

Abstract
Nijmegen breakage syndrome (NBS) is characterised by microcephaly, developmental delay, characteristic facial features, immunodeficiency and radiosensitivity. Nbs1, the protein defective in NBS, functions in ataxia telangiectasia mutated protein (ATM)-dependent signalling likely facilitating ATM phosphorylation events. While NBS shares overlapping characteristics with ataxia telangiectasia, it also has features overlapping with ATR-Seckel (ATR: ataxia-telangiectasia and Rad3-related protein) syndrome, a subclass of Seckel syndrome mutated in ATR. We show that Nbs1 also facilitates ATR-dependent phosphorylation. NBS cell lines show a similar defect in ATR phosphorylation of Chk1, c-jun and p-53 in response to UV irradiation- and hydroxyurea (HU)-induced replication stalling. They are also impaired in ubiquitination of FANCD2 after HU treatment, which is ATR dependent. Following HU-induced replication arrest, NBS and ATR-Seckel cells show similarly impaired G2/M checkpoint arrest and an impaired ability to restart DNA synthesis at stalled replication forks. Moreover, NBS cells fail to retain ATR in the nucleus following HU treatment and extraction. Our findings suggest that Nbs1 functions in both ATR- and ATM-dependent signalling. We propose that the NBS clinical features represent the result of these combined defects.
Keywords: cell cycle checkpoints, DNA damage responses, DNA repair, Nijmegen breakage syndrome, replication fork arrest
Introduction
Nijmegen breakage syndrome (NBS) was identified as a chromosome instability disorder in 1981 (Weemaes et al, 1981). Ataxia telangiectasia (A-T) and NBS are characterised by chromosome instability particularly involving chromosomes 7 and 14, cellular and clinical hypersensitivity to ionising radiation (IR), characteristic radioresistant DNA synthesis, humoral and cellular immune defects and marked cancer predisposition (Taalman et al, 1983; Taylor et al, 1993; Shiloh, 1997; International Nijmegen Breakage Syndrome Study Group, 2000). NBS has therefore been described as an A-T-like disorder (Saar et al, 1997; Wegner et al, 1999). Despite overlapping features, NBS and A-T are clinically distinct. NBS patients display microcephaly, developmental delay and characteristic facial features, while A-T is a neurodegenerative disorder with marked and debilitating progressive ataxia. Ataxia-telangiectasia-like disorder (ATLD) is a further A-T-like disorder. ATLD patients show a mild A-T phenotype with no microcephaly or developmental delay (Stewart et al, 1999).
The protein mutated in A-T is ataxia telangiectasia mutated protein (ATM), a phosphoinositol 3-kinase-like kinase (PIKK) (Savitsky et al, 1995). ATM is central to the signalling response to DNA double-strand breaks (DSBs) and phosphorylates multiple damage response proteins, including p53, H2AX, Chk2, RPA, Brca1, BLM, 53BP1, SMC1, Rad17, Rad1 and Rad9 in response to IR (Iliakis et al, 2003; Shiloh, 2003). As a consequence of these phosphorylation defects, A-T cells display radiosensitivity and cell cycle checkpoint defects after exposure to IR (for reviews, see Lavin and Khanna, 1999; Khanna et al, 2001; Shiloh, 2003). ATM is not activated in the initial response to UV irradiation or hydroxyurea (HU), an agent that causes replication stalling.
Nbs1 and Mre11, the proteins deficient in NBS and ATLD, respectively, function as components of the Mre11/Rad50/Nbs1 (MRN) complex, which colocalises with γ-H2AX at the site of DSBs (Carney et al, 1998; Varon et al, 1998; Paull et al, 2000). NBS and ATLD cells show impaired IR-induced phosphorylation, demonstrating a role for MRN in facilitating ATM-dependent signalling (Matsuura et al, 1998; Buscemi et al, 2001; Girard et al, 2002; Kim et al, 2002; Nakanishi et al, 2002; Yazdi et al, 2002; Gatei et al, 2003). Consistent with such a role, MRN was recently shown to stimulate the kinase activity of ATM in vitro towards its substrates p53, Chk2 and histone H2AX, and Nbs1 was shown to directly contact ATM (Lee and Paull, 2004). Recent data have provided evidence for a role for MRN as a damage sensor acting upstream of ATM although there is also compelling evidence for a downstream role (Carson et al, 2003; Uziel et al, 2003; Horejsi et al, 2004). Notwithstanding the precise point at which MRN functions, the requirement of MRN for ATM-dependent phosphorylation likely underlies the similar radiosensitivity and cell cycle checkpoint defects of A-T, ATLD and NBS cell lines.
Other studies have provided evidence for a wider role of MRN. While ATM is nonessential in mice and humans, Nbs1- and Mre11-defective mice are embryonic lethal (Yamaguchi-Iwai et al, 1999). The microcephaly and developmental delay in NBS patients also suggests an additional function for Nbs1. There is also evidence that Mre11/Rad50/Xrs2 (MRX), the Saccharomyces cerevisiae homologue of MRN (Haber, 1998), promotes both nonhomologous end-joining and homologous recombination (Bressan et al, 1999; Petrini, 1999) although direct evidence for a role of MRN in these processes in mammalian cells has not been forthcoming. Two studies using yeast have demonstrated that MRX is required for Mec1/Rad3-dependent S-phase checkpoint activation although a third study did not observe a role for Rad50 in the response to HU (D'Amours and Jackson, 2001; Hartsuiker et al, 2001; Chahwan et al, 2003). Additionally, MRN is deposited on chromatin in an S-phase-specific manner and localises to single-stranded DNA (ssDNA) arising in HU-treated cells (Mirzoeva and Petrini, 2001), and loss of MRX/MRN components leads to chromosome breakage during replication (Yamaguchi-Iwai et al, 1999; Costanzo et al, 2001). Taken together, indirect evidence for an additional role of the MRN complex in the response to DNA damage in mammalian cells is strong, although the precise function is still unclear.
Recently, we identified a hypomorphic mutation in ataxia-telangiectasia and Rad3-related protein (ATR) in two related Seckel syndrome patients (O'Driscoll et al, 2003). Seckel syndrome is genetically and clinically heterogenous and we have designated this subset of Seckel syndrome ATR-Seckel (O'Driscoll et al, 2004). ATR-Seckel patients have features that overlap NBS, namely pronounced microcephaly, developmental delay and characteristic facial features (Goodship et al, 2000). ATR, like ATM, is a PIKK family member and plays a central role in a parallel damage response pathway (Bentley et al, 1996; Zou et al, 2002). However, while ATM responds to the presence of DNA DSBs, ATR appears to be activated by ssDNA, arising at stalled replication forks or generated during processing of bulky lesions (Costanzo et al, 2003; Zou and Elledge, 2003). ATR phosphorylates many of the same damage response proteins as ATM, including Nbs1, which localises to stalled replication forks (Shiloh, 2001; Zhou et al, 2002). These findings prompted us to examine if Nbs1 is also required for ATR-dependent signalling. To explore further the contribution of ATR signalling to clinical phenotype, we also examined cell lines from ATLD patients, who do not display Seckel-like features. Strikingly, we observed similar cellular defects in ATR-Seckel and NBS cell lines including impaired substrate phosphorylation, impaired G2/M checkpoint arrest and an impaired ability to restart replication at stalled replication forks. Our findings strongly suggest that Nbs1 functions both in ATR- and ATM-dependent signalling. We propose that NBS clinical features represent the result of these combined defects.
Results
NBS and ATR-Seckel lymphoblastoid cell lines are similarly impaired in phosphorylation induced by replication stalling
We examined phosphorylation of Chk1 and c-jun by Western blotting in NBS, ATR-Seckel, ATLD and A-T lymphoblastoid cell lines (LBLs) following treatment with HU and aphidicolin (APH), respectively, agents that causes replication fork stalling. While Chk1 and c-jun phosphorylation was clearly observed in treated wild type (WT) (GM1958), A-T (GM3189) and ATLD1/2 LBLs, it was not seen in an ATR-Seckel LBL (DK0064) nor in two NBS LBLs (LB112 and LB195) (Figure 1A). Chk1 and c-jun phosphorylation were also examined by immunofluorescence (IF) following 2 h treatment with HU. 30–40% of control, A-T and ATLD LBLs gave an intense fluorescent signal after HU using either anti-p-Chk1 or anti-p-c-jun antibodies. In contrast, there was no increase in positive cells in NBS or ATR-Seckel LBLs (Figure 1B). These data suggest that NBS cells are impaired in these two ATR-dependent phosphorylation events following replication fork stalling.
Figure 1.
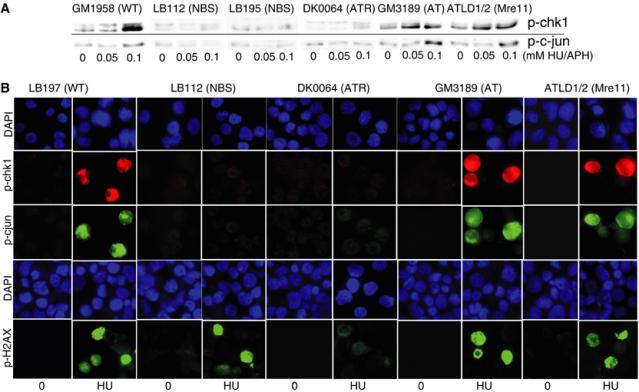
NBS LBLs shows reduced Chk1 and c-jun phosphorylation following replication stalling. (A) Control (GM1958), NBS (LB112 and LB195), ATR-Seckel (DK0064), A-T (GM3189) and ATLD1/2 LBLs were treated with 0.05 and 0.1 mM HU for 2 h and examined by Western blotting using anti-p-Chk1 antibodies (top panel). The same LBLs were treated with 0.05 and 0.1 mM APH for 2 h and examined by Western blotting using anti-p-c-jun antibodies. Damage-induced phosphorylation was not observed in NBS or ATR-Seckel cells. (B) The indicated LBLs were treated with 2 mM HU for 2 h and examined by IF using anti-p-Chk1, p-c-jun and p-H2AX antibodies. Control, A-T and ATLD cells show approximately 40% positive cells corresponding to the percentage of cells entering S phase during this period. The number of strongly positive cells using LB112, LB195 or DK0064 cells was markedly reduced using p-c-jun and p-Chk1 antibodies. Results for GM1958 (control) and LB195 (NBS) are not shown but are identical to those obtained with LB197 and LB112, respectively. Using the p-H2AX antibody, the two NBS lines gave a signal similar to control cells, while ATR-Seckel cells show substantially decreased H2AX phosphorylation.
Several studies have demonstrated different genetic requirements for the phosphorylation of H2AX by ATM and ATR compared to other substrates (Ward and Chen, 2001; Foray et al, 2003; Stiff et al, 2004). We also examined HU-induced H2AX phosphorylation in NBS cells. In contrast to the results obtained with Chk1 and c-jun, we observed normal H2AX phosphorylation in the NBS lines, which was in contrast to the marked reduction in DK0064 cells (Figure 1B).
NBS and ATR-Seckel fibroblast cell lines are similarly impaired in UV-induced phosphorylation events
Previously, we showed that F02-98, an ATR-Seckel fibroblast, is impaired in the phosphorylation of H2AX, p53, Rad17 and Nbs1 following exposure to UV (5 J m−2) while showing a normal ability to phosphorylate the same substrates in response to IR (O'Driscoll et al, 2003). For these experiments, we use plateau phase primary fibroblasts, which have few (<1%) detectable replicating cells when assessed by BrdU labelling (data not shown). Under such conditions, activation of ATR occurs at single strand regions generated by nucleotide excision repair (NER) (O'Driscoll et al, 2003). Phosphorylation of Chk1, p53 and c-jun was examined 2 h following exposure to 2, 5 and 10 J m−2 by IF. With all three substrates, we observed impaired phosphorylation in the two NBS cell lines (347BR and CZD82CH) similar to that observed in F02-98 cells (Figure 2A–C). A-T and ATLD cells gave a normal response. Following IR, we observed decreased p53 phosphorylation in ATLD cells using similar techniques (Figure 2G). In all cases, residual phosphorylation is observed in the NBS and ATR-defective lines, which is more marked following exposure to higher UV doses. We also examined phosphorylation using a recently derived phospho-SQ/TQ antibody, which recognises phosphorylation of the consensus ATR/ATM motif (Uziel et al, 2003) (Figure 2D). The ability of these antibodies to recognise ATR-dependent phosphorylation events is shown by the UV-dose-dependent increase in fluorescent intensity and the reduced response in F02-98 cells. The signal was reduced to similar extents in NBS and F02-98 cell lines. The residual signal could be due to other stress-induced phosphorylation events since these antibodies are nonspecific.
Figure 2.
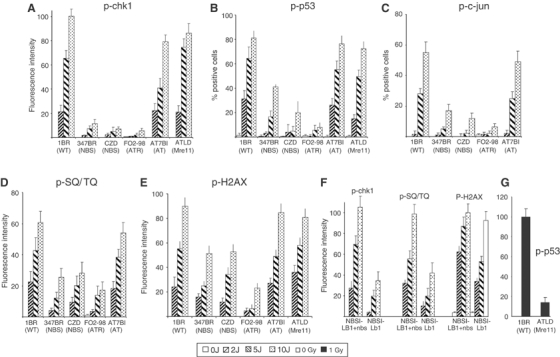
NBS primary fibroblasts are impaired in UV-induced ATR-dependent phosphorylation of Chk1, c-jun and p53. Noncycling primary human fibroblasts from control (1BR), NBS (347BR and CZD82CH), ATR-Seckel syndrome (F02-98), A-T (AT7BI) and ATLD1/2 patients were treated with UV as indicated and analysed by IF using the indicated phosphospecific antibodies 2 h post-irradiation. (A) p-Chk1, (B) p-p53, (C) p-c-jun, (D) phospho-SQ/TQ antibodies and (E) p-H2AX. (F) Results using NBS-ILB1 fibroblasts uncomplemented or complemented with full-length Nbs1 (p95). (G) Impaired phosphorylation of p-p53 in ATLD1/2 fibroblasts exposed to 1 Gy γ-rays. Previously, we observed decreased α-p53 phosphorylation after IR in NBS cells using similar techniques (Girard et al, 2002). For panels A–F, positive cells showed a lawn of fluorescence rather than discrete foci as observed using γ-irradiation. For panels A, D, E and F, the nuclear fluorescence intensity of captured images was quantified using Simple PCI software. For panels B and C, the percentage of positively staining cells was measured. The results represent the mean and s.d. of three experiments. Similar results were obtained using either technique. The ATR dependency is demonstrated by the marked reduction in phosphorylation in F02-98 cells. The specificity of the Chk1 and c-jun antibodies for IF is verified by the similar results obtained by Western blotting shown in Figure 1. The specificity of the p53 antibody was verified by demonstrating the lack of a signal in p53−/− mouse embryonic fibroblasts (Supplementary Figure 1).
We also examined H2AX phosphorylation in UV-treated fibroblasts. We observed a mild reduction in H2AX phosphorylation in both NBS lines in contrast to a more marked reduction in F02-98 cells (Figure 2E).
To verify that the impaired phosphorylation is due to diminished Nbs1, we examined NBS-ILB1, a transformed NBS fibroblast and NBS-ILB1 expressing full-length Nbs1 (NBS-ILB1+Nbs1). Expression of Nbs1 cDNA fully complemented the decreased UV-induced phosphorylation of Chk1 and SQ/TQ observed in NBS-ILB1 cells (anti-p53 and c-jun antibodies were not examined since NBS-ILB1 cells show a high background signal likely due to their transformed phenotype) (Figure 2F). UV-induced H2AX phosphorylation, although only partly decreased in NBS-ILB1 similar to the impact observed in NBS primary fibroblasts, was increased upon Nbs1 expression. The ability of ATR cDNA to complement the phosphorylation defect of F02-98 fibroblasts and DK0064 LBLs has been previously described (O'Driscoll et al, 2003; Alderton et al, 2004).
NBS cells fail to ubiquitinate FANCD2 following treatment with HU
Recently, it was shown that following treatment with HU and mitomycin C, ATR phosphorylation of FANCD2 is required for the ubiquitination of FANCD2 implicating ATR in the Fanconi aneamia (FA) pathway (Andreassen et al, 2004; Pichierri and Rosselli, 2004). Thus, decreased FANCD2 ubiquitination is observed in ATR-Seckel cells. Here, we demonstrate that two NBS LBLs (LB195 and LB112) show a similar defect in FANCD2 ubiquitination to ATR-Seckel LBLs (DK0064) following exposure to HU (Figure 3). We conclude that Nbs1, like ATR, plays a role in FANCD2 ubiquitination.
Figure 3.
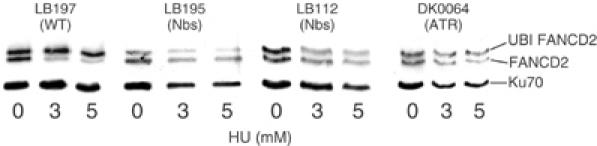
NBS cells are impaired in FANCD2 ubiquitination. LBLs from control (LB197), NBS (LB195 and LB112) and ATR-Seckel (DK0064) were treated with 0, 3 and 5 mM HU for 24 h and examined by Western blotting using anti-FANCD2 antibodies. The presence of ubiquitinated FANCD2 is evident by the mobility shift. As observed previously, approximately 50% of the protein is endogenously ubiquitinated (Andreassen et al, 2004). In control cells, this increases to approximately 80–90% after HU treatment. No change in the level of ubiquitinated FANCD2 is observed in two NBS or ATR-Seckel cell lines. Ku70 is shown as a loading control. The apparent decreased level of total FANCD2 after treatment in some cell lines is due to decreased protein levels shown by the loading control.
NBS and ATR-defective LBLs fail to arrest at the G2/M checkpoint following exposure to UV
One consequence of ATR-dependent phosphorylation is activation of G2/M cell cycle checkpoint arrest. As a procedure to examine G2/M arrest, we monitored the number of mono- and binucleate cells in the presence of cytochalasin B at 72 h post-treatment with different DNA-damaging agents using the cytokinesis block proliferation index (CBPI) assay (Fenech and Morley, 1985). Binucleate cells represent cells that have progressed through the G2/M checkpoint and mitosis but have failed to divide into daughter cells due to the presence of cytochalasin B, an inhibitor of cytokinesis. Greater than 50% of untreated cells are binucleate, demonstrating that they have progressed through mitosis during the 72 h incubation (Figure 4A). Following treatment of control LBLs (GM2188 and GM1958) with HU, APH or UV, the percentage of binucleate cells decreases around two-fold indicating the presence of a G2/M checkpoint. Similar results were obtained with an A-T (AO) and ATLD1/2 LBL (Figure 4A and not shown). No appreciable decrease in the number of binucleate cells is observed in the two NBS LBLs nor in DK0064 (ATR-Seckel) cells following exposure to HU, APH or UV, demonstrating that G2/M arrest following these treatments is ATR and Nbs1 dependent (Figure 4A).
Figure 4.
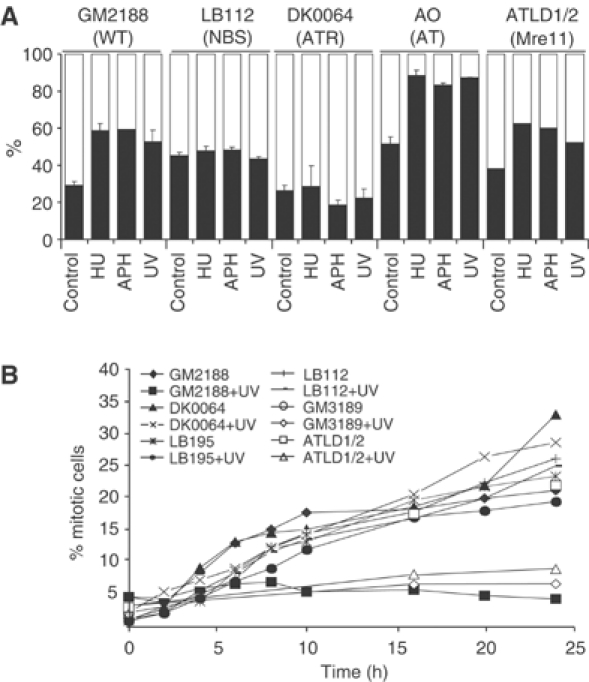
NBS and ATR-Seckel cells fail to arrest at the G2/M checkpoint following UV treatment. (A) The CBPI checkpoint assay. Cells were treated as indicated for 2 h followed by addition of cytochalasin B, an inhibitor of cytokinesis. After 72 h, cells were scored for the percentage of binucleate/mononucleate cells. The results represent the mean and s.d. of three experiments except for ATLD where n=1. Two experiments using LB195 cells gave similar results to those obtained with the LB112 cells. Results for LB197 (not shown) are identical to those obtained with GM2188. Black bars represent mononucleates and the white bar represents binucleates. (B) G2/M checkpoint arrest monitored by mitotic index. Cells were untreated or irradiated with 2.5 J m−2, incubated for varying times up to 24 h in the presence of 1.5 μM nocodazole and the percentage of mitotic cells counted. The results shown are from a single experiment. For all lines, similar results were obtained in three experiments where the mitotic index was monitored at 24 h post-treatment.
We also examined the time course of G2/M arrest by measuring the mitotic index in nocodozole-arrested cells either untreated or at varying times post UV treatment. Control (GM2188), A-T (GM3189) and ATLD1/2 LBLs show a robust UV-induced G2/M checkpoint arrest evident by a decreased mitotic index (Figure 4B). In contrast, UV treatment of the two NBS LBLs (LB195 and LB112) and the ATR-Seckel LBL (DK0064) failed to impact upon the mitotic index, demonstrating the lack of any appreciable G2/M arrest.
ATR and NBS cells show impaired recovery of DNA synthesis at stalled replication forks
Another role of ATR is to maintain the integrity of stalled replication forks. Consequently, once the block to replication is removed, DNA synthesis at the stalled fork can rapidly resume. We examined ATR-Seckel, NBS, A-T and ATLD cells for recovery of DNA synthesis after replication stalling using a recently described technique that visualises DNA replication using fluorescently labelled antibodies specific to halogenated derivatives of deoxyuridine (dU) (Dimitrova and Gilbert, 2000; Feijoo et al, 2001). Replication forks were marked by pulse labelling with CldU, arrested by treating with APH for 2 h, and fork progression allowed to restart by removal of APH in the presence of IdU. Control cells show extensive replication recovery represented by the overlap of CldU (red) and IdU (green) labelling (Supplementary Figure 3). This was quantified by measuring the % of CldU-labelled cells with >80% overlap of CldU and IdU labelling (Figure 5). Very few cells showed a partial overlay. In both the ATR and NBS cells, there was a marked decrease in IdU-labelled cells, demonstrating that replication has not restarted at the stalled replication forks during the period analysed (Figure 5 and Supplementary data). A-T and ATLD cells showed a normal response.
Figure 5.
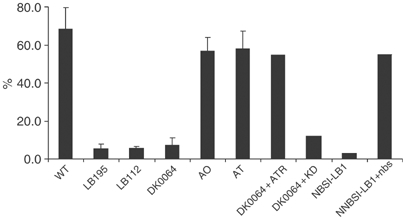
ATR and NBS cells show impaired recovery following replication stalling. Replication forks were marked by pulse labelling for 20 min with CldU and then arrested by treating with APH for 2 h. Prior to removal of APH, cells were treated with IdU for 20 min so that fork progression after replication stalling can be visualised. After removal of APH, cells are allowed to swell in hypotonic solution for 10 min at room temperature during which time replication recovery can take place. Control cells show extensive replication recovery represented by the overlap of CldU (red) and IdU (green) labelling (Supplementary Data). This was quantified by measuring the % of CldU-labelled cells with >80% overlap of CldU and IdU labelling. The results represent the mean and s.d. of three experiments. Decreased recovery was observed in the ATR-Seckel and NBS cell lines and was complemented by expression of ATR and Nbs1, respectively.
Recovery from replication fork stalling was also diminished in NBS-ILB1 cells and recovered in cells expressing Nbs1. Expression of full-length ATR but not kinase-dead ATR also restored replication recovery to DK0064 cells (Figure 5).
RPA but not ATR is present at the sites of DNA damage in NBS cells
To gain mechanistic insight into the role of Nbs1 in ATR signalling, we examined if the recruitment of RPA, an upstream step required for ATR recruitment, was impaired in NBS cells (Zou and Elledge, 2003). We examined RPA recruitment in noncycling primary human fibroblasts following exposure to UV. Under normal repair proficient conditions, incision is the rate-limiting step of NER and the resulting single strand tract is rapidly filled in (Squires et al, 1982). To enhance our ability to detect RPA recruitment, we treated cells with 20 J m−2 UV in the presence of cytosine β-D-arabinofuranoside (Ara-C) and HU, which inhibits the repair synthesis but not the excision step of NER. Under these conditions, chromatin-bound RPA was observed by IF following permeabilisation and extraction in UV-irradiated but not unirradiated cells (Figure 6A). A similar signal was observed in 347BR (NBS), F02-98 (ATR-Seckel) and ATLD1/2 cells. Similar results were obtained in LBLs 2 h post-treatment with HU (Supplementary Figure 2). These experiments were carried out in parallel to the experiments shown in Figures 1 and 2, where phosphorylation of Chk1, p53 and c-jun was seen to be 3- to 10-fold reduced in NBS and ATR-Seckel cells. Thus, we conclude that the recruitment of RPA does not require Nbs1 or ATR. The role of Nbs1 in ATR activation must, therefore, lie downstream of RPA recruitment and is unlikely to represent a role in end processing.
Figure 6.
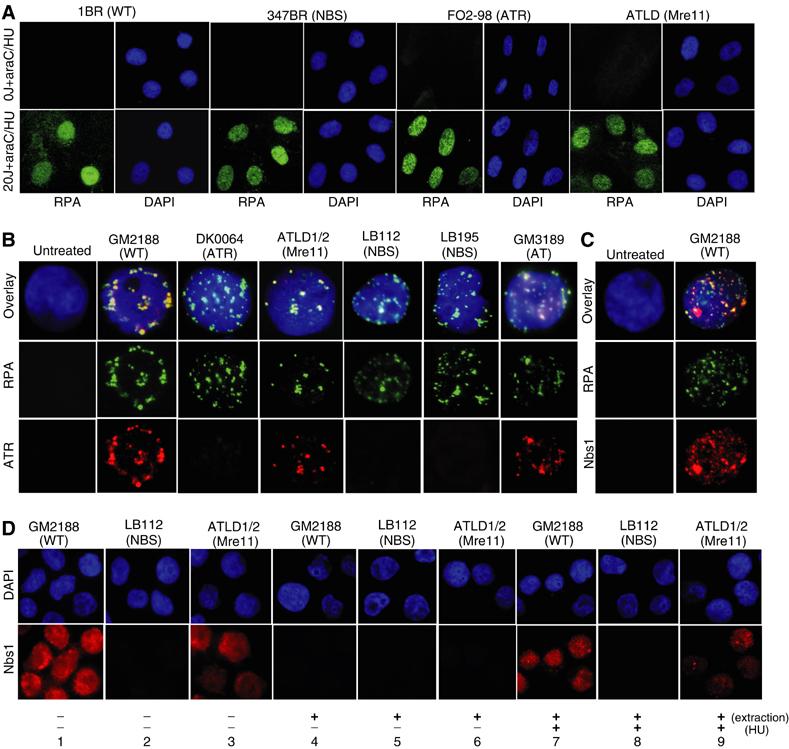
While RPA is recruited normally to the sites of damage in NBS and ATR-Seckel cells, ATR foci are not detectable. (A) The indicated primary fibroblasts were unirradiated or irradiated with 20 J m−2 followed by incubation in 100 μM Ara-C or 1 mM HU for 1 h prior to permeabilisation, extraction and IF using α-RPA. Ara-C and HU prevent the ‘fill-in' step of NER and thus enhance the checkpoint signal. UV irradiation induces RPA recruitment at damage sites in all cell lines. (B, C) The indicated LBLs were untreated or treated with 5 mM HU for 2 h prior to stringent permeabilisation and extraction (0.5% NP40, 1 min) and IF using α-RPA, α-ATR or α-Nbs1 (C). Only untreated control cells are shown. All other untreated cells gave a similar lack of signal. Merged images are shown in the DAPI panels. (D) The indicated cell lines were either untreated or treated with 2 mM HU and processed without extraction or gently permeabilised with extraction (0.1% Triton X-100, 30 s). Milder extraction conditions were used in this panel compared to those used in panels B and C.
Next, we examined whether ATR is localised to the damage sites in NBS cells. We were unable to detect ATR retention in control fibroblasts following HU treatment. However, strong permeabilisation and extraction conditions allowed us to see the nuclear retention of ATR and RPA as foci in WT LBLs following HU (Figure 6B). No such foci were visible in untreated cells (Figure 6B). The ATR foci were visible in around 30% of the RPA-positive cells most likely due to the strong extraction conditions employed. The ATR foci showed near-complete overlap with the RPA foci (Figure 6B). A-T and ATLD LBLs showed a similar retention of ATR and overlap with RPA after HU treatment. Strikingly, no retention of ATR was observed in either of the two NBS LBLs examined. These data provide strong evidence that Nbs1 is required for recruitment or retention of ATR after replication fork stalling.
Next we examined whether Nbs1 colocalised to the RPA (and therefore ATR) foci formed after replication fork stalling. Following HU treatment and strong extraction of WT LBLs, we observed an increase in Nbs1 retained in the nucleus with clearly visible foci evident in around 10–20% of the cells (Figure 6C). Although the signal was more diffuse than that obtained with RPA, we observed evidence of colocalisation with RPA. To examine this response in ATLD LBLs, we first examined the expression and localisation of Nbs1. Without extraction, we observed decreased Nbs1 expression that localised entirely to the nucleus in undamaged cells (Figure 6D, panels 1–3). Decreased Nbs1 expression in ATLD is consistent with previous Western blotting analysis (Stewart et al, 1999). Following treatment with HU and extraction, Nbs1 was retained in the nucleus in ATLD cells (Figure 6, panels 7–9). No retention was observed in untreated cells (Figure 6, panels 4–6). The decreased signal precluded an assessment of colocalisation with RPA.
Discussion
Here, we demonstrate that four NBS cell lines (two fibroblast lines and two LBLs) show reduced ATR-dependent phosphorylation, fail to effect G2/M arrest after treatment with a range of agents known to activate ATR and fail to resume DNA synthesis at stalled replication forks. This phenotype overlaps with that displayed by a cell line defective in ATR and is not observed in A-T cells. Thus, we conclude that Nbs1 facilitates ATR-dependent phosphorylation and G2/M checkpoint arrest as well as ATM-dependent phosphorylation and checkpoint arrest. There is further circumstantial evidence that the MRN complex plays a role after replication stalling. Studies in yeast have demonstrated a role for MRX in the Rad3/Tel1 signalling pathway following HU treatment (D'Amours and Jackson, 2001; Chahwan et al, 2003). Additionally, studies in Xenopus have shown that absence of the Mre11 protein leads to DSB formation during replication (Costanzo et al, 2001). One explanation for these latter findings is that MRN functions in a DSB repair pathway to handle lesions generated at the replication fork. Our findings here suggest an alternative model, namely that Nbs1 has a role in ATR-dependent checkpoint signalling. We propose that this is at least one factor contributing to its role in the response to replication stalling.
It is notable that despite the similar response of NBS and ATR-deficient cell lines in most assays, H2AX phosphorylation was only mildly decreased in NBS fibroblasts and occurred normally in NBS LBLs contrasting with the marked decrease observed in ATR-Seckel fibroblasts and LBLs. Although this difference was mild for the fibroblasts, it was marked for the LBLs and expression of Nbs1 increased H2AX phosphorylation in the transformed NBS cell line. Previous studies have also reported different genetic requirements for H2AX phosphorylation relative to other substrates (Ward and Chen, 2001; Foray et al, 2003; Stiff et al, 2004). Although the basis underlying these findings requires further study, it suggests that ATR may be activated efficiently in NBS cells but is more important for the phosphorylation of some substrates versus others. One possible explanation is that Nbs1 may be more important for non-chromatin-bound substrates compared to H2AX, which is chromatin localised and thus an early step in the damage response (see below).
In most assays, we observed a strikingly similar response between the NBS and F02-98 cells, which have a hypomorphic mutation that decreases but does not abolish ATR function. However, residual ATR function is evident after higher doses. The NBS cell lines analysed harbour the common founder 675Δ5 mutation, which allows expression of a truncated C-terminal polypeptide of Nbs1 (p70) (Maser et al, 2001). One explanation for residual ATR-dependent phosphorylation is leakiness of the Nbs1 mutation. An alternative explanation is that Nbs1 enhances but is not absolutely required for ATR phosphorylation similar to the findings for the role of MRN in ATM-dependent phosphorylation (Lee and Paull, 2004).
To gain mechanistic insight into the role of Nbs1 in ATR signalling, we examined early steps in the damage response. We found that Nbs1 colocalises with RPA, which in turn colocalises with ATR at the damage site. These findings are consistent with studies showing that after HU Mre11 localises to the sites of replication stalling and that Nbs1 is phosphorylated in an ATR-dependent manner (Mirzoeva and Petrini, 2001; Franchitto and Pichierri, 2002; O'Driscoll et al, 2003). Indeed, Nbs1 phosphorylation is not observed in ATR-Seckel cells (O'Driscoll et al, 2003). While RPA is recruited normally to damage sites in NBS cells, the nuclear retention of ATR was markedly decreased after replication stalling, suggesting that Nbs1 functions to recruit or retain ATR. Our finding that H2AX phosphorylation is only modestly reduced in NBS cells suggests that ATR may be activated normally but fails to be retained at the damage site. Based on these findings, we propose a working model that Nbs1 serves to amplify the ATR signal by facilitating the retention of ATR at the damage site whereas the initial localisation and activation of ATR may be Nbs1 independent. There is mounting evidence that MRN plays an upstream role in ATM signalling acting as a damage sensor or activator of ATM although there is also evidence for a downstream function (Carson et al, 2003; Uziel et al, 2003; Horejsi et al, 2004) (for a review, see Lavin, 2004). Interestingly, autophosphorylation of ATM, an early step in the damage response, is significantly reduced in ATLD1/2 cell lines and more modestly reduced in NBS cells (Uziel, 2003 #10371). A similar impairment in the nuclear retention of ATM in response to neocarzinostatin is also observed (Uziel, 2003 #10371). The role of Nbs1 in ATR signalling, therefore, has clear parallels to its role in ATM signalling although further work is required to dissect the precise role played in amplifying the signal from the two PIKKs.
In contrast to the marked defect in ATR signalling in NBS cell, the ATLD1/2 cell line showed a normal response, which is distinct from the defect in ATM-dependent signalling observed in ATLD cells (Stewart et al, 1999; Uziel et al, 2003). Additionally, while the nuclear retention of ATM after radiation is diminished in ATLD cells, ATR remained efficiently chromatin bound in ATLD cells (Uziel et al, 2003). Consistent with previous findings, we also observed decreased IR-induced p53 phosphorylation in the ATLD cell line (Stewart et al, 1999). Our findings, therefore, suggest that there may be different requirements for components of the MRN complex for ATM- and ATR-dependent signalling. It is possible that the mutation in Mre11 in ATLD1/2 results in a separation of Mre11 function in ATM versus ATR signalling. More interestingly, Mre11 may be dispensable for ATR signalling.
Although, UV-induced ATR activation has been reported to be replication dependent, we observed UV-induced ATR-dependent phosphorylation of H2AX and additional substrates in nonreplicating G0/G1 fibroblasts using IF both in this study and previously (O'Driscoll et al, 2003; Ward et al, 2004). Moreover, this phosphorylation is not observed in NER-deficient XP-A-deficient cells (O'Driscoll et al, 2003). We attribute this response to induction of ATR by single strand gaps generated during NER. Since the incision step is rate limiting in NER, the single strand gaps generated will be transient (Squires et al, 1982). The ability to detect such phosphorylation may be dependent upon the cell type and conditions employed.
NBS patients are characterised by immunodeficiency, cancer predisposition, microcephaly, development delay and characteristic facial features (International Nijmegen Breakage Syndrome Study Group, 2000). Some, but not all, of these clinical features overlap with A-T (e.g. immunodeficiency and cancer predisposition) (Shiloh, 1997). Neither A-T nor ATLD patients manifest the latter three characteristics, displaying instead progressive neurodegeneration. Strikingly, microcephaly, developmental delay and characteristic facial features are also seen in ATR-Seckel patients (Goodship et al, 2000; O'Driscoll et al, 2003). Thus, we propose that these features of NBS may be a result of impaired ATR signalling, while impaired ATM signalling and/or defective repair may contribute to the immunodeficiency observed in NBS. The distinct clinical features of ATLD and NBS is striking but unexplained and, we propose, arises either because Mre11 itself or the mutation in the ATLD patients does not impact upon ATR signalling. An additional syndrome to consider in this context is FA, which also overlaps clinically with NBS. Recently, studies have shown that ATR is required for FANCD2 ubiquitination (Andreassen et al, 2004; Pichierri and Rosselli, 2004). Since FANCD2 ubiquitination is required for FA activation, this couples ATR signalling to activation of the FA pathway (Gregory et al, 2003). Here, we show that NBS is also required for FANCD2 ubiquitination in response to replication stalling, strongly suggesting that Nbs1 also plays a role in the FA pathway. While the radiosensitivity of NBS cells has been known for many years, it has recently been shown that NBS cells also display crosslinking agent sensitivity, a hallmark of FA cells (Nakanishi et al, 2002). Several patients who were originally diagnosed as FA patients but proved upon further analysis to be NBS have recently been described, further demonstrating the overlap in clinical features (Nakanishi et al, 2002; Gennery et al, 2004). Our findings here provide a basis for this overlap.
In conclusion, we provide evidence that cell lines derived from NBS patients have impaired ATR-dependent signalling, cell cycle checkpoint arrest and impaired ability to recover from replication stalling. At a mechanistic level, we provide evidence that Nbs1 is required for the efficient retention of ATR at the damage site. This provides a novel function for Nbs1 and an explanation for the microcephaly and developmental delay, which is characteristic of NBS patients but not observed in ATLD or A-T patients.
Materials and methods
Cells and cell culture conditions
1BR3, AT7BI and F02-98 are primary fibroblasts from a control, A-T and an ATR-Seckel patient, respectively. 347BR and CZD82CH are NBS primary fibroblast lines homozygous for the 675Δ5 mutation (Girard et al, 2002). ATLD1/2 has a truncating mutation in Mre11 (Stewart et al, 1999). CZD82CH and ATLD1/2 cells were kindly supplied by Drs J Hall and M Taylor, respectively. Cells were cultured in minimum essential medium supplemented with 20% fetal calf serum (FCS), penicillin and streptomycin. LB197, GM1958 and GM2188 are control EBV transformed lymphoblastoid cell lines (LBL); AO and GM3189 are A-T LBLs; LB195 and LB112 are NBS LBLs. DK0064 is an ATR-defective LBL derived from the same patient as F02-98 cells. LBLs were grown in RPMI medium supplemented with 15% FCS, penicillin and streptomycin. NBS-ILB1 cells are transformed NBS fibroblasts from M Zdzienicka. NBS-ILB1+Nbs1 cells are retrovirally complemented with full-length human Nbs1. WT and p53−/− mouse embryonic fibroblasts were kindly supplied by D Barnes.
Treatment with DNA-damaging agents
Irradiation was carried out using a UVC source (0.6 J m−2 s−1). APH, HU, cytochalasin B and Ara-C were purchased from Sigma-Aldrich (Poole, UK).
Antibodies
α-p53Ser15 (rabbit polyclonal), α-SQ/TQ and α-chk1Ser317 antibodies were purchased from Cell Signaling Technology (Beverly, MA). α-H2AXSer139 and α-histone H3Ser10 antibodies were from Upstate Technology (Buckingham, UK). α-RPA (Ab2, p34 subunit) antibodies were from Oncogene research products (Darmstadt, Germany). α-cjunSer63 antibodies were from Santa Cruz (Santa Cruz, CA). α-FANCD2 antibodies were from Novus Biologicals (Littleton, Colorado). Anti-rabbit, anti-rat and anti-mouse secondary antibodies were purchased from Dako (Glostrup, Denmark). α-IdU and α-CldU antibodies were from Beckton Dickinson (San Jose, CA) and Abcam (Cambridge, UK). Note that both antibodies are raised against BrdU but crossreact only with the indicated halogenated nucleotide.
Immunofluorescence
Cells were fixed in 3% paraformaldehyde and 2% sucrose phosphate-buffered saline (PBS) for 10 min at room temperature and permeabilised in 20 mM HEPES pH 7.4, 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose and 0.5% Triton X-100 (Sigma-Aldrich, Poole, UK) for 2 min at 4°C. Coverslips were washed in PBS prior to immunostaining. Primary antibody incubations were performed for 40 min at 37°C at 1:100 dilutions (1:800 for α-γ-H2AX) in PBS supplemented with 2% bovine serum fraction V albumin (BSA) (Sigma-Aldrich, Poole, UK) and followed by washing in PBS. Incubations with α-mouse TRITC and FITC or with α-rabbit FITC secondary antibodies (Sigma-Aldrich, Pool, UK) were performed at 37°C at 1:100 in 2% BSA for 20 min. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, Poole, UK) for 10 min at 4°C. Coverslips were mounted in Vectashield (Vector Laboratories, Peterborough, UK). The error bars represent the standard deviation of the mean. A minimum of three experiments were carried out where error bars are shown. When indicated, fibroblasts were extracted with the standard permeabilisation buffer (0.2% Triton X-100) for 2 min before fixation. LBLs were extracted either mildly with permeabilisation buffer containing 0.1% Triton X-100 for 30 s, or more stringently with buffer containing 0.5% NP-40 for 1 min, followed by standard fixation and then three washes with this buffer.
Quantification of fluorescence intensity was performed on blind captured images using standardised capture settings, image processing and analysis performed on Simple PCI software. Variations in immunostaining between experiments were controlled for by standardising to the background in untreated cells. At least 100 cells were quantified for each time point.
FANCD2 ubiquitination
Well-proliferating LBLs were treated continuously for 24 h with 3 or 5 mM HU. Cells were pelleted and washed twice with PBS and then resuspended in 50 μl PBS. An equal volume of 2 × sample buffer (100 mM Tris–HCl pH 6.8, 4% SDS, 12% β-mercaptoethanol) was added, the sample vortexed and then boiled for 5 min. Samples were electrophoresed on a 6% SDS–PAGE gel followed by standard Western blot analysis.
RPA recruitment
Cells were washed with PBS, then exposed to 20 J m−2 UVC and incubated at 37°C for 1 h in medium supplemented with 100 μM Ara-C and 1 mM HU. RPA was detected by the standard indirect IF procedure, except that before PFA fixation the cells were permeabilised for 2 min (standard buffer but with 0.2% Triton X-100) and then washed three times with PBS.
G2/M checkpoint arrest
The CBPI assay. The CBPI procedure was as previously described with modification (Fenech and Morley, 1985; Gutierrez-Enriquez and Hall, 2003). In brief, cells were preincubated for 2 h with 0.1 μM APH, 0.2 mM HU or UV irradiated (2.5 J m−2 UV) and then washed twice with 10 ml of complete medium. Following incubation for 72 h in the presence of cytochalasin B (5 μg/ml), cells were processed for IF except that before fixation, they were swollen in 75 mM KCl for 10 min. Carnoy's fixative (3:1, methanol:acetic acid) was used in place of PFA. Cells were cytospun onto poly-L-lysine-coated slides in DAPI/acridine orange (2 μg/ml) solution.
Analysis by mitotic index. Cells were exposed to 2.5 J m−2 UV and incubated overnight in complete medium containing 1.5 μM nocodazole, followed by processing for IF as detailed above. Mitotic cells were detected by α-histone H3Ser10 antibodies and cells were counterstained with DAPI.
Replication fork stability assay
The procedure followed was as described previously with modification (Dimitrova and Gilbert, 2000; Feijoo et al, 2001). Cells were labelled with CldU (50 μM) for 20 min, pelleted, washed with PBS and resuspended in complete medium with 10 μM APH and incubated for 2 h. Cells were then preincubated with IdU (50 μM) for 20 min in the presence of 10 μM APH. Cells were pelleted and then swollen in 75 mM KCl for 10 min. This processing time allows fork reinitiation and IdU incorporation. IF was performed as described except that after permeabilisation cells were incubated with 2 M HCl for 30 min to denature the DNA. Cells were blocked for 1 h with 10% FCS in PBS and then incubated with both primary antibodies overnight at 4°C. The first label was detected with red secondary antibodies and the second with green secondary antibodies.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Acknowledgments
We thank Drs J Hall, M Taylor, K Sperling and M Zdzienicka for providing cells and Dr A D'Andrea for providing unpublished information. Work in the PAJ laboratory is supported by the Medical Research Council, the Human Frontiers Science Programme, the Primary Immunodeficiency Association, the Leukaemia Research Fund, the Department of Health and EU grant (FIGH-CT-200200207).
References
- Alderton GK, Joenje H, Varon R, Borglum AD, Jeggo PA, O'Driscoll M (2004) Seckel syndrome exhibits cellular features demonstrating defects in the ATR signalling pathway. Hum Mol Genet 13: (in press) [DOI] [PubMed] [Google Scholar]
- Andreassen PR, D'Andrea AD, Taniguchi T (2004) ATR couples FANCD2 monoubiquitination to the DNA damage response. Genes Dev 18: 1958–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley NJ, Holtzman DA, Flaggs G, Keegan KS, DeMaggio A, Ford JC, Hoekstra M, Carr AM (1996) The Schizosaccharomyces pombe rad3 checkpoint gene. EMBO J 15: 6641–6651 [PMC free article] [PubMed] [Google Scholar]
- Bressan DA, Baxter BK, Petrini JH (1999) The Mre11–Rad50–xrs2 protein complex facilitates homologous recombination-based double-strand break repair in Saccharomyces cerevisiae. Mol Cell Biol 19: 7681–7687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscemi G, Savio C, Zannini L, Micciche F, Masnada D, Nakanishi M, Tauchi H, Komatsu K, Mizutani S, Khanna K, Chen P, Concannon P, Chessa L, Delia D (2001) Chk2 activation dependence on Nbs1 after DNA damage. Mol Cell Biol 21: 5214–5222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR III, Hays L, Morgan WF, Petrini JHJ (1998) The hMre11/hRad50 protein complex and Nijmegen breakage-syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 93: 477–486 [DOI] [PubMed] [Google Scholar]
- Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD (2003) The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J 22: 6610–6620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahwan C, Nakamura TM, Sivakumar S, Russell P, Rhind N (2003) The fission yeast Rad32 (Mre11)–Rad50–Nbs1 complex is required for the S-phase DNA damage checkpoint. Mol Cell Biol 23: 6564–6573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo V, Robertson K, Bibikova M, Kim E, Grieco D, Gottesman M, Carroll D, Gautier J (2001) Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Mol Cell 8: 137–147 [DOI] [PubMed] [Google Scholar]
- Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J (2003) An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell 11: 203–213 [DOI] [PubMed] [Google Scholar]
- D'Amours D, Jackson SP (2001) The yeast Xrs2 complex functions in S phase checkpoint regulation. Genes Dev 15: 2238–2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova DS, Gilbert DM (2000) Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat Cell Biol 2: 686–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, Smythe C (2001) Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol 154: 913–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenech M, Morley AA (1985) Measurement of micronuclei in human lymphocytes. Mutat Res 147: 29–36 [DOI] [PubMed] [Google Scholar]
- Foray N, Marot D, Gabriel A, Randrianarison V, Carr A, Perricaudet M, Ashworth A, Jeggo P (2003) A subset of ATM and ATR-dependent phosphorylation events requires the BRCA1 protein. EMBO J 22: 2860–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchitto A, Pichierri P (2002) Bloom's syndrome protein is required for correct relocalization of RAD50/MRE11/NBS1 complex after replication fork arrest. J Cell Biol 157: 19–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatei M, Sloper K, Sorensen C, Syljuasen R, Falck J, Hobson K, Savage K, Lukas J, Zhou BB, Bartek J, Khanna KK (2003) Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J Biol Chem 278: 14806–14811 [DOI] [PubMed] [Google Scholar]
- Gennery AR, Slatter MA, Bhattacharya A, Barge D, Haigh S, O'Driscoll M, Coleman R, Abinun M, Flood TJ, Cant AJ, Jeggo PA (2004) The clinical and biological overlap between Nijmegen breakage syndrome and Fanconi anaemia. Clin Immunol 113: 214–219 [DOI] [PubMed] [Google Scholar]
- Girard P-M, Riballo E, Begg A, Waugh A, Jeggo PA (2002) Nbs1 promotes ATM dependent phosphorylation events including those required for G1/S arrest. Oncogene 21: 4191–4199 [DOI] [PubMed] [Google Scholar]
- Goodship J, Gill H, Carter J, Jackson A, Splitt M, Wright M (2000) Autozygosity mapping of a Seckel syndrome locus to chromosome 3q22.1–q24. Am J Hum Genet 67: 498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RC, Taniguchi T, D'Andrea AD (2003) Regulation of the Fanconi anemia pathway by monoubiquitination. Semin Cancer Biol 13: 77–82 [DOI] [PubMed] [Google Scholar]
- Gutierrez-Enriquez S, Hall J (2003) Use of the cytokinesis-block micronucleus assay to measure radiation-induced chromosome damage in lymphoblastoid cell lines. Mutat Res 535: 1–13 [PubMed] [Google Scholar]
- Haber JE (1998) The many interfaces of Mre11. Cell 95: 583–586 [DOI] [PubMed] [Google Scholar]
- Hartsuiker E, Vaessen B, Carr AM, Kohli J (2001) Fission yeast Rad50 stimulates sister chromatid recombination and links cohesion with repair. EMBO J 20: 6660–6671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horejsi Z, Falck J, Bakkenist CJ, Kastan MB, Lukas J, Bartek J (2004) Distinct functional domains of Nbs1 modulate the timing and magnitude of ATM activation after low doses of ionizing radiation. Oncogene 23: 3122–3127 [DOI] [PubMed] [Google Scholar]
- Iliakis G, Wang Y, Guan J, Wang H (2003) DNA damage checkpoint control in cells exposed to ionizing radiation. Oncogene 22: 5834–5847 [DOI] [PubMed] [Google Scholar]
- International Nijmegen Breakage Syndrome Study Group (2000) Nijmegen breakage syndrome. The International Nijmegen Breakage Syndrome Study Group. Arch Dis Child 82: 400–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna KK, Lavin MF, Jackson SP, Mulhern TD (2001) ATM, a central controller of cellular responses to DNA damage. Cell Death Differ 8: 1052–1065 [DOI] [PubMed] [Google Scholar]
- Kim ST, Xu B, Kastan MB (2002) Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev 16: 560–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin MF (2004) The Mre11 complex and ATM: a two-way functional interaction in recognising and signaling DNA double strand breaks. DNA Repair (Amst) 3: 1515–1520 [DOI] [PubMed] [Google Scholar]
- Lavin MF, Khanna KK (1999) ATM: the protein encoded by the gene mutated in the radiosensitive syndrome ataxia-telangiectasia. Int J Radiat Biol 75: 1201–1214 [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT (2004) Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304: 93–96 [DOI] [PubMed] [Google Scholar]
- Maser RS, Zinkel R, Petrini JH (2001) An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat Genet 27: 417–421 [DOI] [PubMed] [Google Scholar]
- Matsuura K, Balmukhanov T, Tauchi H, Weemaes C, Smeets D, Chrzanowska K, Endou S, Matsuura S, Komatsu K (1998) Radiation induction of p53 in cells from Nijmegen breakage syndrome is defective but not similar to ataxia-telangiectasia. Biochem Biophys Res Commun 242: 602–607 [DOI] [PubMed] [Google Scholar]
- Mirzoeva OK, Petrini JH (2001) DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol Cell Biol 21: 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Ranganathan V, New HV, Moreau LA, Stotsky M, Mathew CG, Kastan MB, Weaver DT, D'Andrea AD (2002) Interaction of FANCD2 and NBS1 in the DNA damage response. Nat Cell Biol 4: 913–920 [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Gennery AR, Seidel J, Concannon P, Jeggo PA (2004) An overview of three disorders associated with genetic instability: LIG4 syndrome, RS-SCID and ATR-Seckel syndrome. DNA Repair 3: 1227–1235 [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA (2003) A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 33: 497–501 [DOI] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 10: 886–895 [DOI] [PubMed] [Google Scholar]
- Petrini JH (1999) The mammalian Mre11–Rad50–nbs1 protein complex: integration of functions in the cellular DNA-damage response. Am J Hum Genet 64: 1264–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichierri P, Rosselli F (2004) The DNA crosslink-induced S-phase checkpoint depends on ATR–CHK1 and ATR–NBS1–FANCD2 pathways. EMBO J 23: 1178–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saar K, Chrzanowska KH, Stumm M, Jung M, Nurnberg G, Wienker TF, Seemanova E, Wegner RD, Reis A, Sperling K (1997) The gene for the ataxia-telangiectasia variant, Nijmegen breakage syndrome, maps to a 1 cM interval on chromosome 8q21. Am J Hum Genet 60: 605–610 [PMC free article] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, Sanal O, Lavin MF, Jaspers NGJ, Taylor MR, Arlett CF, Miki T, Weissman SM, Lovett M, Collins FS, Shiloh Y (1995) A single ataxia telangiectasia gene with a product similar to PI 3-kinase. Science 268: 1749–1753 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (1997) Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu Rev Genet 31: 635–662 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2001) ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev 11: 71–77 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155–168 [DOI] [PubMed] [Google Scholar]
- Squires S, Johnson RT, Collins ARS (1982) Initial rates of DNA incision in UV-irradiated human cells. Differences between normal, xeroderma pigmentosum and tumour cells. Mutat Res 95: 389–404 [DOI] [PubMed] [Google Scholar]
- Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM (1999) The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99: 577–587 [DOI] [PubMed] [Google Scholar]
- Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA (2004) ATM and DNA-PK function redundantly to phosphorylate H2AX following exposure to ionising radiation. Cancer Res 64: 2390–2396 [DOI] [PubMed] [Google Scholar]
- Taalman RDFM, Jaspers NGJ, Scheres JMJC, De Wit J, Hustinx TWJ (1983) Hypersensitivity to ionising radiation, in vitro, in a new chromosomal breakage disorder, the Nijmegen breakage syndrome. Mutat Res 112: 23–32 [DOI] [PubMed] [Google Scholar]
- Taylor AMR, McConville CM, Woods CG, Byrd PJ, Hernandez D (1993) Clinical and cellular heterogeneity in ataxia-telangiectasia. In Nato ASI Series, Gatti RA, Painter RB (eds) Vol. H77 Ataxia-Telangiectasia, pp 209–231. Berlin, Heidelberg: Springer-Verlag [Google Scholar]
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y (2003) Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 22: 5612–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, Beckmann G, Seemanova E, Cooper PR, Nowak NJ, Stumm M, Weemaes CMR, Gatti RA, Wilson RK, Digweed M, Rosenthal A, Sperling K, Concannon P, Reis A (1998) Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 93: 467–476 [DOI] [PubMed] [Google Scholar]
- Ward IM, Chen J (2001) Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 276: 47759–47762 [DOI] [PubMed] [Google Scholar]
- Ward IM, Minn K, Chen J (2004) UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J Biol Chem 279: 9677–9680 [DOI] [PubMed] [Google Scholar]
- Weemaes CMR, Hustinx TWJ, Scheres JMC, van Munster PJJ, Bakkeren JAJM, Taalman RDFM (1981) A new chromosomal instability disorder: the Nijmegan breakage syndrome. Acta Paediatr Scand 70: 557–564 [DOI] [PubMed] [Google Scholar]
- Wegner RD, Chrzanowska K, Sperling K, Stumm M (1999) Ataxia telangiectasia variants (Nijmegen breakage syndrome). In Primary Immunodeficiency Diseases, a Molecular and Genetic Approach, Ochs HD, Smith CIE, Puck JM (eds) pp 324–334. New York: Oxford University Press [Google Scholar]
- Yamaguchi-Iwai Y, Sonoda E, Sasaki MS, Morrison C, Haraguchi T, Hiraoka Y, Yamashita YM, Yagi T, Takata M, Price C, Kakazu N, Takeda S (1999) Mre11 is essential for the maintenance of chromosomal DNA in vertebrate cells. EMBO J 18: 6619–6629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY, Qin J (2002) SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev 16: 571–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XY, Wang X, Hu B, Guan J, Iliakis G, Wang Y (2002) An ATM-independent S-phase checkpoint response involves CHK1 pathway. Cancer Res 62: 1598–1603 [PubMed] [Google Scholar]
- Zou L, Cortez D, Elledge SJ (2002) Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev 16: 198–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA–ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3