA-Egr-1 construct. Using the same method, an irrelevant pAd-siRNA-Ctl construct was performed with the following sequence: 5′-TCCCAAGGCATAAGGCTGAGCTGATTCAAGAG ATCAGCTCAGCCTTATGCCTTT-3′.
The pCDNA-Gadd45a and pCDNA-Gadd45b vectors have been kindly provided by Drs AJ Fornace Jr and TA Libermann.
Quantitative real-time one-step RT–PCR and Western blot
RNAs were extracted using Trizol reagent (Invitrogen) and Rneasy protect minikit (Quiagen) either 1 or 3 h after irradiation, to measure Egr-1, Gadd45a and Gadd45b expression. mRNA expression level was quantified by real-time one-step RT–PCR using the SYBR-Green PCR Master Mix (Applied Biosystem) according to the manufacturer's instructions. The relative amount of each gene to GAPDH internal control and the fold stimulation was calculated by using the equation 2−ΔΔCT, where ΔCT=CTgene−CTGAPDH and ΔΔCT=ΔCTstimulated condition−ΔCTunstimulated condition. Each gene was amplified using the appropriate specific primers (sequences available upon request). The results are the average of three separate experiments.
For the Western blot analysis, total proteins were extracted 3 h after irradiation with buffer (20 mM HEPES, 350 mM NaCl, 500 μM EDTA, 1 mM MgCl2, 20% glycerol, 1% NP-40), boiled and separated by 7.5% SDS–polyacrylamide gel electrophoresis (PAGE). The proteins were transferred to Immobilon P membranes (Millipore Corporation, Bedford, MA), and reacted with primary rabbit polyclonal antibodies to Egr-1 (Sc-110), mouse monoclonal antibody to αERK2 D-2(Sc-1647) (Santa Cruz Biotechnology Inc., Santa Cruz, CA) and anti-rabbit and anti-mouse secondary antibodies conjugated to horseradish peroxidase for enhanced chemiluminescence detection of the signals (Amersham, Grand Island, NY).
Chromatin immunoprecipitation assay
Live cells were treated with 1% formaldehyde for 5 min at 37°C and 30 min at 4°C. The cells were then harvested and suspended in the lysis buffer (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.1% NP-40, protease inhibitors). After 10 min incubation on ice, the cells were centrifuged (5000 r.p.m., 5 min at 4°C) to pellet the nuclei. Nuclei were then suspended in the nuclei lysis buffer (50 mM Tris–HCl pH 8.1, 10 mM EDTA, 1% SDS, protease inhibitors) and incubated on ice for 10 min. The chromatin was sheared by sonication (average size 0.5 kb) and samples were microfuged at 14 000 r.p.m. for 10 min. The sheared chromatin was then immunoprecipitated using specific antibodies directed against either Egr-1 (Santa Cruz Biotechnology, sc-189) or p65, and a nonimmune serum as a negative control. After immunoprecipitation, the crosslink was reverted by heat treatment (67°C overnight and proteinase K digestion). The captured genomic fragments were then recovered by phenol–chloroform extraction. Identification of the captured Egr-1 or Gadd45a and Gadd45b regulatory sequences was performed by PCR analysis using respectively the following primers: 5′-GGCTCCCGGCTTGGAACCAG-3′ and 5′-CCGGCACCTCCATCCTGCAC-3′; 5′-GGCGGAAGGTGGTTGGCTGA-3′ and 5′-AGCTCAGGCCCTGGCGCTCT-3′; 5′-GGCATTCGCGGTCACCTACCC-3′ and 5′-ATTGGGCGTGGCCTCAGTGG-3′. A total of 30 cycles of PCR were performed and the amplified products were analyzed on a 2% agarose gel.
Nuclear extracts and electrophoretic mobility shift assay
Nuclear extracts from keratinocytes were prepared as described (Dignam et al, 1983). DNA binding reactions were performed in a volume of 25 μl of 10 μg nuclear protein in the appropriate binding buffer (NF-κB 5 × buffer: Tris 50 mM pH 7.5, NaCl 500 mM, EDTA 5 mM, glycerol 20%, salmon sperm DNA 0.4 mg/ml, DTT 5 mM; Egr-1 5 × buffer: HEPES 50 mM pH 7.9, glycerol 50%, DTT 5 mM, KCl 250 mM, MgCl2 12.5 mM, salmon sperm DNA 0.4 mg/ml). In all, 200 000 cpm of 32P-radiolabeled double-stranded DNA probe (NF-κB: GCGACCCGGAAATGCCATATAA and Egr-1 244 bp fragment located in position −64/+181 from the transcription start point of Gadd45a promoter) were used per reaction. The binding reaction was preincubated with competitors (homologous cold probe, mutated cold probe and antibodies) for 15 min at 4°C and then incubated with the radiolabeled probe for 20 min at room temperature. DNA/protein complexes were resolved on 5% polyacrylamide gels and detected by autoradiography.
Acknowledgments
We thank Professor Ron Hay (University of St Andrews) and Dr Eileen D Adamson (The Burnham Institute, Cancer Research Center, La Jolla, CA) for the gift of the immortalized RelA−/− and Egr-1−/− and matching WT MEFs. We are grateful to Drs AJ Fornace Jr and TA Libermann for providing plasmids. We thank Dr Rene Feyereisen for helpful comments on the manuscript. This work was supported by grants from Association pour la Recherche sur le Cancer (ARC), Société de Recherche en Dermatologie (SRD) and Institut National de la Santé et de la Recherche Medicale (INSERM).
References
- Abdulkadir SA, Qu Z, Garabedian E, Song SK, Peters TJ, Svaren J, Carbone JM, Naughton CK, Catalona WJ, Ackerman JJ, Gordon JI, Humphrey PA, Milbrandt J (2001) Impaired prostate tumorigenesis in Egr1-deficient mice. Nat Med 7: 101–107 [DOI] [PubMed] [Google Scholar]
- Ahmed MM, Venkatasubbarao K, Fruitwala SM, Muthukkumar S, Wood DP Jr, Sells SF, Mohiuddin M, Rangnekar VM, Nair P, Maddiwar NG, Jacob RJ (1996) EGR-1 induction is required for maximal radiosensitivity in A375-C6 melanoma cells. Role of EGR-1 in thapsigargin-inducible apoptosis in the melanoma cell line A375-C6. J Biol Chem 271: 29231–29237 [DOI] [PubMed] [Google Scholar]
- Armstrong BK, Kricker A (2001) The epidemiology of UV induced skin cancer. J Photochem Photobiol B 63: 8–18 [DOI] [PubMed] [Google Scholar]
- Barkett M, Gilmore TD (1999) Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene 18: 6910–6924 [DOI] [PubMed] [Google Scholar]
- Baron V, De Gregorio G, Krones-Herzig A, Virolle T, Calogero A, Urcis R, Mercola D (2003) Inhibition of Egr-1 expression reverses transformation of prostate cancer cells in vitro and in vivo. Oncogene 3: 4194–4204 [DOI] [PubMed] [Google Scholar]
- Brash DE (1997) Sunlight and the onset of skin cancer. Trends Genet 13: 410–414 [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550–553, Epub 2002 Mar 21 [DOI] [PubMed] [Google Scholar]
- Campbell KJ, Rocha S, Perkins ND (2004) Active repression of antiapoptotic gene expression by RelA(p65) NF-kappa B. Mol Cell 13: 853–865 [DOI] [PubMed] [Google Scholar]
- Cleaver JE, Crowley E (2002) UV damage, DNA repair and skin carcinogenesis. Front Biosci 7: d1024–d1043 [DOI] [PubMed] [Google Scholar]
- Dajee M, Lazarov M, Zhang JY, Cai T, Green CL, Russell AJ, Marinkovich MP, Tao S, Lin Q, Kubo Y, Khavari PA (2003) NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature 6: 639–643 [DOI] [PubMed] [Google Scholar]
- Dazard JE, Gal H, Amariglio N, Rechavi G, Domany E, Givol D (2003) Genome-wide comparison of human keratinocyte and squamous cell carcinoma responses to UVB irradiation: implications for skin and epithelial cancer. Oncogene 22: 2993–3006 [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11: 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit V, Mak TW (2002) NF-kappaB signaling. Many roads lead to madrid. Cell 111: 615–619 [DOI] [PubMed] [Google Scholar]
- Harada T, Morooka T, Ogawa S, Nishida E (2001) ERK induces p35, a neuron-specific activator of Cdk5, through induction of Egr1. Nat Cell Biol 3: 453–459 [DOI] [PubMed] [Google Scholar]
- Hildesheim J, Bulavin DV, Anver MR, Alvord WG, Hollander MC, Vardanian L, Fornace AJ Jr (2002) Gadd45a protects against UV irradiation-induced skin tumors, and promotes apoptosis and stress signaling via MAPK and p53. Cancer Res 62: 7305–7315 [PubMed] [Google Scholar]
- Hofer G, Grimmer C, Sukhatme VP, Sterzel RB, Rupprecht HD (1996) Transcription factor Egr-1 regulates glomerular mesangial cell proliferation. J Biol Chem 271: 28306–28310 [DOI] [PubMed] [Google Scholar]
- Hollander MC, Kovalsky O, Salvador JM, Kim KE, Patterson AD, Haines DC, Fornace AJ Jr (2001) Dimethylbenzanthracene carcinogenesis in Gadd45a-null mice is associated with decreased DNA repair and increased mutation frequency. Cancer Res 61: 2487–2491 [PubMed] [Google Scholar]
- Huang RP, Fan Y, Boynton AL (1999) UV irradiation upregulates Egr-1 expression at transcription level. J Cell Biochem 73: 227–236 [DOI] [PubMed] [Google Scholar]
- Jin S, Fan F, Fan W, Zhao H, Tong T, Blanck P, Alomo I, Rajasekaran B, Zhan Q (2001) Transcription factors Oct-1 and NF-YA regulate the p53-independent induction of the GADD45 following DNA damage. Oncogene 20: 2683–2690 [DOI] [PubMed] [Google Scholar]
- Kasibhatla S, Brunner T, Genestier L, Echeverri F, Mahboubi A, Green DR (1998) DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol Cell 1: 543–551 [DOI] [PubMed] [Google Scholar]
- Kearsey JM, Coates PJ, Prescott AR, Warbrick E, Hall PA (1995) Gadd45 is a nuclear cell cycle regulated protein which interacts with p21Cip1. Oncogene 11: 1675–1683 [PubMed] [Google Scholar]
- Khachigian LM, Lindner V, Williams AJ, Collins T (1996) Egr-1-induced endothelial gene expression: a common theme in vascular injury. Science 271: 1427–1431 [DOI] [PubMed] [Google Scholar]
- Kulms D, Schwarz T (2002a) Independent contribution of three different pathways to ultraviolet-B-induced apoptosis. Biochem Pharmacol 64: 837–841 [DOI] [PubMed] [Google Scholar]
- Kulms D, Schwarz T (2002b) Molecular mechanisms involved in UV-induced apoptotic cell death. Skin Pharmacol Appl Skin Physiol 15: 342–347 [DOI] [PubMed] [Google Scholar]
- Li D, Turi TG, Schuck A, Freedberg IM, Khitrov G, Blumenberg M (2001) Rays and arrays: the transcriptional program in the response of human epidermal keratinocytes to UVB illumination. FASEB J 15: 2533–2535, Epub 2001 Sep 17 [DOI] [PubMed] [Google Scholar]
- Muthukkumar S, Nair P, Sells SF, Maddiwar NG, Jacob RJ, Rangnekar VM (1995) Role of EGR-1 in thapsigargin-inducible apoptosis in the melanoma cell line A375-C6. Mol Cell Biol 15: 6262–6272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl HL (1999) Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18: 6853–6866 [DOI] [PubMed] [Google Scholar]
- Perez-Castillo A, Pipaon C, Garcia I, Alemany S (1993) NGFI-A gene expression is necessary for T lymphocyte proliferation. J Biol Chem 268: 19445–19450 [PubMed] [Google Scholar]
- Riggs PK, Rho O, DiGiovanni J (2000) Alteration of Egr-1 mRNA during multistage carcinogenesis in mouse skin. Mol Carcinogen 27: 247–251 [DOI] [PubMed] [Google Scholar]
- Smith ML, Chen IT, Zhan Q, Bae I, Chen CY, Gilmer TM, Kastan MB, O'Connor PM, Fornace AJ Jr (1994) Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science 266: 1376–1380 [DOI] [PubMed] [Google Scholar]
- Soehnge H, Ouhtit A, Ananthaswamy ON (1997) Mechanisms of induction of skin cancer by UV radiation. Front Biosci 2: D538–D551 [DOI] [PubMed] [Google Scholar]
- Sukhatme VP, Cao XM, Chang LC, Tsai-Morris CH, Stamenkovich D, Ferreira PC, Cohen DR, Edwards SA, Shows TB, Curran T, Le Beau MM, Adamson ED (1988) A zinc finger-encoding gene coregulated with c-fos during growth and differentiation, and after cellular depolarization. Cell 53: 37–43 [DOI] [PubMed] [Google Scholar]
- Takao J, Yudate T, Das A, Shikano S, Bonkobara M, Ariizumi K, Cruz PD (2003) Expression of NF-kappaB in epidermis and the relationship between NF-kappaB activation and inhibition of keratinocyte growth. Br J Dermatol 148: 680–688 [DOI] [PubMed] [Google Scholar]
- Vailly J, Gagnoux-Palacios L, Dell'Ambra E, Romero C, Pinola M, Zambruno G, De Luca M, Ortonne JP, Meneguzzi G (1998) Corrective gene transfer of keratinocytes from patients with junctional epidermolysis bullosa restores assembly of hemidesmosomes in reconstructed epithelia. Gene Therapy 5: 1322–1332 [DOI] [PubMed] [Google Scholar]
- Vairapandi M, Balliet AG, Hoffman B, Liebermann DA (2002) GADD45b and GADD45g are cdc2/cyclinB1 kinase inhibitors with a role in S and G2/M cell cycle checkpoints induced by genotoxic stress. J Cell Physiol 192: 327–338 [DOI] [PubMed] [Google Scholar]
- Virolle T, Adamson ED, Baron V, Birle D, Mercola D, Mustelin T, de Belle I (2001) The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat Cell Biol 3: 1124–1128 [DOI] [PubMed] [Google Scholar]
- Virolle T, Coraux C, Ferrigno O, Cailleteau L, Ortonne JP, Pognonec P, Aberdam D (2002) Binding of USF to a non-canonical E-box following stress results in a cell-specific derepression of the lama3 gene. Nucleic Acids Res 30: 1789–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virolle T, Krones-Herzig A, Baron V, De Gregorio G, Adamson ED, Mercola D (2003) Egr1 promotes growth and survival of prostate cancer cells. Identification of novel Egr1 target genes. J Biol Chem 4: 11802–11810 [DOI] [PubMed] [Google Scholar]
- Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ Jr, Liebermann DA, Bottinger EP, Roberts AB (2003) Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J Biol Chem 278: 43001–43007, Epub 2003 Aug 21 [DOI] [PubMed] [Google Scholar]
- Zhan Q, Antinore MJ, Wang XW, Carrier F, Smith ML, Harris CC, Fornace AJ Jr (1999) Association with Cdc2 and inhibition of Cdc2/Cyclin B1 kinase activity by the p53-regulated protein Gadd45. Oncogene 18: 2892–2900 [DOI] [PubMed] [Google Scholar]