

CREB-H plays a key role in regulating secretion in metabolic pathways, particularly triglyceride homeostasis. Phosphorylation at a conserved serine motif, the P-motif, provides an integrated control mechanism of CREB-H function, coupling intercompartmental transport in the cytoplasm with stabilization of the active form in the nucleus.
Abstract
CREB-H, an ER-anchored transcription factor, plays a key role in regulating secretion in metabolic pathways, particularly triglyceride homeostasis. It controls the production both of secretory pathway components and cargoes, including apolipoproteins ApoA-IV and ApoC-II, contributing to VLDL/HDL distribution and lipolysis. The key mechanism controlling CREB-H activity involves its ER retention and forward transport to the Golgi, where it is cleaved by Golgi-resident proteases, releasing the N-terminal product, which traffics to the nucleus to effect transcriptional responses. Here we show that a serine-rich motif termed the P-motif, located in the N-terminus between serines 73 and 90, controls release of the precursor transmembrane form from the ER and its forward transport to the Golgi. This motif is subject to GSK-3 phosphorylation, promoting ER retention, while mutation of target serines and drug inhibition of GSK-3 activity coordinately induce both forward transport of the precursor and cleavage, resulting in nuclear import. We previously showed that for the nuclear product, the P-motif is subject to multiple phosphorylations, which regulate stability by targeting the protein to the SCFFbw1a E3 ubiquitin ligase. Thus phosphorylation at the P-motif provides integrated control of CREB-H function, coupling intercompartmental transport in the cytoplasm with stabilization of the active form in the nucleus.
INTRODUCTION
The endoplasmic reticulum (ER) is the site of protein synthesis, folding, quality control, and secretion. It is also the site of production of many other components, including steroids, lipids, and fatty acids, and thus plays a particularly important role in integrating numerous metabolic signals and homeostatic responses to diverse internal and external cues (Tabas and Ron, 2011; Walter and Ron, 2011; Basseri and Austin, 2012; Cnop et al., 2012; Fu et al., 2012; Guerriero and Brodsky, 2012). A key arm of these homeostatic control processes is the activation of ER-resident transcription factors by environmental stimuli that act through intercompartmental trafficking and regulated intramembrane proteolysis (RIP). These pathways ultimately lead to the nuclear import of the active factor, promoting adaptive transcriptional responses of selected target genes (Brown and Goldstein, 1997; Brown et al., 2000; Tirasophon et al., 1998; Wang et al., 2000; Ye et al., 2000; Rawson, 2002).
The prototypical example of these pathways is the one involving homeostatic responses to fluctuation in cholesterol and fatty acid levels controlled by the ER-anchored transcription factors sterol response element–binding protein-1 (SREBP1) and SREBP2. SREBPs contain two transmembrane domains that anchor them in the ER in a complex with other factors, including SREBP cleavage–activating protein (SCAP) and Insig (Nohturfft et al., 1999; Brown et al., 2002; Yang et al., 2002; Sun et al., 2005). Low levels of cholesterol are sensed by SCAP, and a SCAP-SREBP complex dissociates from Insig, allowing its forward transport to the Golgi apparatus. At the Golgi, SREBP is sequentially cleaved by two site-specific proteases, site 1 protease (S1P) and site 2 protease (S2P), releasing the N-terminal active cleaved product into the cytoplasm for subsequent translocation into the nucleus. SREBP-1c, the predominant isoform in liver and adipocytes, is also activated by insulin signaling through several kinases, including GSK-3, and the AKT/mTORC1 pathway, which activates both transcriptional expression and processing of SREBP-1c (Hegarty et al., 2005; Sundqvist et al., 2005; Yellaturu et al., 2009; Owen et al., 2012; Dong et al., 2015).
A new, distinct class of ER-anchored transmembrane transcription factors (the cyclic AMP response element–binding protein-3 [CREB3] family) has been identified (Lu et al., 1997; Honma et al., 1999; Cao et al., 2002; Omori et al., 2002; Raggo et al., 2002; Storlazzi et al., 2003; Bailey and O’Hare, 2007; Kondo et al., 2007; Lee, 2012). These represent an ancient lineage, with homologues in many evolutionary branches (Barbosa et al., 2013). In humans, the CREB3 family comprises five members—CREB3/Luman, CREB3L1/OASIS, CREB3L2/BBF2H7, CREB3L3/CREB-H, and CREB3L4/CREB4—which are expressed with different degrees of tissue specificity. These factors regulate many key physiological processes, with demonstrated roles in processes including metabolic homeostasis, differentiation, bone formation, cartilage formation, and inflammation (Zhang et al., 2006; Murakami et al., 2009; Saito et al., 2009; Vecchi et al., 2009; Lee et al., 2010, 2011; Asada et al., 2012; Saito et al., 2012). Similarly to SREBP, the CREB3 proteins are synthesized as ER-resident precursors and undergo regulated intercompartmental transport and RIP, resulting in the release of their transcriptionally active N-terminal domains to promote distinct transcriptional programs (Bailey and O’Hare, 2007). However, although their roles in various pathways have been clearly demonstrated and certain stimuli, including differentiation-induced ER stress, cytokines, and metabolic fluctuations, are known to promote CREB3 processing (Murakami et al., 2009; Vecchi et al., 2009; Lee et al., 2010, 2011), much less is known about the key protein determinants and regulatory mechanisms controlling their intercompartmental transport, the process that ultimately controls activity.
Here we investigate CREB-H, a CREB3 family member mainly expressed in the liver and the intestine. Both the RNA expression levels and activation of CREB-H at the protein level are modulated by metabolic signals, including fasting, insulin, fatty acids, and a high-fat diet (Danno et al., 2010; Gentile et al., 2010; Lee et al., 2010; Lee, 2012; Chanda et al., 2011; Nakagawa et al., 2014). CREB-H has been reported to be involved in the regulation of transcription of acute-phase response proteins during inflammation (Zhang et al., 2006), in gluconeogenesis through the up-regulation of G6Pase and PEPCK (Lee et al., 2010), the production and secretion of apolipoproteins (Lee et al., 2011; Zhang et al., 2012; Barbosa et al., 2013; Xu et al., 2014), and the control of expression of genes modulating fatty acid and triglyceride levels (Lee et al., 2011; Zhang et al., 2012). In mice fed on a high-fat diet, knockout of CREB-H results in fatty liver and hypertriglyceridemia (Zhang et al., 2012). In humans, mutations in CREB-H were associated with autosomal dominant hypertriglyceridemia (Lee et al., 2011; Zhang et al., 2012; Cefalu et al., 2015). Conversely, overexpression of CREB-H in mice results in increased lipolysis, hepatic ketogenesis, insulin sensitivity, and energy expenditure and protects against diet-induced hyperinsulinemia and hyperglycemia, as well as significantly reduces plasma triglycerides (Nakagawa et al., 2014). The key targets most efficiently activated by CREB-H are the apolipoproteins, in particular ApoA-IV, ApoA5, ApoA-I, and ApoC-II, which then are incorporated or modulate chylomicron and lipoprotein particle assembly and function (Lee et al., 2011; Zhang et al., 2012; Barbosa et al., 2013; Xu et al., 2014). However, although some studies reported increased transcript levels of CREB-H and increased processing of the protein under metabolic cues, very little is known about the detailed mechanisms and signaling pathways involved.
Previous work from our laboratory showed that the CREB-H precursor is a glycosylated, short-lived protein and that a conserved cytosolic determinant (the ER-retention motif [ERM]), immediately proximal to the transmembrane domain, was a key determinant necessary for the ER retention of the protein (Bailey and O’Hare, 2007; Llarena et al., 2010). Furthermore, examining expression of the cleaved mature N-terminal product in the nucleus, we showed that a highly conserved serine-rich determinant termed the P-motif, with a core sequence of DSGxS and located around residues 73–81, acted as a phosphodegron and was subject to phosphorylation by GSK-3 and CKII kinases. This phosphorylation promoted binding of the F-box factor Fbw1a and subsequent proteasomal degradation. Our results showed, first, that nuclear CREB-H was constitutively phosphorylated, leading to its rapid turnover, and second, that suppression of phosphorylation increased nuclear stability. We proposed that regulated phosphorylation/dephosphorylation of the nuclear product acted as one part of the overall regulation of the CREB-H homeostatic cycle (Barbosa et al., 2015).
In this study, we examine the role of the P-motif in the context of the full-length precursor form of the protein. Consistent with our earlier results, we find that the full-length form is multiply phosphorylated, including in the P-motif. Of importance, serine-to-alanine substitutions within the motif resulted in pronounced constitutive ER–Golgi transport in the absence of any exogenous stimuli. This was accompanied by increased proteolytic cleavage and nuclear transport, increased transcriptional activity, and increased secretion of the key physiological target, ApoA-IV. Furthermore, chemical inhibition of GSK-3 suppressed CREB-H phosphorylation, specifically of serines 73 and 81 within the P-motif, and in vitro assays confirmed direct phosphorylation of serine 73 by GSK-3β. Of importance, GSK-3 inhibition resulted in a pronounced increase in the ER–Golgi transport of the parental protein and its subsequent proteolytic processing. These results, combined with our previous data, provide evidence for a unifying model in which the P-motif acts as a dual control system coordinately regulating both intercompartmental cytoplasmic transport of the precursor form and nuclear stability of the cleaved product for the homeostatic modulation of CREB-H activity.
RESULTS
Phosphorylation of full-length CREB-H within the P-motif
Although CREB-H has been shown to be a key factor in the homeostatic control of metabolic pathways, including synthesis and export of triglycerides and apolipoproteins, the signaling pathways and mechanisms involved in its selective ER localization and forward trafficking to the Golgi are poorly understood. The general principles of CREB-H localization are shown in Figure 1a. We previously identified a determinant in CREB-H (the ERM) adjacent to the transmembrane domain on the cytosolic side (Figure 1a). This motif is critical for ER retention, and its deletion results in pronounced constitutive transport of CREB-H to the Golgi (Bailey et al., 2007; Llarena et al., 2010). More recently, in analyzing determinants of stability of the cleaved nuclear form, we identified a serine-rich motif in the N-terminus that is phosphorylated and is important in regulating the overall levels and transcriptional activity of the processed product in the nucleus (Barbosa et al., 2015). This serine-rich motif (termed the P-motif) is illustrated in Figure 1a (labeled P), with the expanded amino acid sequence indicating highly conserved residues within the motif (white lettering on black background). Asterisks indicate serines whose mutation influences the stability of the nuclear form (Barbosa et al., 2015). These serines are extremely well conserved in CREB-H homologues from numerous species (Bailey et al., 2007; Barbosa et al., 2015).
FIGURE 1:

FL CREB-H is multiply phosphorylated within the P-motif. (a) Diagram illustrating the precursor form of CREB-H anchored in the ER membrane. Unidentified signals (indicated by a question mark) induce release from the ER, transport to the Golgi, and subsequent processing by S1P and S2P proteases. Bottom, subdomains of CREB-H: bZIP, the basic-zipper DNA-binding domain; TM, the transmembrane domain; and the S1P and S2P sites. P, the P-motif, with the amino acid sequence expanded below. Residues that are very highly conserved across all CREB-H homologues are indicated by white lettering on a black background (Barbosa et al., 2015). Key serine residues are indicated by asterisks. (b) COS cells were transfected with the expression vector for FL CREB-H and treated without or with BFA (final concentration 1 µg/ml, added 1 h before harvesting). Total cell extracts were analyzed by SDS–PAGE and Western blotting. A mock-transfected control sample is shown in lane 1. Lanes 4 and 5 show a shorter exposure of lanes 2 and 3. CREB-H FL precursor migrates as multiple species (solid bars), as discussed in the text. (c) Dephosphorylation of CREB-H. Soluble extracts in nondenaturing buffer from cells expressing the precursor form of CREB-H were incubated at 37°C with (lane 1) or without phosphatase inhibitors (lane 2) or with added λ-phosphatase again with (lane 4) or without phosphatase inhibitors (lane 3). The total population of CREB-H was detected using the anti-SV5 mouse primary antibody, and phosphorylation of serine 73 was simultaneously detected using the specific anti-pS73 antibody (Barbosa et al., 2015). CREB-H species were simultaneously detected by appropriate fluorochrome- labeled anti-mouse and anti-rabbit secondary antibodies using a LI-COR laser scanning system as described in Materials and Methods. Individual channels are shown separately, as well as together in the merged channel.
We sought to investigate whether the P-motif played any additional role in regulating the abundance and location of the full-length (FL) protein at the ER or its transport to the Golgi. Previous analysis indicated that CREB-H migrates in SDS–PAGE gel as a series of closely spaced bands (Llarena et al., 2010; Barbosa et al., 2013, 2015). Although CREB-H is known to be glycosylated (Bailey et al., 2007; Chan et al., 2010), and differential glycosylation could influence migration, it seemed reasonable to propose that the multiple CREB-H bands could also be due to multiple phosphorylated species. The typical appearance of CREB-H after transient transfection of the wild-type (wt) parental form in COS cells is shown in Figure 1b. Transfected cells were incubated without (lane 2) or with brefeldin A (BFA; lane 3, BFA added 60 min before harvesting). A shorter exposure of the blot is shown in lanes 4 and 5 to aid inspection. FL CREB-H migrates as a series of species (solid lines), whereas after BFA treatment, multiple cleaved products (Cl) are observed, as previously described (Llarena et al., 2010; Barbosa et al., 2013). We showed that the cleavage product labeled “a” represents the fully processed form, cleaved by S1P and S2P, whereas the more slowly migrating product, labeled “b” represents a mixture of an intermediate form cleaved by S1P only together with additional, S1P plus S2P fully processed, phosphorylated species (Llarena et al., 2010).
To examine whether the closely migrating species of the FL precursor form were due to phosphorylation, we first performed a phosphatase assay on extracts of cells expressing the wt SV5 epitope-tagged form (Figure 1c). Soluble cell extracts were made from transiently transfected cells and incubated at 37°C with or without phosphatase inhibitors (lanes 1 and 2) or in the presence of added λ-phosphatase, again with or without phosphatase inhibitors (lanes 3 and 4). Samples were subject to SDS–PAGE electrophoresis, blotted, and then probed with anti-SV5 to detect the total CREB-H species or simultaneously probed on the same blot with a phosphospecific antibody that selectively identifies CREB-H species phosphorylated specifically on serine 73 (anti-pS73; Barbosa et al., 2015). In extracts incubated with phosphatase inhibitors (lane 1), multiple FL CREB-H species were detected with the anti-SV5 antibody (at least four upper bands with an additional faint lower band indicated by the arrow). On incubation without phosphatase inhibitors there was a slight shift in the ratio of the band intensities to faster-migrating species, likely due to intrinsic phosphatase activity in the extracts (lane 2). With the addition of λ-phosphatase, there was a significant loss of the upper species and a corresponding pronounced increase of the intensity of the lower species (lane 3). This shift in migration was virtually eliminated by the addition of phosphatase inhibitors (lane 4). These data indicate that the more slowly migrating species represent phosphorylated CREB-H, and the species remaining after phosphatase treatment represent either λ-phosphatase–resistant forms or dephosphorylated but differentially modified forms—for example, glycosylated.
Simultaneous analysis with anti-pS73 demonstrated, first, the presence of S73 phosphorylation of FL CREB-H. We noted that the phosphospecific antibody preferentially detected the most slowly migrating upper bands, as seen by comparison of the individual channels and the merged image (Figure 1c, lane 1). Second, treatment with phosphatase almost completely abolished detection with anti-pS73, whereas detection by anti-SV5 clearly remained (lane 3). Again, the loss of pS73 was substantially reversed by inclusion of phosphatase inhibitors (lane 4). Virtually identical results were obtained with another phosphospecific antibody (Barbosa et al., 2015), anti-pS81 (unpublished data). Taken altogether, our results strongly support the conclusion that the precursor form of CREB-H is subject to multiple phosphorylation events, including, although not limited to, serines 73 and 87 in the P-motif.
Increased stability, constitutive cleavage, and nuclear import of P-motif mutants
To examine whether the P-motif was involved in the regulation of ER retention and Golgi transport of FL CREB-H, we created mutants in which serines were substituted to alanines within the motif in various combinations. We previously described two distinct regions of the P-motif (Barbosa et al., 2015), the 3S and DSG regions (Figure 2a). Phosphosite prediction algorithms indicate that these sites represent good consensus fits for GSK-3 phosphorylation, with interdigitated consensus sites for CKII phosphorylation. We also showed that in the cleaved nuclear form, these sites can be phosphorylated by GSK and CKII, and whereas individual serine substitution had modest effects, combined mutation resulted in loss of phosphorylation by these kinases, together with significantly increased steady-state levels and half-lives of the cleaved protein (Barbosa et al., 2015).
FIGURE 2:

Serine-to-alanine substitutions in the P-motif promotes stabilization and cleavage. (a) Schematic showing the position of the P-motif with the wt sequence and mutants expanded below. Serines that were substituted by alanines are numbered and indicated by underlining. (b) Comparison of expression in liver cells (Huh 7) of wt CREB-H and the various P-motif mutants (3S+DSG, DSG, 3S). Labeling is as for Figure 1. Bottom, actin as a sample loading control. (c) Same analysis as in b, showing a longer exposure of the cleaved products. (d) Quantitative estimates of protein abundance. Blots were scanned in the appropriate channel using a LI-COR Odyssey laser scanning system, background subtracted, volume integrated, and normalized for loading using actin abundance in the separate channel. Fold increases in overall levels of the mutant proteins are compared with wt levels, which were standardized to 1. (e) Soluble extracts of cells transfected with the wt or the 3S+DSG mutant were fractionated and equal cell equivalents of the nuclear fractions analyzed by Western blotting for cleaved CREB-H. Lamin a/c, which fractionated exclusively to the nuclear fraction, was used as a loading control.
We therefore constructed equivalent mutants in the background of the FL precursor form of CREB-H. The 3S mutant has three serines (73, 77, and 81) mutated to alanine, the DSG mutant has two serines (87 and 90) mutated to alanine, and the 3S+DSG mutant contains a combination of both 3S and DSG mutants with the five serines of the P-motif mutated to alanine (Figure 2a). Comparison of the expression levels of these mutants with the wt protein demonstrates a clear phenotype (Figure 2b). There was a significant increase in steady-state levels when the DSG motif alone was mutated and a more moderate but reproducible increase when the 3S motif was mutated (Figure 2b, lanes 1–4). When both regions were mutated together in the 3S+DSG variant, there was a very striking increase in abundance. Laser scanning densitometry of the blots demonstrated ∼8-, 4-, and 2.5-fold increases in abundance for the 3S+DSG, DSG, and 3S mutants respectively (Figure 2d).
We also noted that mutation of the DSG motif resulted in the appearance of additional, slower-migrating species (Figure 2b, lane 3). These species are readily seen in this analysis, which used fluorochrome-labeled secondary antibodies and laser scanning for detection, resulting in increased resolution of the bands (Figure 2b, lane 3, bands labeled 1 and 2; in other analyses reported here using standard chemiluminescence, these bands were observed but not always well resolved). This upward shift in migration of the DSG mutant was reversed by simultaneous mutation of the 3S sites (3S+DSG), resulting in high levels of the mutant protein, migrating detectably faster than the DSG form and slightly faster than the wt form (Figure 2b, lanes 1–3). The 3S mutant alone also showed a slightly faster migration than the wt form. These results are consistent with a possible interpretation that phosphorylation at the DSG site may suppress phosphorylation within the 3S site. In this explanation, DSG-site mutation alone results in both increased levels of protein and increased relative phosphorylation within the 3S site, whereas additional mutation of the 3S site further stabilizes the protein but in the now-hypophosphorylated, faster-migrating form. Whatever the precise explanation for the appearance and then loss of these individual species, what is clear from our results taken together was that the FL form of CREB-H is multiply phosphorylated, serines S73 and S87 within the P-motif are phosphorylated, and substitution of the 3S and in particular the DSG motif increases steady-state levels of the protein.
Furthermore, for each of the mutants, we observed a corresponding increase in the appearance of the constitutively cleaved products (Figure 2, b, and longer exposure, c). We noted also that the relative shift upward in migration of the FL form in the DSG mutant and reversal of this migration shift by simultaneous 3S mutation were reflected in migration patterns of the cleavage products. Thus the 3S+DSG mutant exhibited the highest levels of constitutive cleavage products, with the DSG mutant producing lower levels with slower mobility and the 3S mutant the lowest levels (although still modestly increased from those seen with the wt parental form (Figure 2, lanes 2–5). Quantitative analysis of the increased abundance of nuclear form was more difficult due to the extremely low levels of constitutive cleavage of the parental wt form (Figure 2d). However, it was clear that the DSG and 3S mutants were present in increased amounts in the FL form, and they produced increased but lower abundance and variable amounts of the cleaved products, which were most readily observed for the combined 3S+DSG mutant.
We next examined whether the constitutive cleavage products were present in the nucleus by both immunofluorescence analysis (see later discussion) and fractionation and Western blotting (Figure 2e). We obtained consistent results by both methods, although for fractionation and Western blotting, due to the lower overall levels and recoveries of the separate mutants, the results are shown for only the combined 3S+DSG mutant. As anticipated from the total levels, we observed considerably higher levels of cleaved products in the nucleus compared with wt, which were virtually undetectable in the absence of BFA treatment (Figure 2e, lanes 2 and 3). In this example, only modest amounts of wt cleavage product were observed in the nucleus after BFA treatment, with the mutant species exhibiting slightly increased mobility.
These results support the interpretation that the increased appearance of cleavage products from the P-motif mutants represented increased abundance, increased Golgi transport, and subsequent S1P/S2P cleavage. It would therefore be anticipated that mutation of the S1P/S2P sites in the background of the P-motif mutation would prevent appearance of the cleaved products. Each of the mutants was therefore transferred into the background of a mutant containing substitutions R361A and P337L, which we previously showed to completely prevent S1P and S2P cleavage, respectively (Bailey and O’Hare, 2007; Llarena et al., 2010). The results, especially for the 3S+DSG mutant, confirmed that the appearance of constitutive cleavage products was indeed S1P/S2P dependent, reinforcing the proposal that the P-motif mutation resulted in increased Golgi transport and cleavage (Figure 3a, lanes 3 and 7).
FIGURE 3:

Increased abundance, S1P/S2P-dependent cleavage, and half-life of P-motif mutants. (a) Expression profiles from cells expressing of CREB-H wt and mutant 3S+DSG or DSG either in the normal background (lanes 1–4, S1P+/S2P+) or in the background of a mutant containing R361A/P337L substitutions that prevent cleavage by S1P and S2P (lanes 5–8, S1P−/S2P−). Control mock-transfected samples are shown in lanes 1 and 5. The increased steady-state level of the FL species of the mutants can be readily observed, together with the constitutive levels of the cleavage product (Cl) most readily detected for the 3S+DSG mutant. The appearance of cleavage products was abolished by S1P/S2P mutation. (b) Cells transfected with each of the wt or P-motif mutants in the S1P/S2P-ve background were treated with 100 μg/ml cycloheximide (+CHX) for different times up to 240 min. Samples were harvested at the indicated time points and analyzed by Western blot using the anti–SV5 epitope antibody and the Dylight 680–conjugated secondary antibody as described in Material and Methods. Typical results from comparative analyses performed at least four times. Note that for ease of inspection, the exposure for the wt is longer than that for the mutant. All quantitative data were obtained from raw scans. (c) The relative levels of each of the variants for these half-life experiments were calculated after background subtraction and volume integration. Values at each time point are means from values obtained from four independent experiments. Quantitative estimates for the half-lives were obtained using GraphPad Prism software from linear regression using one-phase decay analysis.
Finally, to estimate their relative stabilities, each of the wt and mutant proteins was analyzed for abundance after cycloheximide treatment, using quantitative Western blotting and laser scanning densitometry (Figure 3, b and c). Consistent with results on the increases in steady-state levels, the results (Figure 3c) showed an increase in half-life of the 3S mutant (154 min) over the wt (127 min) and more significant increases for the DSG (225 min) and 3S+DSG mutants (264 min).
Constitutive transport of P-motif mutants to the Golgi
Although the effect of the P-motif mutation on the abundance of FL CREB-H was clear, considering that these mutations stabilized the nuclear form (Barbosa et al., 2015), the appearance of the cleavage products could be due to their increased stability, increased forward transport to the Golgi, or a combination of both processes.
We therefore next addressed localization of wt and the P-motif mutants by immunofluorescence. In particular, we sought to examine whether, as anticipated from the results of the fractionation experiments (Figure 2e), we would observe increased nuclear localization with the P-motif mutants. Moreover, to specifically examine localization of the FL protein in the absence of consideration of the increased stability of the nuclear product, we also examined the wt and P-motif mutant forms in the background of the S1P−/S2P− mutant. In this way, any alteration in localization—in particular, increased forward transport to the Golgi—could be more specifically assessed in the absence S1P/S2P cleavage, which by virtue of cleavage and nuclear import of the N-terminal products, would have a tendency to reduce relative association with the Golgi.
The results of CREB-H localization in relation to the trans-Golgi network marker TGN46 are shown in Figure 4 and demonstrate several points. First, in the context of normal S1P/S2P-positive cleavage, the wt CREB-H was largely cytosolic (associating with the ER marker calreticulin; unpublished data; but see also Stirling and O’Hare, 2006; Bailey et al., 2007; Llarena et al., 2010). There was minimal colocalization with the Golgi marker (Figure 4a, i) and little or no detectable nuclear protein. Both the DSG and 3S mutants showed slightly increased association with TGN46, although the majority of these mutants remained largely in an ER distribution. Nevertheless, distinct levels of nuclear protein were observed with each mutant above the background seen with the wt protein (Figure 4a, ii and iii, arrows). The result with the combined DSG+3S mutant was most striking, with almost complete loss of normal ER pattern, substantial association with TGN46, and high levels of nuclear protein (Figure 4a, iv, arrows).
FIGURE 4:

Serine substitution in the P-motif promotes constitutive ER–Golgi transport. (a) Subcellular localization of the precursor form of wt and DSG, 3S, and 3S+DSG mutants. (i–iv) Variants with intact cleavage sites for S1P and S2P. (v–viii) Variants with mutated cleavage sites. The S1P/S2P background status is indicated as wt or (S1P-/S2P−) at the left-hand side. Cells were transiently transfected and after 48 h fixed in 4% paraformaldehyde and processed as described in Materials and Methods for immunofluorescence using anti-SV5 epitope (red channel) and anti-TGN46 (green channel) primary antibodies. (b) Higher-magnification image for the typical Golgi pattern obtained for 3S+DSG mutant in the context of the S1P−/S2P− background. The 3S+DSG mutant in this context showed almost quantitative constitutive localization in the Golgi and the absence of any nuclear product.
Mutation of the S1P and S2P cleavage sites had little effect on localization of the wt protein (Figure 4a, v), consistent with previous results (Bailey and O’Hare, 2007; Llarena et al., 2010). However, the increased nuclear accumulation of the P-site mutants was completely abolished by disruption of the S1P/S2P cleavage sites (Figure 4a, vi and vii). Furthermore, whereas the individual 3S-(S1P−/S2P−) and DSG-(S1P−/S2P−) mutants were moderately increased in Golgi association over the wt (S1P−/S2P−) version, the combined DSG+3S-(S1P−/S2P−) mutant exhibited a striking change in localization, with prominent Golgi association together with complete loss of nuclear protein. In many cells, Golgi association represented almost the entire population of the mutant protein (Figure 4a, viii), more readily seen in Figure 4b, which shows an additional field with several cells expressing the mutant CREB-H, all exhibiting a consistent pattern of strong colocalization with TGN46 and complete loss of nuclear protein. Quantitative analysis of numerous cells for the 3S+DSG mutant in both S1P+/S2P+ and S1P−/S2P− backgrounds is given in Supplemental Figure S1. Supplemental Figure S1a shows representative images for classification purposes. The summary data (Supplemental Figure S1b) show, first, pronounced nuclear accumulation of the 3S+DGS (S1P+/S2P+) mutant, with >30% of cells showing an exclusively nuclear pattern (pattern 1, N), a similar percentage with majority nuclear plus cytoplasmic localization (pattern 3, N > C), and <20% showing a cytoplasmic-only pattern (pattern 2, C). In contrast, for the S1P−/S2P− version, >60% showed a cytoplasmic pattern (frequently with exclusive TGN46 associated), a minority showed some nuclear plus cytoplasmic localization, and none showed an exclusively nuclear protein. The results from Western blotting analysis of abundance, cleavage, and fractionation, taken together with these results from immunofluorescence analyses, strongly support the following conclusions: the 3S and DSG regions separately contribute to stability and localization of CREB-H, with combined mutations having a major effect; the cleavage and nuclear import of the P-motif mutants are completely dependent on the Golgi-associated proteases S1P and S2P; and the P-motif is a critical component of localization, such that its mutation results in efficient release and forward transport to the Golgi. In other words, the presence of the P-motif is important for ER retention and thus for regulation of the localization and activation of CREB-H.
GSK inhibitors suppresses phosphorylation, promoting Golgi transport, cleavage, and nuclear import of CREB-H
Within the P-motif, serines S73, S77, and S81 all match the GSK-3 kinase consensus sequence S/TxxxS/T (Doble and Woodgett, 2003; Cohen and Goedert, 2004). Furthermore, we previously demonstrated that the P-motif was indeed a site for GSK-3 phosphorylation of the cleaved product in the nucleus (Barbosa et al., 2015). Therefore we next sought to investigate whether GSK-3 also played a role in the observed phosphorylation of FL precursor CREB-H and in particular the P-motif, which was clearly involved in transport and processing of CREB-H in the cytoplasm.
We first tested the effect of the GSK-3 inhibitor LiCl on the expression profile of CREB-H (Figure 5a). LiCl, or NaCl as control, was added to CREB-H–transfected cells for various times (1, 2, or 4 h) before harvesting and analysis by SDS–PAGE and Western blotting. Treatment with LiCl had several specific effects. First, there was an overall increase in the abundance of twofold to threefold, more obvious at 2–4 h (compare lanes 4–6 and 7–9). Second, combined with this increase in abundance, we observed a relative shift in CREB-H mobility, to faster-migrating species (indicated by the two upper arrows on the right) and a loss of the more slowly migrating forms, consistent with a loss of phosphorylation. Third, in cells treated with LiCl but not with NaCl, we also observed the production of the CREB-H cleavage product (Figure 5a, Cl, arrows on right-hand side, lanes 3, 6, and 9), consistent with the proposal that LiCl induces both loss of phosphorylation and increased processing to the cleaved form.
FIGURE 5:

GSK-3 inhibition induces cleavage of CREB-H and reduces phosphorylation at serines 73 and 81. (a) COS-1 cells transfected with the vector expressing the precursor form of CREB-H under the control of a TK promoter were untreated or treated with 20 mM of either NaCl or LiCl for 1 h (lanes 1–3), 2 h (lanes 4–6), or 4 h (lanes 7 and 8). Extracts of equal cell equivalents were analyzed by Western blot using the anti-SV5 antibody and anti–γ-tubulin as a loading control. (b, c) Extracts were analyzed by Western blot for the presence of phosphate at serines 73 and 81 of CREB-H using the anti-SV5, anti-pS73 (b, lanes 1–7), and anti-pS81 (c, lanes 1–7) antibodies as described in Materials and Methods. FL refers to the bands of the precursor form of CREB-H and Cl to the cleavage product of CREB-H. Asterisk indicates a nonspecific band detected by the antibody.
We next examined the effect of LiCl specifically on phosphorylation within the P-motif, using the phosphospecific antibodies anti-pS73 and anti-pS81, probing Western blots as before but now simultaneously using the anti-SV5 antibody (total CREB-H) and the anti-pS73 and anti-pS81 antibodies (Figure 5, b and c). Within 1 h of treatment with LiCl, we observed a specific and selective loss in phosphorylation at both serines 73 and 81 (Figure 5, b and c, lanes 1 and 2). After 2 and 4 h of LiCl treatment (lanes 4 and 6), the total level of CREB-H had increased, and the mobility shifted with a relative increase in faster-migrating forms (SV5, green channel and white arrows). Although the phosphorylated species appear to have recovered, in fact, from quantitative measurement of the ratios of total versus pS73 or pS81 signals, the relative levels of phosphorylation at S73 and S81 remained suppressed (see also Supplemental Figure S2). Figure 5, b and c, bottom, again demonstrates the appearance of the cleavage product of CREB-H in LiCl-treated cells. These cleaved products could not be detected with the anti-phosphospecific antibodies due to the comparatively lower absolute levels of the cleaved products. Nevertheless, in keeping with the results for the FL precursor, we expect that LiCl also suppresses phosphorylation of the cleaved product, and indeed we showed this to be the case in experiments with overexpressed cleaved CREB-H in isolation (Barbosa et al., 2015).
To confirm and extend these results, we next used the more potent and specific GSK-3 inhibitor CHIR99021 (CHIR, Figure 6). Similarly to the results obtained with LiCl, treatment with CHIR resulted in an overall increase in abundance of CREB-H, cleavage of the precursor, and production of the cleaved product (Figure 6a, lanes 2 and 3). We also compared the effect of CHIR with inhibitors of additional kinases (Figure 6, b and c), carrying out simultaneous analysis of total CREB-H and phosphorylation at serine 73 (pS73). Treatment with either CHIR or SB16763 (SB), the two selective GSK-3 inhibitors, resulted in a specific suppression of phosphorylation at S73 coupled with the appearance of the cleavage product (Figure 6, b and c, lanes 2 and 6). Although not always detectable, treatment with CHIR resulted in a slight downward shift in the overall migration pattern of the FL CREB-H species (Figure 6, a and b, SV5, lane 2), resulting in the appearance of faster-migrating species (white arrows, SV5, lane 2). Of interest, although we routinely observed an increase in the abundance of CREB-H when cells were treated with CHIR, this was not the case with SB16763, which, if anything, resulted in a decrease in the FL precursor, although still inducing appearance of the Cl product (lanes 1, 2, and 6). An increase in CREB-H abundance was observed for cells treated with the PI3K inhibitor LY29400, but without any suppression in phosphorylation at S73. No changes were detected after treatment with wortmannin (a nonspecific inhibitor of PI3K) or tetrabromocinnamic acid (TBCA; a selective casein kinase 2 inhibitor). The effect of LY29400 has not been investigated further.
FIGURE 6:

The selective GSK-3 inhibitor CHIR99021 induces cleavage of CREB-H and inhibits phosphorylation at serine 73. (a) Transfected COS-1 cells expressing the precursor form of CREB-H under control of a TK promoter were untreated (–) or treated (+) with 10 µM CHIR99021 (6 h) after an incubation of 16 h in 5% serum. Equal cell equivalents were analyzed by Western blot with the anti-SV5 antibody. FL indicates the precursor form of CREB-H and Cl the cleavage product. (b, c) Cells expressing CREB-H, as described, were either untreated (–) or treated with CHIR99021 (10 μM, CHIR), LY294002 (10 μM, LY), wortmannin (0.2 µM, Wort), TBCA (10 μM), or SB216763 (20 μM) for 6 h. Extracts were analyzed by Western blot using anti-SV5 antibody and simultaneously with anti-pS73 antibody. Image in b shows the precursor form of the protein; c shows higher-sensitivity scans for detection of the cleavage product.
We next investigated the effect of CHIR on localization of the wt precursor form of CREB-H and its S1P−/S2P− mutated form (Figure 7). In untreated control cells (Figure 7, a and b, –CHIR), wt CREB-H exhibited typical ER localization, with some minor nuclear localization and some colocalization with the Golgi marker TGN46 (Figure 7a, I). This limited localization outside the normal ER pattern may be the result of the treatment of cells in lower serum (controls in the absence of CHIR were performed in low serum, as were CHIR treatments). Nonetheless, when cells were treated with CHIR, there was a striking change in localization, and in the majority of cells, CREB-H was found in significantly increased amounts in the Golgi together with prominent localization in the nucleus (Figure 7a, II, arrows). The effect of CHIR on CREB-H localization was even more dramatic when analyzing the uncleavable S1P−/S2P− variant. In untreated cells, this mutant exhibits the typical ER localization pattern, with a small fraction colocalizing in the Golgi compartment (Figure 7b, III), again possibly likely due to low serum treatment. In contrast, after CHIR treatment, the majority of cells showed a virtually exclusive perinuclear pattern, strongly colocalizing with the TGN46 Golgi marker (Figure 7b, IV) and a complete absence of the CHIR-induced nuclear population seen with the cleavable parental form.
FIGURE 7:
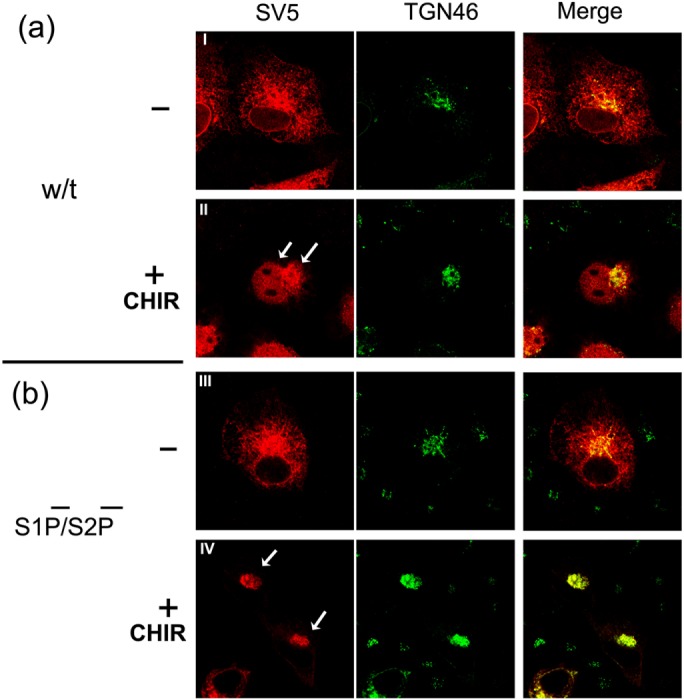
Inhibition of GSK-3 with CHIR99021 stimulates ER–Golgi trafficking of CREB-H. (a, b) Subcellular localization of CREB-H in normal S1P/S2P background (a) or S1P/S2P-ve backgrounds (b). COS-1 cells were transfected with a plasmid expressing wt CREB-H or the S1P−/S2P− form under the control of a TK promoter. Medium was replaced at 32 h with medium containing 5% serum for 16 h, followed by treatment without (I and III) or with CHIR99012 (II and IV) added at 10 μM for 6 h. Cells were fixed in 4% paraformaldehyde and stained with anti-SV5 (red channel) and anti-TGN46 (green channel) primary antibodies. Arrows indicate features as discussed in the text.
In summary, the P-motif contains consensus sites for GSK-3 phosphorylation. Serine substitution within the motif or inhibition of GSK-3 activity promotes concomitant alterations in CREB-H migration patterns in SDS–PAGE, suppression of phosphorylation (including S73), and increased transport, processing, and nuclear localization of the protein. Taken together, therefore, these results strongly reinforce the conclusion that inhibition of GSK-3 kinase promotes CREB-H ER–Golgi traffic and subsequent processing via suppression of P-motif phosphorylation, including at least serine 73. The corollary to this is that active GSK normally promotes ER retention of CREB-H and the suppression of its transport and activation pathway.
GSK phosphorylation of full-length CREB-H
We next examined direct phosphorylation of CREB-H by GSK-3. To that end, we first extracted proteins from cells expressing CREB-H under denaturing conditions to dissociate any interacting proteins and inactivate any cellular enzymes. Extracts were then diluted to appropriate buffer conditions and incubated without or with phosphatase to remove any preexisting CREB-H phosphorylation, then immunoprecipitated with anti-SV5 to capture CREB-H, blotted, and subsequently probed with anti-SV5 for total CREB-H or anti-pS73 as a specific readout for P-site–phosphorylated species (Figure 8, top). Consistent with the earlier results (Figure 1), phosphatase treatment of CREB-H resulted in a loss of the upper species and a downward shift in overall migration patterns (SV5, lanes 2 and 4) compared with samples without added phosphatase (SV5, lanes 1 and 3 vs. 2 and 4). In samples treated with phosphatase, there was an almost complete loss of phosphorylated S73 as detected by the phosphospecific antibody (pS73, lanes 2 and 4). These immunoprecipitated samples were then incubated without or with GSK-3β kinase (Figure 8, bottom, lanes 1 and 2 and lanes 3 and 4, respectively). In the absence of added GSK-3β kinase (lanes 1 and 2), the samples without or with prior phosphatase treatment did not change in migration pattern or in the status of serine 73 phosphorylation (bottom, lanes 1 and 2). However, when the immunoprecipitated CREB-H was incubated with GSK-3β kinase, we observed a very substantial increase in S73 phosphorylation in the sample that had been subject to prior phosphatase treatment (Figure 8, bottom, lane 2 vs. lane 4). Moreover the newly phosphorylated pS73 species (red channel) migrated more with the upper species, as seen in the merged channel (lane 4). Although GSK-3β had phosphorylated CREB-H on S73 (and possibly elsewhere), the overall migration pattern of the protein (detected by anti-SV5) did not visibly shift upward, indicating that only a minor proportion of the overall population was phosphorylated. Nevertheless, these results demonstrate that GSK-3β phosphorylates CREB-H in vitro and does indeed phosphorylate serine 73.
FIGURE 8:
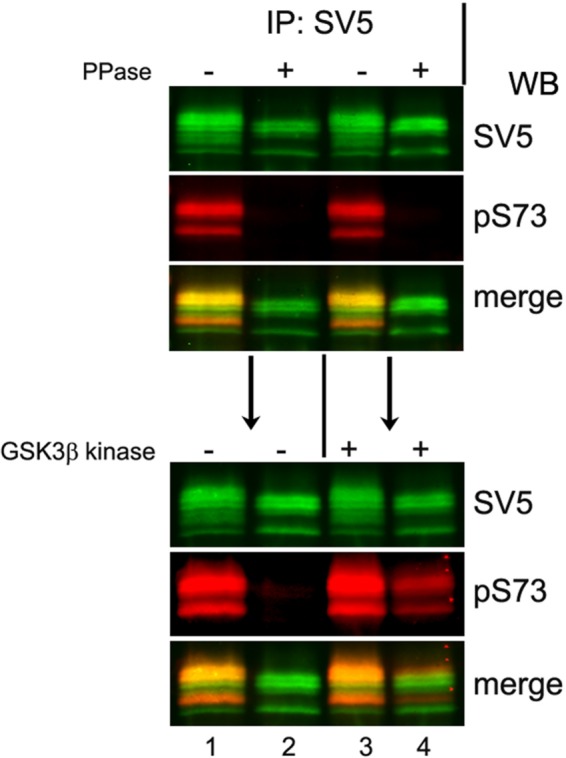
GSK-3β kinase directly phosphorylates the P-motif of CREB-H. GSK-3β directly phosphorylates the precursor form of CREB-H in vitro. COS cells transfected with a plasmid expressing wt CREB-H under the control of the TK promoter were harvested and lysed under denaturing conditions as described in Materials and Methods. Samples were diluted into the appropriate buffer and incubated without or with phosphatase (PPase) as indicated. CREB-H was then subsequently immunopurified from these extracts, further incubated without (lanes 1 and 2) or with (lanes 3 and 4) purified GSK-3β kinase, and finally analyzed by Western blot with anti-SV5 epitope and phosphospecific anti-pS73 primary antibodies.
Constitutive activation of transcription of a key target, ApoA-IV, by substitution of the P-motif in full-length CREB-H
We next investigated the effect of the GSK-3 inhibitors on CREB-H–promoted secretion of a known prominent target gene, ApoA-IV (Lee et al., 2011; Zhang et al., 2012; Barbosa et al., 2013, 2015), with the expectation that GSK inhibition would increase ApoA-IV secretion. However, although GSK-3 inhibitors unequivocally induced CREB-H translocation to the Golgi and the nucleus, the results measuring ApoA-IV secretion in response to these inhibitors were inconclusive and generally failed to demonstrate increased activity. We believe that this may be due to pleiotropic effects of GSK-3 inhibitors on any number of downstream cellular functions with which this multifunctional kinase is associated (Doble and Woodgett, 2003). An alternative approach using knockdown of GSK-3 by small interfering RNA yielded either insufficient knockdown GSK-3 in our cells or limited activation of ApoA-IV secretion. Therefore, in another approach, we examined the effect of the P-motif mutations on CREB-H transcriptional activity and secretion of the native ApoA-IV. First, the promoter was fused to luciferase in pGL3-ApoA-IV-LUC and the target construct cotransfected with wt FL CREB-H or the various P-site mutants (Figure 9a). Whereas limited activation of Apo A-IV could be detected for wt CREB-H, in comparison, we observed extremely potent activation (>300-fold) for the 3S+DSG variant. The DSG and 3S variants also promoted very significant activation (∼200- and 100-fold, respectively). Of importance, when the S1P and S2P cleavage sites were mutated (S1P−/S2P− variants of wt, 3S+DSG, and DSG), activation was almost completely abolished, with only weak residual activity, which may result from independent or nonspecific cleavage. These results are entirely consistent with those from localization studies, demonstrating the increased Golgi and nuclear localization of the P-site mutants and the major reduction in nuclear transport upon S1P/S2P-site inactivation.
FIGURE 9:

Serine substitution in the P-motif enhances transcriptional activation and secretion of ApoA-IV. (a) HepG2 cells were cotransfected in triplicate with 50 ng of pGL3-ApoAIVpr-LUC and 500 ng of plasmids expressing wt or P-motif mutants in the context of normal or S1P/S2P-ve backgrounds as indicated. After 48 h, cells were extracted and luciferase assays performed as described in Materials and Methods. Values represent mean fold activation over the control samples, with error bars indicating SDs. (b, c) HepG2 cells were transfected with plasmids expressing wt and P-motif mutants of CREB-H. Medium was replaced at 48 h with serum-free medium for a further 24 h. The medium was then collected from equivalent cell numbers, and secreted proteins were precipitated and analyzed by Western blot for ApoA-IV as described in Materials and Methods. (b) Cellular protein extracts harvested and analyzed for detection of the CREB-H variants (with rabbit anti–γ-tubulin as a loading control). (c) Corresponding levels of secreted ApoA-IV in the medium. FL indicates the CREB-H precursor form and Cl the cleavage product.
Second, we compared the wt and the P-motif mutants in activation studies of the endogenous native ApoA-IV gene in HepG2 cells (Figure 9, b and c) by measuring secretion of the protein product. To that end, we transfected HepG2 cells with the different phosphorylation variants and after 48 h removed the medium and continued incubation in fresh serum-free medium serum for 24 h. We then analyzed cell extracts for expression of the various CREB-H proteins (Figure 9b) and the corresponding medium for levels of secreted ApoA-IV protein (Figure 9c). The results (Figure 9b), first, for CREB-H expression, migration patterns, and cleavage were completely consistent with those described earlier (Figure 2). Second, consistent with results of ApoA-IV promoter activation, the 3S+DSG variant promoted high levels of secretion of ApoA-IV protein (Figure 9c, lane 3) compared with the modest levels for wt CREB-H (lane 2), which were only marginally above that seen in controls from empty vector–transfected cells (lane 1). The DSG and the 3S variant again showed reduced activity compared with the combined mutant but were significantly above those seen for the wt protein (lanes 1–5). Of importance for the 3S+DSG variant, mutation of the S1P/S2P sites (lane 6) resulted in almost complete abolition of ApoA-IV secretion. In this experiment for the 3S+DSG(S1P−/S2P−) mutant, we noted the appearance of a product migrating above the normal cleaved band obtained for the 3S+DSG mutant. This product was not routinely observed and may represent a nonspecific cleavage product, because we did not detect nuclear species, significant luciferase activation, or ApoA-IV secretion.
Although each line of evidence, including the presence of consensus GSK sites within the P-motif, the consequences of P-motif mutation for CREB-H localization and cleavage, the corresponding effects of GSK-3 inhibition on localization and cleavage, and finally the effect of P-motif mutation on target gene activation, supports our proposal, taken together, these data provide robust support for the proposal that CREB-H is phosphorylated by GSK-3 and that such phosphorylation regulates CREB-H ER–Golgi intercompartmental trafficking, cleavage, and ultimately nuclear activity.
DISCUSSION
CREB-H plays a key role in lipid and triglyceride metabolism (Lee et al., 2010, 2011; Lee, 2012; Zhang et al., 2012; Barbosa et al., 2013; Xu et al., 2014). Several distinct physiological stimuli, including fasting, high-fat diet, saturated fatty acids, and insulin, have been reported to increase transcription of CREB-H and promote its activation. However, we have comparatively little mechanistic understanding of how these metabolic cues regulate CREB-H, especially the key pathway of intercompartmental trafficking from the ER to the Golgi and associated cleavage and nuclear import, which together ultimately control its activity.
In this work, we demonstrate that a serine-rich region in the N-terminus of CREB-H, the P-motif, is a key determinant by which CREB-H activity is controlled. We provide evidence that this motif is involved in the regulation of the ER–Golgi traffic and consequential cleavage and nuclear localization. We propose that phosphorylation by GSK-3 at the P-motif promotes CREB-H ER retention and suppresses its transport and processing and present a model for dual integrated control, coupling regulation of intercompartmental transport within the cytoplasm with stabilization of the transcriptionally active form in the nucleus.
Multiple lines of evidence provide robust support for these conclusions. First, we show that the precursor form of CREB-H is constitutively phosphorylated, including at least serines 73 and 81 of the P-motif. Second, serine-to-alanine substitutions in the 3S region of the P-motif result in a migration shift of the protein and the appearance of the processed product. Simultaneous substitution of serines in the DSG region of the P-motif causes a substantial increase in the stability of the protein. Third, localization studies demonstrate that serine substitution in the P-motif results in S1P/S2P-dependent cleavage and nuclear import. Fourth, when assessing localization in the context of the S1P/S2P uncleavable mutant, the 3S+DSG mutant shows extremely efficient and specific localization in the Golgi, strongly supporting the proposal that this mutant no longer undergoes default ER localization and traffics to the Golgi. Fifth, treatment of cells with GSK-3 inhibitors results in the loss of phosphorylation at serines 73 and 81, combined with the appearance of the cleavage product. CHIR99021 treatment, in particular, induced CREB-H localization to the Golgi and nucleus, with the S1P/S2P uncleavable mutant accumulating almost quantitatively in the Golgi. Sixth, in in vitro assays, immunopurified CREB-H could be phosphorylated by GSK-3β kinase at serine 73. Finally, consistent with the induced transport and nuclear import of CREB-H, the phosphorylation mutants of CREB-H exhibit significantly increased transcriptional activation of the ApoA-IV promoter, and cells expressing these different variants also show increased secretion of endogenous ApoA-IV protein.
CREB-H ER–Golgi transport
The conclusion that phosphorylation at the P-motif regulates ER–Golgi transport of CREB-H must be integrated into a model encompassing two previous observations from our work—first, that another determinant, the membrane-proximal ERM determinant is a key determinant involved in ER retention, and second, that phosphorylation also regulates stability of the nuclear form of the protein.
With regard to the first aspect, previous work from our laboratory identified the ERM, adjacent to the transmembrane domain on the cytosolic side, as a key determinant promoting ER retention (Figure 10, ERM). Deletion of the ERM promotes efficient constitutive transport of CREB-H to the Golgi, independently of the status of the P-motif (Bailey and O’Hare, 2007; Bailey et al., 2007; Llarena et al., 2010). Thus we propose a model in which these two regions—the P-motif and the ERM—are involved together in controlling intracellular localization of CREB-H. Although there are several possible (not mutually exclusive) models to integrate our findings, in one model, the P-motif and ERM motif are interconnected, with the P-motif providing a regulatory role, controlled by phosphorylation/dephosphorylation cycles, that modulates function at the ERM, promoting its interaction with auxiliary factors that control ER retention. This could occur via formation of a compound, physically integrated P-motif/ERM site or by inducing a specific presentation of the ERM as an independent site.
FIGURE 10:

Integrated model for the role of the P-motif in phosphorylation, trafficking, and nuclear stability of CREB-H by insulin signaling. The FL precursor form of CREB-H is anchored at the ER membrane, indicating the ER luminal domain, transmembrane domain, and the N-terminal domain facing the cytosol. The small open circles containing a cross illustrate the two sites for cleavage by the Golgi proteases S1P and S2P. In the N-terminal domain, features include the ERM motif, previously identified as an ER-retention motif that encompasses determinants involved in S2P cleavage; the DNA-binding domain, illustrated by a cylinder; and the P-motif, shown as a target site for GSK-3 phosphorylation and subject to activation by insulin signaling. When GSK-3 is active (1), CREB-H is constitutively phosphorylated at the P-motif. In model a, this promotes the function of a physically independent ERM motif and its association with a proposed ER-retention factor (labeled factor X). In model b, the phosphorylated P-motif interacts with the ERM determinant to form a physically integrated determinant that recruits factor X. When GSK-3 is suppressed—for example, by insulin signaling (2)—the P-motif loses its phosphates, the ERM interaction is also suppressed, factor X dissociates, and CREB-H is released from the ER and transported to the Golgi, where it will undergo processing, releasing the transcriptional active N-terminus of the protein in the cytoplasm (3). In a coordinated manner, the now-underphosphorylated N-terminal domain is stabilized due to reduced targeting to SCF-mediated degradation (4), promoting CREB-H activity, until it is eventually degraded or rephosphorylated when the duration of the signaling events has waned due to homeostatic correction, restoring both retention of precursor CREB-H in the ER and turnover of the previously active nuclear form.
Thus, via constitutive GSK-3 activity, the phosphorylated P-motif and interaction with the ERM region (whether direct or indirect) would promote binding of cell factor(s) involved in ER-retention. On suppression of GSK-3 activity by extracellular stimuli, including, for example, insulin, or by activation of specific phosphatases or de novo synthesis of CREB-H in the absence of phosphorylation, the unphosphorylated P-motif would not communicate with the ERM, and ER-retention factors would not be recruited. CREB-H would then be recognized and incorporated by the vesicular transport machinery, potentially in a constitutive manner, for trafficking to the Golgi. In a variant of this model, ER retention would be the default, not requiring specific retention factors. Instead, only the unphosphorylated motif would recruit positively acting transport factors that promote forward transport to the Golgi. Although possible, this model fits less well with our observations, in particular that, for the wt protein, ER localization is the default, and, by deletion of the ERM, the result is very efficient forward transport to the Golgi. Although further complexity will inevitably arise (see later discussion), our model thus provides a mechanistic understanding for the previously demonstrated effect of insulin in promoting CREB-H cleavage (Zhang et al., 2012)
Integrating the CREB-H P-motif in ER–Golgi transport and nuclear stabilization
Previously, examining the mature cleaved form of CREB-H, we identified the P-motif as including a DSG-type phosphodegron for the Fbw1a F-box protein and SCF-mediated degradation of the nuclear protein (Barbosa et al., 2015). Here we observe a significant stabilization of the precursor form of CREB-H when we substitute the serines to alanine in the DSG region of the P-motif, implying a similar role of this region at the ER. Similar dual-compartmental regulation by an F-box protein has also been reported for SREBP1c, for which Fbw7a controls the levels of both the precursor in the cytoplasm and nuclear form of the protein (Sundqvist et al., 2005; Dong et al., 2015). We considered the effect of the P-motif mutation—in particular, the DSG variant—on the stability of the nuclear product and the possibility that the increased transport and appearance of the cleaved product were mainly due to its increased stability rather than increased transport out of the ER. This was ruled out by our analysis of the P-motif mutants in the background of uncleavable S1P−/S2P− mutant. Thus the P-motif plays a role in both cytoplasmic transport and stabilization of the nuclear product.
Of interest, the DSG mutant exhibited elevated levels of the nuclear product compared with the 3S mutant, which showed greater Golgi localization in the context of the S1P−/S2P− mutant. This suggests that different serines within the motif are differentially involved in transport of the precursor or stability. For example, the 3S mutant might exit the ER more efficiently but has a faster turnover in the nucleus, resulting in overall lower levels. The 3S region is characterized by a consensus sequence recognized by GSK-3, S/TxxS/TP (Doble and Woodgett, 2003; Cohen and Goedert, 2004), in which serines 73, 77, and 81 each represents a potential target for GSK-3 phosphorylation. Clearly, the combination of serine substitutions in both motifs (3S+DSG) had a profound effect on transport and cleavage, with the majority of the protein being localized in the Golgi and nucleus, and in the context of an S1P−/S2P− mutant accumulating substantially at the Golgi.
Certain features of our results require further investigation. Chemical inhibition of GSK-3 had a more pronounced effect in CREB-H transport and processing than serine-to-alanine substitutions in the 3S region of the P-motif. One possible explanation might be that besides the serine 73, 77, or 81 of CREB-H, GSK-3 can also phosphorylate another serine or threonine in CREB-H or GSK-3 can inhibit/phosphorylate another protein or proteins that are implicated in CREB-H ER–Golgi transport. Although additional serines in the DSG region seem obvious alternative candidate sites, our previous in vitro assays did not show any decrease in GSK-3 phosphorylation for the DSG mutant and instead found that a different kinase, CKII kinase, phosphorylated it upon priming by GSK-3 phosphorylation at the 3S motif (Barbosa et al., 2015). Therefore, for CREB-H located at the ER, it is possible that phosphorylation in the 3S motif might prime for phosphorylation at the DSG motif.
The precise mechanism of regulation of intercompartmental transport by phosphorylation remains to be elucidated. For example, given that we previously showed that CREB-H is subject to retrotranslocation and ER-associated degradation (ERAD; Bailey et al., 2007), it could be that phosphorylation is required to retain the protein in that pathway and that loss of phosphorylation causes stabilization, which, as a consequence, increases forward transport to the Golgi. Alternatively, loss of phosphorylation could, as the primary event, drive the protein into the vesicular transport pathway, and stabilization results from a redirection away from ERAD. However, we note that proteasome inhibition does not of itself result in significant Golgi accumulation. The opposite is the case: it results in accumulation in the deglycosylated state in the cytoplasm (Bailey et al., 2007). In addition, overexpression of the wild-type protein, employing routinely used CMV promoters, also does not result in significant Golgi accumulation. Therefore our results show that phosphorylation and dephosphorylation are key to transport, with the data clearly demonstrating that both the 3S and the DSG serines/regions of the P-motif contribute to the retention of the protein at the ER by rendering the protein subject to ERAD and/or inhibiting its vesicular transport.
The model has clear similarities to that reported for SREBPs, which are regulated by multiple positive and negative phosphorylation events in response to metabolic cues, including cholesterol, fatty acids, and insulin (Xu et al., 1999; Horton et al., 2002; Eberle et al., 2004; Bengoechea-Alonso and Ericsson, 2009). GSK-3 phosphorylates SREBP-1a, promoting retention in the ER and targeting both the precursor and the nuclear cleaved form for proteasomal degradation (Kim et al., 2004; Sundqvist et al., 2005; Li et al., 2011; Dong et al., 2015). Insulin, in addition to transcriptional activation of SREBP1, promotes its transport and processing (Hegarty et al., 2005). The simplest model therefore is that insulin, possibly via PI3-K/AKT suppression of GSK phosphorylation, results in loss of ER retention, increased transport, and cleavage, although this may also occur via additional kinase pathways, including AMP-activated protein kinase (Hegarty et al., 2005; Yellaturu et al., 2005, 2009; Li et al., 2011). Regulation of SREBP activation in response to various metabolic signatures clearly involves multiple kinase pathways, including, in addition to those already mentioned, the mammalian target of rapamycin complex (Krycer et al., 2010; Owen et al., 2012). Furthermore, apart from phosphorylation of SREBP itself, insulin represses expression of certain of the factors involved in ER retention, including Insig-2, providing additional routes that could liberate the SREBP-1c/SCAP complex for transport to the Golgi (Yabe et al., 2003; Yellaturu et al., 2009; Dong et al., 2012). Therefore it is conceivable, if not likely, that similar multimodal regulation by insulin might occur for CREB-H, in which insulin regulates phosphorylation at the P-motif through multiple kinases. Similarly to the SREBP-1c, insulin signal could also target the proposed ER retention factor (i.e., equivalent to SCAP). It is also possible that insulin could also target either a phosphatase that dephosphorylates CREB-H or another signal that regulates de novo synthesis of CREB-H to yield a dephosphorylated protein.
Furthermore, like SREBPs, the regulation of CREB-H activity will undoubtedly be complex, and apparently opposing stimuli have been reported to produce similar effects on CREB-H. Thus, on one hand, fatty acids, insulin, and an atherogenic, high-fat diet stimulate CREB-H liver expression and processing. On the other hand, fasting has also been reported to stimulate CREB-H expression and activity (Danno et al., 2010; Gentile et al., 2010; Lee et al., 2010; Zhang et al., 2012; Kim et al., 2014; Nakagawa et al., 2014). Thus, as for SREBP, metabolic regulation of CREB-H is likely to be complex and multifactorial, involving positive and negative homeostatic controls that respond to complex overlapping environmental cues.
Nevertheless, the present study provides the first mechanistic insight into the regulation of another type of transcription factor, CREB-H, which is now recognized, along with SREBPs, to be an important homeostatic regulator of triglyceride and lipid metabolism. We identify specific residues within a key motif that exerts coordinated dual control at both cytoplasmic trafficking levels and in nuclear stability, and we propose an integrated homeostatic circuit for regulation of its activity. Further work can be directed at identification of factors that differentially interact with CREB-H either in a phosphorylation-dependent manner or requiring the presence of the ERM in order to dissect how apparently distinct metabolic cues influence P-motif phosphorylation, transport, cleavage, and stability.
MATERIALS AND METHODS
Cells
HepG2 cells and Huh7 cells were grown on collagen-coated plates in MEM containing 0.1 mM nonessential amino acids (NEAA), 10% fetal bovine serum (FBS), 2 mM l-glutamine, 1 mM sodium pyruvate, and penicillin and streptomycin at 100 U/ml and 100 μg/ml, respectively (Pen/Strep). COS-1 cells were grown in DMEM supplemented with 10% newborn calf serum (NBCS) and Pen/Strep. Huh7 cells were cultured in DMEM, supplemented with 10% FBS, 0.1 mM NEAA, and Pen/Strep. For standard assays, HepG2, Huh7, and COS-1 cells were cultured at 37˚C in a 5% CO2 environment under standard conditions.
Chemicals and inhibitors
Protein stability and half-life were assayed in Huh7 cells using cycloheximide (Sigma- Aldrich, Gillingham, United Kingdom) at a concentration of 100 µg/ml. Brefeldin A (Sigma-Aldrich) was prepared as 10 mg/ml stock in methanol and added to cells at a final concentration of 1 μg/ml for 30 or 60 min. MG132 was prepared at a concentration of 20 mM in dimethyl sulfoxide (DMSO) and added to cells at a final concentration of 10 µM. Kinase Inhibitors CHIR99021 (Tocris Biosciences, Bristol, United Kingdom), LY29400 (Calbiochem, Nottingham, United Kingdom), and TBCA (Calbiochem) were prepared at 10 mM in DMSO and added at 10 µM. SB216763 (Tocris Biosciences) was prepared at 20 mM in DMSO and added at 20 μM. Wortmannin (Calbiochem) was prepared at 10 mM and added at 0.2 μM. For the treatment with kinase inhibitor, cells were incubated in 5% serum before treatment.
Plasmids
A series of plasmids expressing the SV5 epitope–tagged truncated nuclear mutant form of CREB-H in which individual or combined serines were substituted by alanines was constructed by site-directed mutagenesis or PCR-based mutagenesis as described in Barbosa et al. (2015). From these CREB-H truncated variants of the various mutations—3S (S73, 77, 81A; pSB33), DSG (S87, 90A; pDJB136), and 3S+DSG (S73, 77, 81, 87, 90A; pSB31)—we constructed the corresponding full-length mutants used here. To that end, we introduced the DNA region that codes for the transmembrane domain and C-terminus ER luminal domain of CREB-H into each of the plasmids containing the different serine substitutions. This was achieved by replacing the region between the ClaI and XbaI restriction sites for the corresponding fragment from the plasmid expressing the parental SV5 epitope–tagged CREB-H FL protein (pDJB123) as previously described (Bailey et al., 2007; Llarena et al., 2010). This yielded full-length CREB-H variants 3S (S73, 77, 81A; pSC14), DSG (S87, 90A; pDJB135), and 3S+DSG (S73, 77, 81, 87, 90A; pSB35). We subsequently generated the corresponding S1P−/S2P− mutants of these serine variants using the same methodology but with the fragment introduced derived from the plasmid expressing CREB-H S1P−/S2P− protein (pDJB146; Bailey et al., 2007; Llarena et al., 2010). The resulting S1P−/S2P− variants were named pSC21 (3S), pSB32 (DSG), and pSB34 (3S+DSG).
The luciferase reporter vector for one of the key CREB-H targets, ApoA-IV, contained the promoter region from −2399 to +85 and has been previously described (Barbosa et al., 2015).
Transfections
Where more robust expression was required for biochemical analysis (e.g., for in vitro kinase phosphorylation assays or detection with anti-phosphospecific antibodies), transfections were routinely performed in COS-1 cells as previously described (Llarena et al., 2010; Barbosa et al., 2013, 2015) using Genejammer (Agilent Technologies, Cheshire, United Kingdom), FuGENE (Promega, Southampton, United Kingdom), or calcium phosphate transfection reagents according to the manufacturer’s instructions. Considering that CREB-H is efficiently expressed in the liver (as well as the small intestine), in certain experiments as indicated, HepG2 or Huh7 liver cells were employed, although these generally yielded less well resolved images for colocalization studies in the ER and Golgi than the flatter, larger COS cells. In addition, because we obtained somewhat higher levels of CREB-H in Huh7 cells than HepG2 cells, we used Huh7 cells in more demanding assays such as those for half-life using cycloheximide. Independent of cell line used, we routinely transfected 0.5–1 μg of the appropriate expression vector with amounts of DNA normalized using pUC19 carrier DNA where needed.
Phosphatase treatment
Cells transfected with expression vectors for wt CREB-H FL protein were washed in ice-cold phosphate-buffered saline (PBS) and harvested by being scraped from the dishes, pelleted, and resuspended in λ-phosphatase buffer (New England Biolabs, Hitchin, United Kingdom) containing 0.4% NP-40, 0.5% Triton X-100, protease inhibitors (1× Complete Protease; Roche), and 1 mM phenylmethylsulfonyl fluoride (PMSF). As controls, two aliquots of the sample were treated with orthovanadate (10 mM) and sodium fluoride (20 mM) and incubated at 37°C either with or without added phosphatase for 1 h to reflect the starting material before incubation with phosphatase alone. Parallel samples were then incubated with 400 U of λ-phosphatase and incubated at 37°C for 1 h. The reactions were terminated by addition of SDS sample buffer to 1× and boiling for 5 min. Samples were analyzed by SDS–PAGE and Western blotting.
Western blot analysis
SDS–PAGE and Western blot analysis were performed exactly as described previously (Barbosa et al., 2015). Antibodies used were as follows. Primary antibodies were mouse anti-V5, (1:10,000; Life Technologies), rabbit anti–actin 20-36 (1:2500; Sigma-Aldrich), mouse anti–actin AC-40 (1:2500; Sigma-Aldrich), rabbit anti–γ-tubulin (1:2500; Sigma-Aldrich), anti–ApoA-IV 1D6B6 (1:2000; Cell Signaling, Leiden, Netherlands), rabbit phospho-GSK-3α/β(Ser-21/9) D17D2 (1:2000; Cell Signaling), and anti–CREB-H pS73 and pS81 (1:100; Barbosa et al., 2015). Secondary antibodies were conjugated with horseradish peroxidase (for detection by conventional chemiluminescence) or with Dylight680 or Dylight800 (Fisher Scientific, Lutterworth, United Kingdom) for detection by laser scanning using the LI-COR Odyssey Image system. The latter method was routinely employed for CREB-H detection using the phosphospecific antibodies because it allowed simultaneous analysis, on one blot, of total CREB-H species with the anti-mouse antibody to the epitope tag in one channel and the phosphorylated species with the anti-rabbit antibody to the phosphorylated peptide in a second channel.
Cell fractionation
Transfected cells were washed in ice-cold PBS, collected by scrapping in PBS, pelleted, and washed a further two times. Cell pellets were then gently resuspended in 1× PBS, 1% Triton, 0.5 mM sodium fluoride, 0.1 mM sodium vanadate, 0.5 mM PMSF, and 1× Complete, and proteins were extracted for 10 min at 4°C. Extracts were centrifuged at 3000 rpm at 4°C. The supernatant cytosolic fractions were made to 2% SDS in sample buffer and the samples boiled for 5 min. The pellets containing the nuclear fraction were washed once in the cell lysis buffer and resuspended in SDS sample buffer.
Transcription activation assays
HepG2 cells seeded on collagen-coated 24-well plates were transfected in triplicate with expression vectors for CREBH FL, CREB-H S1P−/S2P−, or mutants together with the target luciferase reporter vector, pGL3-hApoA-IV-LUC. For each assay, 50 ng of target luciferase vector, pGL3-hApoA-IV-LUC, was cotransfected with 500 ng of the activator vector or with 500 ng of pUC19 (control) to equalize for the DNA amount. Cell lysates were prepared 48 h after transfection by addition of Glo Lysis buffer (Promega). Firefly luciferase activity was assayed using the Bright-Glo assay system as described by the manufacturer, and activity was measured using a Victor luminescence reader (PerkinElmer). Assays were routinely performed in triplicate and readings calculated as fold increase relative to control.
Analysis of ApoA-IV secretion
ApoA-IV levels were analyzed as described previously (Barbosa et al., 2013, 2015). Medium was harvested from the cells and proteins precipitated with 15% trichloroacetic acid. Precipitates were centrifuged at 15,000 × g for 15 min and pellets washed twice with 100% ice-cold acetone or ethanol. Pellets were air-dried and subsequently resuspended in 2× SDS sample buffer. ApoA-IV levels were analyzed by SDS–PAGE and Western blotting as described earlier.
Immunofluorescence analysis
Immunofluorescence assays were performed exactly as described previously (Barbosa et al., 2013, 2015). Primary antibodies used were rabbit anti-SV5 (1:1000; Covance) and sheep anti-TGN46 (1:200; AbD Serotec, Kidlington, United Kingdom). Fluorochrome (Alexa 488, Alexa 594)–conjugated secondary antibodies of appropriate specificity (Fisher Scientific, Loughborough, United Kingdom) were diluted 1:200 in PBS/10% NBCS and added for 45 min. After washing, cells were mounted in Mowiol and visualized using a Zeiss LSM 410 or 510 Meta confocal microscope imaging system and a Zeiss Plan-Apochromat (63×, 1.4 numerical aperture) lens. Images for each channel were captured sequentially with fourfold or eightfold averaging at an image size of 512 × 512 or 1024 × 1024 pixels. Composite illustrations were prepared using Adobe software. Example images shown are representative of numerous images gathered for each test construct and condition.
In vitro GSK-3β kinase assays
COS-1 cells in 100-cm dishes were transfected with 6 μg of the expression vector for wt CREB-H (pDJB123) and harvested 48 h later. After two washes with 1× PBS buffer, the cell pellets were resuspended in a denaturing buffer (50 mM Tris, pH 7.5, 1% SDS, 5 mM dithiothreitol [DTT]) and boiled for 10 min. Lysates were then diluted in buffer containing 50 mM Tris, pH 7.5, 250 mM NaCl, 0.5% NP-40 containing 1× Complete EDTA-free protease inhibitor cocktail (Sigma-Aldrich), and 0.1 mM PMSF and cleared by centrifugation at 13,000 rpm. Samples were then incubated with either phosphatase inhibitors to prevent dephosphorylation (100 μM sodium orthovanadate, 5 mM glycerophosphate) or 400 U of λ-phosphatase (New England Biolabs) and 1 mM MnCl2. The samples were incubated at 37°C for 45 min with agitation and reactions stopped by addition of 5 mM of EDTA. CREB-H was then immunoprecipitated using the mouse anti-SV5 antibody (5 mg/ml) at a 1:100 dilution with overnight incubation, followed by addition of Protein G–Sepharose beads (Sigma-Aldrich) for 2 h. CREB-H–containing beads were centrifuged at low speed and extensively washed in buffer. This procedure resulted in phosphatase-treated or untreated CREB-H captured on beads by immunoprecipitation. Each of these samples was then divided in two. For one aliquot, phosphatase-treated (or untreated) CREB-H was directly eluted by boiling in SDS sample buffer. The other aliquot was incubated with GSK-3β kinase for analysis of GSK-3 phosphorylation. The beads were first equilibrated in the GSK-3β assay buffer (25 mM Tris-HCl, 12 mM MgCl2, 2 mM DTT, 100 μM sodium orthovanadate, 5 mM glycerophosphate). ATP was added at a concentration of 200 μM to all the samples, together with 50 ng of purified GSK-3β (Millipore, Watford, United Kingdom). Reactions were carried out for 45 min at 30°C with agitation and terminated by removing the buffer and boiling the beads in SDS sample buffer. Samples were then analyzed by SDS–PAGE, followed by Western blotting for total CREB-H with anti-SV5 antibody or for phosphorylated forms using anti-pS73 specific antibody as described.
Supplementary Material
Acknowledgments
This work was supported by the Medical Research Council, MR/K017926/1.
Abbreviations used:
- AKT
protein kinase B
- ATF6
activating transcription factor 6
- BFA
brefeldin A
- bZIP
basic and leucine zipper domain
- CBB
Coomassie brilliant blue
- CHX
cycloheximide
- CKII
casein kinase II
- COS-1
SV40 transformed African green monkey kidney cells
- CREB-H
cyclic AMP response element binding factor-H
- Cul1
Cullin 1
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- GSK3
glycogen synthase kinase 3
- LiCl
lithium chloride
- MG132
carbobenzoxy-l-leucyl-l-leucyl-l-leucinal
- NBCS
newborn calf serum
- RIP
regulated intramembrane proteolysis
- S1P
site-1 protease
- S2P
site-2 protease
- SCF
Skp, Cullin, F-box containing complex
- SREBP
sterol regulatory element–binding protein
- UPR
unfolded protein response
- wt
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-01-0075) on April 5, 2017.
REFERENCES
- Asada R, Saito A, Kawasaki N, Kanemoto S, Iwamoto H, Oki M, Miyagi H, Izumi S, Imaizumi K. The endoplasmic reticulum stress transducer OASIS is involved in the terminal differentiation of goblet cells in the large intestine. J Biol Chem. 2012;287:8144–8153. doi: 10.1074/jbc.M111.332593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey D, Barreca C, O’Hare P. Trafficking of the bZIP transmembrane transcription factor CREB-H into alternate pathways of ERAD and stress-regulated intramembrane proteolysis. Traffic. 2007;8:1796–1814. doi: 10.1111/j.1600-0854.2007.00654.x. [DOI] [PubMed] [Google Scholar]
- Bailey D, O'Hare P. Transmembrane bZIP transcription factors in ER stress signaling and the unfolded protein response. Antioxid Redox Signal. 2007;9:2305–2321. doi: 10.1089/ars.2007.1796. [DOI] [PubMed] [Google Scholar]
- Barbosa S, Carreira S, Bailey D, Abaitua F, O'Hare P. Phosphorylation and SCF-mediated degradation regulate CREB-H transcription of metabolic targets. Mol Biol Cell. 2015;26:2939–2954. doi: 10.1091/mbc.E15-04-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa S, Fasanella G, Carreira S, Llarena M, Fox R, Barreca C, Andrew D, O'Hare P. An orchestrated program regulating secretory pathway genes and cargos by the transmembrane transcription factor CREB-H. Traffic. 2013;14:382–398. doi: 10.1111/tra.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basseri S, Austin RC. Endoplasmic reticulum stress and lipid metabolism: mechanisms and therapeutic potential. Biochem Res Int. 2012;2012:841362. doi: 10.1155/2012/841362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengoechea-Alonso MT, Ericsson J. A phosphorylation cascade controls the degradation of active SREBP1. J Biol Chem. 2009;284:5885–5895. doi: 10.1074/jbc.M807906200. [DOI] [PubMed] [Google Scholar]
- Brown AJ, Sun L, Feramisco JD, Brown MS, Goldstein JL. Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol Cell. 2002;10:237–245. doi: 10.1016/s1097-2765(02)00591-9. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- Cao G, Ni X, Jiang M, Ma Y, Cheng H, Guo L, Ji C, Gu S, Xie Y, Mao Y. Molecular cloning and characterization of a novel human cAMP response element-binding (CREB) gene (CREB4) J Hum Genet. 2002;47:373–376. doi: 10.1007/s100380200053. [DOI] [PubMed] [Google Scholar]
- Cefalu AB, Norata GD, Ghiglioni DG, Noto D, Uboldi P, Garlaschelli K, Baragetti A, Spina R, Valenti V, Pederiva C, et al. Homozygous familial hypobetalipoproteinemia: two novel mutations in the splicing sites of apolipoprotein B gene and review of the literature. Atherosclerosis. 2015;239:209–217. doi: 10.1016/j.atherosclerosis.2015.01.014. [DOI] [PubMed] [Google Scholar]
- Chan CP, Mak TY, Chin KT, Ng IO, Jin DY. N-linked glycosylation is required for optimal proteolytic activation of membrane-bound transcription factor CREB-H. J Cell Sci. 2010;123:1438–1448. doi: 10.1242/jcs.067819. [DOI] [PubMed] [Google Scholar]
- Chanda D, Kim DK, Li T, Kim YH, Koo SH, Lee CH, Chiang JY, Choi HS. Cannabinoid receptor type 1 (CB1R) signaling regulates hepatic gluconeogenesis via induction of endoplasmic reticulum-bound transcription factor cAMP-responsive element-binding protein H (CREBH) in primary hepatocytes. J Biol Chem. 2011;286:27971–27979. doi: 10.1074/jbc.M111.224352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med. 2012;18:59–68. doi: 10.1016/j.molmed.2011.07.010. [DOI] [PubMed] [Google Scholar]
- Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- Danno H, Ishii KA, Nakagawa Y, Mikami M, Yamamoto T, Yabe S, Furusawa M, Kumadaki S, Watanabe K, Shimizu H, Matsuzaka T, et al. The liver-enriched transcription factor CREBH is nutritionally regulated and activated by fatty acids and PPARalpha. Biochem Biophys Res Commun. 2010;391:1222–1227. doi: 10.1016/j.bbrc.2009.12.046. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Q, Giorgianni F, Beranova-Giorgianni S, Deng X, O'Meally RN, Bridges D, Park EA, Cole RN, Elam MB, Raghow R. Glycogen synthase kinase-3-mediated phosphorylation of serine 73 targets sterol response element binding protein-1c (SREBP-1c) for proteasomal degradation. Biosci Rep. 2015;36:e00284. doi: 10.1042/BSR20150234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XY, Tang SQ, Chen JD. Dual functions of Insig proteins in cholesterol homeostasis. Lipids Health Dis. 2012;11:173. doi: 10.1186/1476-511X-11-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle D, Hegarty B, Bossard P, Ferre P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie. 2004;86:839–848. doi: 10.1016/j.biochi.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012;15:623–634. doi: 10.1016/j.cmet.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Gentile CL, Wang D, Pfaffenbach KT, Cox R, Wei Y, Pagliassotti MJ. Fatty acids regulate CREBh via transcriptional mechanisms that are dependent on proteasome activity and insulin. Mol Cell Biochem. 2010;344:99–107. doi: 10.1007/s11010-010-0533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegarty BD, Bobard A, Hainault I, Ferre P, Bossard P, Foufelle F. Distinct roles of insulin and liver X receptor in the induction and cleavage of sterol regulatory element-binding protein-1c. Proc Natl Acad Sci USA. 2005;102:791–796. doi: 10.1073/pnas.0405067102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma Y, Kanazawa K, Mori T, Tanno Y, Tojo M, Kiyosawa H, Takeda J, Nikaido T, Tsukamoto T, Yokoya S, Wanaka A. Identification of a novel gene, OASIS, which encodes for a putative CREB/ATF family transcription factor in the long-term cultured astrocytes and gliotic tissue. Brain Res Mol Brain Res. 1999;69:93–103. doi: 10.1016/s0169-328x(99)00102-3. [DOI] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Mendez R, Zheng Z, Chang L, Cai J, Zhang R, Zhang K. Liver-enriched transcription factor CREBH interacts with peroxisome proliferator-activated receptor alpha to regulate metabolic hormone FGF21. Endocrinology. 2014;155:769–782. doi: 10.1210/en.2013-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Song MJ, Yoo EJ, Choe SS, Park SD, Kim JB. Regulatory role of glycogen synthase kinase 3 for transcriptional activity of ADD1/SREBP1c. J Biol Chem. 2004;279:51999–52006. doi: 10.1074/jbc.M405522200. [DOI] [PubMed] [Google Scholar]
- Kondo S, Saito A, Hino S, Murakami T, Ogata M, Kanemoto S, Nara S, Yamashita A, Yoshinaga K, Hara H, Imaizumi K. BBF2H7, a novel transmembrane bZIP transcription factor, is a new type of endoplasmic reticulum stress transducer. Mol Cell Biol. 2007;27:1716–1729. doi: 10.1128/MCB.01552-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krycer JR, Sharpe LJ, Luu W, Brown AJ. The Akt-SREBP nexus: cell signaling meets lipid metabolism. Trends Endocrinol Metab. 2010;21:268–276. doi: 10.1016/j.tem.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Lee AH. The role of CREB-H transcription factor in triglyceride metabolism. Curr Opin Lipidol. 2012;23:141–146. doi: 10.1097/MOL.0b013e3283508fed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Giannikopoulos P, Duncan SA, Wang J, Johansen CT, Brown JD, Plutzky J, Hegele RA, Glimcher LH, Lee AH. The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat Med. 2011;17:812–815. doi: 10.1038/nm.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MW, Chanda D, Yang J, Oh H, Kim SS, Yoon YS, Hong S, Park KG, Lee IK, Choi CS, et al. Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metab. 2010;11:331–339. doi: 10.1016/j.cmet.2010.02.016. [DOI] [PubMed] [Google Scholar]
- Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–388. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llarena M, Bailey D, Curtis H, O'Hare P. Different mechanisms of recognition and ER retention by transmembrane transcription factors CREB-H and ATF6. Traffic. 2010;11:48–69. doi: 10.1111/j.1600-0854.2009.00997.x. [DOI] [PubMed] [Google Scholar]
- Lu R, Yang P, O'Hare P, Misra V. Luman, a new member of the CREB/ATF family, binds to herpes simplex virus VP16-associated host cellular factor. Mol Cell Biol. 1997;17:5117–5126. doi: 10.1128/mcb.17.9.5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Saito A, Hino S, Kondo S, Kanemoto S, Chihara K, Sekiya H, Tsumagari K, Ochiai K, Yoshinaga K, et al. Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat Cell Biol. 2009;11:1205–1211. doi: 10.1038/ncb1963. [DOI] [PubMed] [Google Scholar]
- Nakagawa Y, Satoh A, Yabe S, Furusawa M, Tokushige N, Tezuka H, Mikami M, Iwata W, Shingyouchi A, Matsuzaka T, et al. Hepatic CREB3L3 controls whole-body energy homeostasis and improves obesity and diabetes. Endocrinology. 2014;155:4706–4719. doi: 10.1210/en.2014-1113. [DOI] [PubMed] [Google Scholar]
- Nohturfft A, DeBose-Boyd RA, Scheek S, Goldstein JL, Brown MS. Sterols regulate cycling of SREBP cleavage-activating protein (SCAP) between endoplasmic reticulum and Golgi. Proc Natl Acad Sci USA. 1999;96:11235–11240. doi: 10.1073/pnas.96.20.11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori Y, Imai J, Suzuki Y, Watanabe S, Tanigami A, Sugano S. OASIS is a transcriptional activator of CREB/ATF family with a transmembrane domain. Biochem Biophys Res Commun. 2002;293:470–477. doi: 10.1016/S0006-291X(02)00253-X. [DOI] [PubMed] [Google Scholar]
- Owen JL, Zhang Y, Bae SH, Farooqi MS, Liang G, Hammer RE, Goldstein JL, Brown MS. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc Natl Acad Sci USA. 2012;109:16184–16189. doi: 10.1073/pnas.1213343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raggo C, Rapin N, Stirling J, Gobeil P, Smith-Windsor E, O'Hare P, Misra V. Luman, the cellular counterpart of herpes simplex virus VP16, is processed by regulated intramembrane proteolysis. Mol Cell Biol. 2002;22:5639–5649. doi: 10.1128/MCB.22.16.5639-5649.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson RB. Regulated intramembrane proteolysis: from the endoplasmic reticulum to the nucleus. Essays Biochem. 2002;38:155–168. doi: 10.1042/bse0380155. [DOI] [PubMed] [Google Scholar]
- Saito A, Hino S, Murakami T, Kanemoto S, Kondo S, Saitoh M, Nishimura R, Yoneda T, Furuichi T, Ikegawa S, et al. Regulation of endoplasmic reticulum stress response by a BBF2H7-mediated Sec 23a pathway is essential for chondrogenesis. Nat Cell Biol. 2009;11:1197–1204. doi: 10.1038/ncb1962. [DOI] [PubMed] [Google Scholar]
- Saito A, Kanemoto S, Kawasaki N, Asada R, Iwamoto H, Oki M, Miyagi H, Izumi S, Sanosaka T, Nakashima K, Imaizumi K. Unfolded protein response, activated by OASIS family transcription factors, promotes astrocyte differentiation. Nat Commun. 2012;3:967. doi: 10.1038/ncomms1971. [DOI] [PubMed] [Google Scholar]
- Stirling J, O'Hare P. CREB4, a transmembrane bZip transcription factor and potential new substrate for regulation and cleavage by S1P. Mol Biol Cell. 2006;17:413–426. doi: 10.1091/mbc.E05-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storlazzi CT, Mertens F, Nascimento A, Isaksson M, Wejde J, Brosjo O, Mandahl N, Panagopoulos I. Fusion of the FUS and BBF2H7 genes in low grade fibromyxoid sarcoma. Hum Mol Genet. 2003;12:2349–2358. doi: 10.1093/hmg/ddg237. [DOI] [PubMed] [Google Scholar]
- Sun LP, Li L, Goldstein JL, Brown MS. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J Biol Chem. 2005;280:26483–26490. doi: 10.1074/jbc.M504041200. [DOI] [PubMed] [Google Scholar]
- Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7) Cell Metab. 2005;1:379–391. doi: 10.1016/j.cmet.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchi C, Montosi G, Zhang K, Lamberti I, Duncan SA, Kaufman RJ, Pietrangelo A. ER stress controls iron metabolism through induction of hepcidin. Science. 2009;325:877–880. doi: 10.1126/science.1176639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem. 2000;275:27013–27020. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
- Xu J, Nakamura MT, Cho HP, Clarke SD. Sterol regulatory element binding protein-1 expression is suppressed by dietary polyunsaturated fatty acids. A mechanism for the coordinate suppression of lipogenic genes by polyunsaturated fats. J Biol Chem. 1999;274:23577–23583. doi: 10.1074/jbc.274.33.23577. [DOI] [PubMed] [Google Scholar]
- Xu X, Park JG, So JS, Hur KY, Lee AH. Transcriptional regulation of apolipoprotein A-IV by the transcription factor CREBH. J Lipid Res. 2014;55:850–859. doi: 10.1194/jlr.M045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabe D, Komuro R, Liang G, Goldstein JL, Brown MS. Liver-specific mRNA for Insig-2 down-regulated by insulin: implications for fatty acid synthesis. Proc Natl Acad Sci USA. 2003;100:3155–3160. doi: 10.1073/pnas.0130116100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, Brown MS. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Yellaturu CR, Deng X, Cagen LM, Wilcox HG, Mansbach CM, 2nd, Siddiqi SA, Park EA, Raghow R, Elam MB. Insulin enhances post-translational processing of nascent SREBP-1c by promoting its phosphorylation and association with COPII vesicles. J Biol Chem. 2009;284:7518–7532. doi: 10.1074/jbc.M805746200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellaturu CR, Deng X, Cagen LM, Wilcox HG, Park EA, Raghow R, Elam MB. Posttranslational processing of SREBP-1 in rat hepatocytes is regulated by insulin and cAMP. Biochem Biophys Res Commun. 2005;332:174–180. doi: 10.1016/j.bbrc.2005.04.112. [DOI] [PubMed] [Google Scholar]
- Zhang C, Wang G, Zheng Z, Maddipati KR, Zhang X, Dyson G, Williams P, Duncan SA, Kaufman RJ, Zhang K. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology. 2012;55:1070–1082. doi: 10.1002/hep.24783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.