

Abstract
TrmE is a 50 kDa guanine nucleotide-binding protein conserved between bacteria and man. It is involved in the modification of uridine bases (U34) at the first anticodon (wobble) position of tRNAs decoding two-family box triplets. The precise role of TrmE in the modification reaction is hitherto unknown. Here, we report the X-ray structure of TrmE from Thermotoga maritima. The structure reveals a three-domain protein comprising the N-terminal α/β domain, the central helical domain and the G domain, responsible for GTP binding and hydrolysis. The N-terminal domain induces dimerization and is homologous to the tetrahydrofolate-binding domain of N,N-dimethylglycine oxidase. Biochemical and structural studies show that TrmE indeed binds formyl-tetrahydrofolate. A cysteine residue, necessary for modification of U34, is located close to the C1-group donor 5-formyl-tetrahydrofolate, suggesting a direct role of TrmE in the modification analogous to DNA modification enzymes. We propose a reaction mechanism whereby TrmE actively participates in the formylation reaction of uridine and regulates the ensuing hydrogenation reaction of a Schiff's base intermediate.
Keywords: GTP binding, tetrahydrofolate, tRNA modification, TrmE
Introduction
TrmE is a member of the guanine nucleotide-binding proteins (GNBP), which bind and hydrolyse GTP. It contains a canonical G domain and is conserved in all three kingdoms of life. Normally, G-domain proteins cycle between a GTP-bound state, which represents the active state of the protein, and an inactive, GDP-bound state. The activation and inactivation of GNBP is further controlled by guanine nucleotide exchange factors (GEFs). GEFs catalyse the exchange of GDP to GTP and thereby activate the protein. GTPase activating proteins (GAPs) accelerate the slow intrinsic hydrolysis rate.
In contrast to the family of Ras-like small and the heterotrimeric large G proteins, which regulate many crucial cellular processes like differentiation, cell–cell adhesion and nuclear and vesicular transport by ‘switching' signalling pathways on and off, TrmE is believed to be directly involved in an enzymatic reaction, the modification of the wobble position uridine (U34) in tRNAs in bacteria, yeast and mammalia (i.e. tRNALys, tRNAGlu, tRNA4Leu, tRNA4Arg and probably tRNAGln). Bacterial strains lacking the 50 kDa TrmE protein are deficient in the biosynthesis of tRNA modified at position 5 (Elseviers et al, 1984). TrmE-assisted modification of U34 at the 5 position of the uridine base leads to 5-methylaminomethyl-uridine (mnm5U) in bacteria, 5-carboxymethylaminomethyl-uridine in yeast and 5-taurinomethyl-uridine in human.
The modification at U34 allows interaction with G and A, but restricts base pairing with C and U (Yokoyama et al, 1979; 1985; Yokoyama and Nishimura, 1995). This is extremely important in mixed codon box families (Glu, Gln, Lys, Leu and Arg) for which base pairing of U with C or U would lead to misincorporation of amino acids. Furthermore, the modification influences frameshifting during the translation process (Brierley et al, 1997; Hagervall et al, 1998; Bjork et al, 1999; Urbonavicius et al, 2001).
The modification of U34 requires many different proteins: MnmA catalyses the thiolation of U34 at the 2 position leading to s2U (Sullivan et al, 1985). In the modification pathway, TrmE (also called MnmE), together with the protein GidA (Elseviers et al, 1984; Brégeon et al, 2001), is believed to be involved in the addition of the cmnm group at the 5 position, although the precise role of both proteins in the modification reaction is unknown. In a following modification step, the TrmC protein is believed to catalyse the formation of mnm5U (Hagervall et al, 1987) (Figure 1). The modifications at the 5 and the 2 position of uridine are independent of each other, and thiolation of U34 has been performed in vitro with recombinant proteins (MnmA and IscS) (Lauhon, 2002; Kambampati and Lauhon, 2003).
Figure 1.
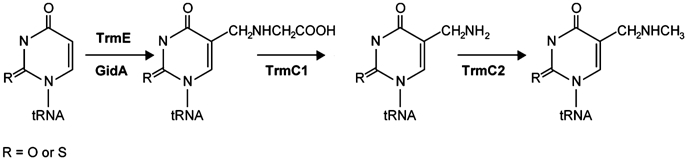
Proposed pathway for the biosynthesis of mnm5U at U34 in tRNA. TrmE and GidA are postulated to be involved in the first modification step for the biosynthesis of 5-carboxymethylaminomethyl-uridine (Elseviers et al, 1984; Brégeon et al, 2001). After cleavage and remethylation, both catalysed by TrmC (according to Hagervall et al, 1987), the final modification mnm5U is achieved. R can be oxygen or sulphur, corresponding to uridine or 2-thiouridine, respectively.
The details of the modification steps that lead to the cmnm5U34 modification are not known. TrmE consists of three regions, an N-terminal region of about 220 amino acids, a central G domain of approximately 160 residues and a C-terminal region of 75 amino acids, which contains a motif highly conserved among the TrmE protein family. The sequence of the N-terminal region does not present homology with any known protein and could be involved in the self-assembly of the full TrmE protein (Cabedo et al, 1999). The G domain, when isolated, conserves the high intrinsic GTPase activity of the intact TrmE molecule, which suggests that removal of the N- and C-terminal regions should not substantially affect its tertiary structure (Cabedo et al, 1999). The C-terminal end contains a highly conserved CxGK motif, which resembles a CaaX box, which in case of the GTP-binding protein Ras is farnesylated in vivo and plays an important role in membrane association and cell signalling (Bourne et al, 1990). But there is no evidence for this motif to be necessary for membrane localization of TrmE. Instead, the conserved cysteine is proposed to be important in the catalysis of the modification reaction; its mutation to serine disrupts the modification of tRNA in vivo (Yim et al, 2003), which leads to the hypothesis that the first step of the modification reaction might be analogous to the C5 modification of pyrimidine catalysed by DNA cytosine-5-methyltransferase (Vilkaitis et al, 2001), where the enzyme uses a cysteine for activation of the C5 position. Studies based on the homologous 5-taurinomethyl modification of U34 in humans showed that taurine is a direct constituent of the modification (Suzuki et al, 2002). Assuming a similar TrmE-mediated modification reaction in human and bacteria (5-taurinomethyl- versus 5-cmnm-uridine) glycine instead of taurine would be incorporated in the Escherichia coli reaction. This would still leave open the question of the nature of the C1 group and the covalent bond formation with glycine. It has been shown very early that S-adenosylmethionine is not the C1 donor in charge (Hagervall et al, 1987).
Mutant alleles of the GidA and TrmE homologues Mto1 and MSS1 in Saccharomyces cerevisiae reveal a respiratory-deficient phenotype (Decoster et al, 1993; Colby et al, 1998). Recent studies on GTPBP3 and Mto1, the human homologues of TrmE and GidA, lead to the suggestion that those proteins may also be involved in several human diseases like the nonsyndromic deafness or different clinical forms of myofibrillar myopathy (MERRF: myoclonic epilepsy; ragged red fibres/MELAS: mitochondrial encephalomyopathy; lactic acidosis; stroke), which are based on mutations in mitochondrial tRNA genes (Li and Guan, 2002; Li et al, 2002; Suzuki et al, 2002).
Since no detailed mechanistic description of the modification reaction exists, and the role of TrmE as a regulatory or catalytic reaction partner is unclear, we decided to get insight into the reaction by solving the structure of TrmE by X-ray crystallography.
Results
Recombinant expression and purification
TrmE from E. coli was expressed in BL21DE3 (TrmE−). The untagged protein was first purified by fractionated ammonium sulphate precipitation, followed by ion exchange chromatography on a Q-Sepharose column and size exclusion chromatography. Since crystals from E. coli TrmE did not diffract under any circumstances, we used it only for the biochemical experiments and turned to a thermophilic protein for structure determination. TrmE from Thermotoga maritima was expressed in Rosetta DE3 bacteria as an N-terminal His-tag fusion protein. It was purified by affinity chromatography via a nickel-nta-sepharose column and size exclusion chromatography using a Superdex S200 column. The construct lacking the N-terminal domain (ΔN-TrmE, G102-K454) from E. coli was expressed as an N-terminal His-tag fusion protein and purified by affinity chromatography.
TrmE binds GDP/GTP with micromolar affinity
During the course of structural studies, we noticed that the affinity of nucleotide was higher than anticipated from previous studies (Cabedo et al, 1999). We thus determined the equilibrium binding parameters of TrmE from E. coli by using fluorescent mant-nucleotides (mant: methylanthraniloyl). As observed for most other GTP-binding proteins, addition of protein to mant-nucleotides produces a large increase in fluorescence (Herrmann and Nassar, 1996). Using a constant concentration of nucleotide and increasing concentration of protein produces a binding isotherm (Figure 2A) that can be fitted to a binding equation. The Kd values for guanosine di- and triphosphate are in the micromolar range, with an affinity for GDP and GppNHp of 0.62 and 5.8 μM, respectively. The difference in affinity between GDP and GTP may actually be smaller since GppNHp is often found to bind with weaker affinity than GTP itself. GMP binds with an affinity lower than 100 μM, which is similar to Ras-like proteins where the affinity to GMP is orders of magnitude lower than that of GDP/GTP (Vetter and Wittinghofer, 2001).
Figure 2.
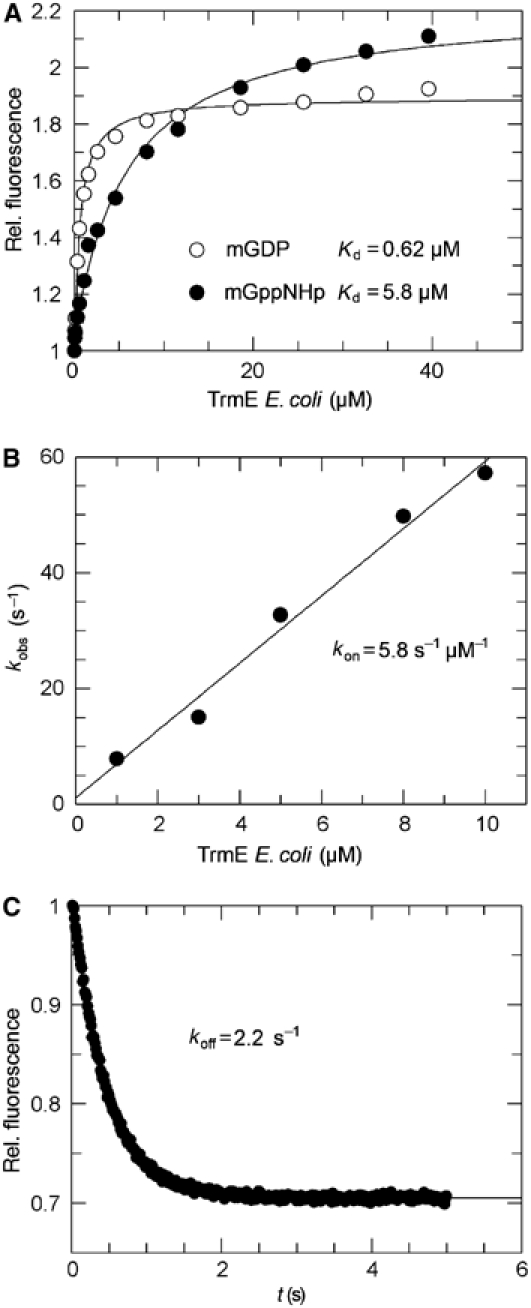
Nucleotide-binding properties of TrmE from E. coli. (A) Determination of the equilibrium dissociation constant Kd for mGDP and mGppNHp by fluorescence equilibrium titration. (B) Determination of the association rate constant for mGDP by stopped flow under pseudo-first-order conditions and (C) the dissociation rate constant for mGDP, as described in Materials and methods. Association and dissociation rates result in a Kd of 0.38 μM for mGDP.
Additionally, kinetic parameters for mGDP binding were determined by stopped flow (Figure 2B and C). The association rate constant was measured by using pseudo-first-order conditions (TrmE in large molar excess over nucleotide). Mant-nucleotides (100 nM) were mixed with increasing concentrations of protein (1–13 μM) and the increase in mant fluorescence was monitored. The fluorescence transients were fitted single exponentially and the observed rate constants kobs were plotted against the protein concentration to give an association rate constant of 5.8 μM−1 s−1 (Figure 2B). The dissociation rate constant koff was measured by displacing mGDP from TrmE by a 200-fold excess of unlabelled nucleotide. The decrease of the fluorescence signal was fitted single exponentially for a rate constant of 2.2 s−1 (Figure 2C). The ratio of koff and kon gives the dissociation constant Kd of 0.380 μM for the TrmE to mGDP interaction, similar to the value found by equilibrium titration.
Previously, an affinity for GTP in the range of 280 μM and an even lower affinity for GDP (>1 mM) were reported, which would be unusually low for a GTP-binding protein. Here we can show that the affinities are indeed much higher than reported and that the nitrocellulose filter binding method might not be appropriate for measuring affinities in the micromolar range, as has been observed before (Lenzen et al, 1998). Our measurements also indicate that the Km for GTP measured previously from the GTP dependence of the GTPase reaction (Cabedo et al, 1999) is most likely not equal to the Kd for GTP.
Overall fold of TrmE
Nucleotide-free TrmE from T. maritima crystallized in the space group P6(2). The crystal structure was solved at 2.3 Å using the single-wavelength anomalous dispersion (SAD) method after Se-Met incorporation. TrmE is a three-domain protein composed of the N-terminal α/β domain, residues 1–118, a central exclusively helical domain formed by residues 119–210 from the middle and the C-terminal residues 381–450, and the G-domain residues 211–380 (Figure 3B).
Figure 3.
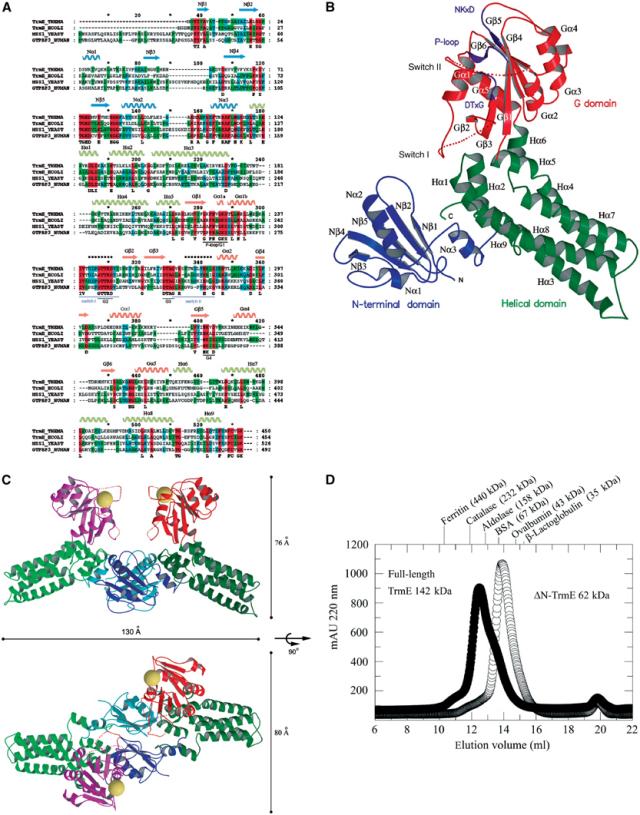
Overall structure of TrmE. (A) Sequence alignment of TrmE from T. maritima (Swissprot accession number Q9WYA4), TrmE from E. coli (Swissprot accession number P25522), MSS1 from S. cerevisiae (Swissprot accession number P32559) and GTPBP3 from Homo sapiens (Swissprot accession number Q8WUW9) with secondary structure assignment determined with DSSP (Kabsch and Sander, 1983). Domains are coloured in blue (N-terminal domain), green (central helical domain) and red (G domain). Flexible regions with weak density are marked with a dashed line (switch I and II). (B) Ribbon presentation of the tertiary structure of TrmE. The N-terminal domain is shown in blue, the central helical domain in green and the G domain in red. The flexible switch regions in the G domain are indicated by dashed lines. The nucleotide-binding motifs and switch regions are marked in purple. (C) Ribbon model of the putative TrmE homodimer in two orientations. Based on the position of the second N-terminal domain (molecule B), the orientation of full-length molecule B was modelled. The homodimerization of TrmE is mainly mediated by the N-terminal domain (blue and light blue). The homodimer has an elongated shape with a size of approximately 130 Å along the longest axis and approximately 76 or 80 Å from the N-terminal domains to the G domain or from G domain to G domain, respectively. Putative nucleotide-binding sites are marked by a sphere. (D) Gel filtration of ΔN-TrmE and full-length TrmE. ΔN-TrmE elutes with a lower apparent molecular mass (62 kDa) than full-length protein (142 kDa). The equilibrium for ΔN-TrmE is on the monomer side, whereas full-length protein is present as homodimer.
The crystallographic asymmetric unit contains two molecules, one of which corresponds to the full-length protein, whereas, surprisingly, the second molecule only contains the N-terminal domain, residues 1–118. Apparently, the second molecule is proteolysed in the course of the crystallization to form the observed structure. Dissolved crystals indeed showed an additional band at approximately 14 kDa corresponding to this degraded fragment (not shown). To show that TrmE is a dimer in solution also, we performed a gel filtration experiment (Figure 3D). This showed that the majority of the full-length protein runs with an apparent molecular mass of 142 kDa, with only a slight shoulder running at 65 kDa. We conclude that TrmE is most likely a dimer in solution and that the larger apparent mass is due to the elongated shape of the dimeric molecule (Figure 3C).
We thus believe (see also below) that the dimerization observed in the crystal is a true representation of the dimer formed by full-length protein in solution. The full-length dimer was modelled by superimposing the full-length structure on top of the N-terminal domain (Figure 3C). The superimposition does not lead to any clashes of structural elements. The whole dimer extends over a length of 130 Å, the width and height is approximately 76 Å. In the dimer, the G domains come into proximity with the putative nucleotide-binding sites facing each other (Figure 3C).
The G domain
The G domain of TrmE has the canonical Ras-like fold (Figure 4A), with no insertion or deletion of secondary structural elements. The 169 residues almost match the number of residues in the minimal G domain (Vetter and Wittinghofer, 2001). TrmE contains at least four of the five conserved nucleotide-binding motifs GxxxxGKS/T or P-loop (Saraste et al, 1990), T, DxxG and NKxD (Bourne et al, 1990; 1991). The totally invariant alanine in the SAK/L (G5) motif of Ras and Gα proteins is less well conserved. The G domain in the X-ray structure is only loosely connected to the residual domains of TrmE, which may explain why the G domain alone can be expressed and exhibits a GTPase activity similar to that of the full-length protein (Cabedo et al, 1999).
Figure 4.
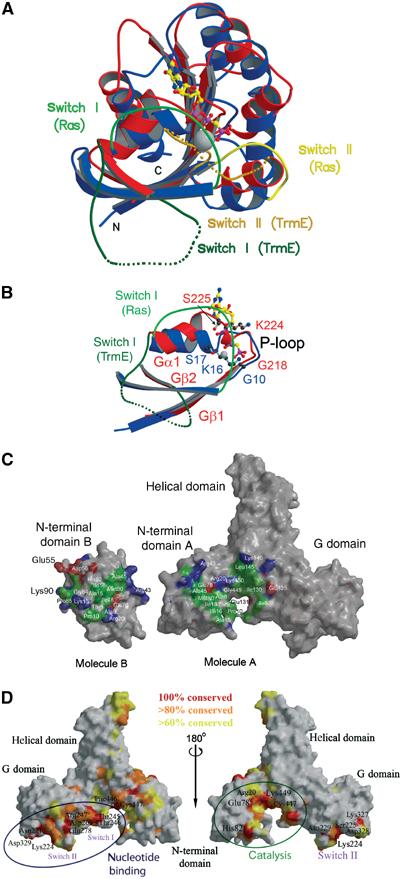
Structural details. (A) Superimposition of the G domains of TrmE and Ras (PDB 121P). The G domain of TrmE has the canonical fold of Ras without additional secondary structure elements. A total of 118 of the 169 C-α positions can be aligned with a maximal r.m.s.d. of 3.6 Å. The G domain of TrmE is shown in red and Ras in blue. (B) Superimposition of the P-loop region of TrmE and Ras. The GKS motif of TrmE is misoriented and an additional 3,10-like helix is formed, which occupies the putative position of the α- and β-phosphate of the nucleotide. Helix Gα1 is moved by 25° compared to the orientation in Ras. Colour coding is according to panel A. (C) Surface representation of TrmE calculated with GRASP (Nicholls et al, 1991), highlighting the interface between monomers. Residues forming the interface (distance <3.5 Å between molecules A and B) are coloured according to biochemical properties. Hydrophobic, acidic and basic residues involved in the interaction are shown in green, red and blue. (D) Conserved surface-exposed residues in a surface representation of TrmE. Totally conserved residues are coloured in red, 80% conserved in orange and 60% conserved in yellow (based on alignment of 79 TrmE sequences). Large conserved areas are in the nucleotide-binding region and at the dimerization interface of the N-terminal domain. Additionally, the tip of the helical domain and the region on the opposite side of the dimer interface are well conserved.
Although we did not succeed in obtaining diffraction quality crystals of the nucleotide-bound form of TrmE, the nucleotide-binding site can clearly be inferred from the comparison with Ras (Figure 4A). Superimposition of the G domains of Ras and TrmE leads to a root mean square deviation (r.m.s.d.) of 1.2 Å for 70 residues, which form the central β-sheet core of the G domain. In all, 118 of the 169 C-α positions could be aligned with a maximal r.m.s.d. of 3.6 Å. The helices flanking the central β-sheet core on both sides show larger deviations. In the G1/P-loop region, which contacts the β- and γ-phosphate of the nucleotide, the r.m.s.d. is in the range of 2.7 Å.
Superposition of the P-loop region of TrmE and Ras (see Figure 4B) reveals a large displacement of the GKS motif, while Gβ1 and residue G218 of TrmE still align very well with Ras. An additional 3,10-like helix is formed, which occupies the position of the α- and β-phosphate. Additionally, helix Gα1 of TrmE is moved by approximately 25°, thereby shifting the orientation of switch I and most probably also the position of Gβ2. The binding of nucleotide would break up the 3,10-like helix and allow the canonical interaction of the P-loop residues with the nucleotide and the magnesium ion.
The positions of the DxxG (contacting γ-phosphate and Mg2+) and NKxD (recognition of the guanine ring) superimpose very well with Ras (r.m.s.d. <1.8 Å). In Ras-like and heterotrimeric G proteins, switch regions have been found to change their structure with the nature of the bound nucleotide. In nucleotide-free TrmE, the switch regions are highly flexible and only weak density was visible in the electron density map. In analogy to other G-domain structures, switch I and II are expected to be stably connected to the core of the G domain upon GTP binding. Since effector proteins sense the nucleotide state of GTP-binding proteins, switch I and II are involved in effector binding in regulatory G proteins. If TrmE has a regulatory role in the modification reaction, we would expect that the switch regions participate in effector binding.
N-terminal domain
The N-terminal domain is composed of five β-strands and three α-helices. The antiparallel β-sheet is arranged in a Greek Key motif. Helix Nα1 is inserted between Nβ2 and Nβ3. Helices Nα2 and Nα3 are located C-terminally of Nβ5, of which helix Nα3 links the N-terminal domain to the central helical domain. Molecule B in the TrmE structure corresponds to a second N-terminal domain, which forms a tight dimer with molecule A.
To show that the N-terminal domain mainly mediates dimer formation, we constructed a protein that lacks the N-terminal domain (ΔN-TrmE) by deleting residues 1–101. The ΔN-TrmE construct was analysed on a gel filtration (Figure 3D) column and eluted with an apparent molecular mass of 62 kDa, much lower than that observed for the full-length protein (142 kDa). There is a slight shoulder at the position of the expected dimer, indicating that after deletion of the N-terminal domain, the monomer–dimer equilibrium is almost totally on the monomer side, while full-length protein shows the opposite behaviour. The tendency for TrmE to form higher aggregates has been shown before by gel filtration (Cabedo et al, 1999; Yamanaka et al, 2000).
The dimerization interface determined from the X-ray structure spans an area of 3260 Å2, using a 1.5 Å ball to probe the surface (Figure 4C). Of this, approximately 1720 Å2 is formed by the interactions of the N-terminal domains alone. The N-terminal domain (molecule B) additionally interacts with the central helical core domain of molecule A, via an additional area of 1540 Å2. Using a model of the full-length TrmE dimer as shown in Figure 3C, the total surface would cover approximately 4800 Å2 (3260+1540 Å2), a strong indication (although no proof) for a constitutive dimer.
Homodimerization involves a number of residues (Figure 4C), where the inner part of the interface is formed by more hydrophobic and the outer rim by mostly charged residues. The hydrophobic core residues are not well conserved (Figure 3A), which is not surprising since they involve a number of main-chain/main-chain or main-chain/side-chain interactions, such as between Ala45 and Arg43, and between Ala8 and Ala15/Ile16. The polar residues Lys13B, Lys90B and Asp56B stabilize the dimer formation by charged interaction with Glu131A, Glu135A and Lys146A.
Central helical domain
The central helical domain consists of nine α-helices. The first part with helices Hα1–Hα5 (residues 119–210) and the second part with helices Hα6–Hα9 (residues 381–450) flank the G domain on both sides. Helices Hα3, Hα4, Hα7 and Hα8 form a long four-helix bundle, which stabilizes the core structure of the domain. The domain contains a totally conserved C-terminal FCV/I/LGK motif (Figures 3A and 4D). As shown by Yim et al (2003), mutation of this totally conserved Cys447 (Cys451 in TrmE from E. coli) residue blocks the modification reaction in vivo. We find the C-terminal loop to be stabilized by interaction with helices Nα3, Hα1 and Hα7.
The position of the C-terminal loop in TrmE is additionally stabilized by several highly conserved interactions. As shown in Figure 5C, the highly conserved Glu78A in the dimer interface stabilizes the conserved Arg20A, which in turn interacts with the main-chain carbonyl group of Gly449. The C-terminus is also stabilized by side-chain interaction with Thr109A.
Figure 5.
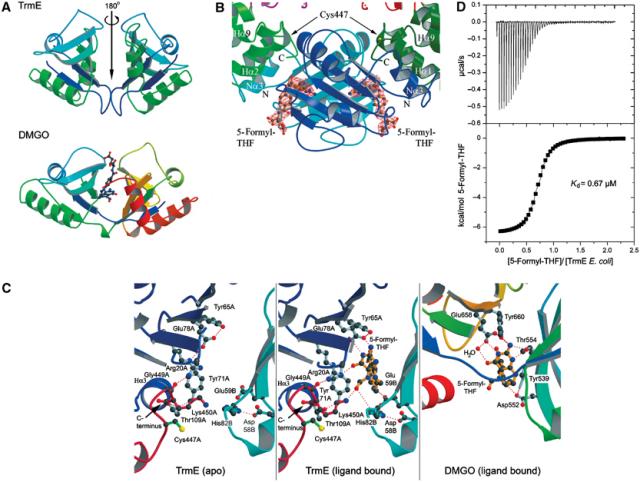
THF binding and catalysis. (A) Comparison of the topology of the N-terminal domain dimer with the THF-binding domain of DMGO (PDB: 1PJ7; Leys et al, 2003). Colour coding is according to the primary structure. The THF-binding site in DMGO is encoded on a single polypeptide, while homodimerization would be required to create a similar THF-binding site in TrmE. Dimerization would also create a second, symmetry-related THF-binding site. (B) The two THF-binding sites in the TrmE dimer. The binding sites are located at the periphery of the N-terminal dimerization interface. On top of the ligand, at a distance of 11 Å, the catalytic cysteine residue is located. The bound 5-formyl-THF molecules in TrmE are surrounded by an Fo−Fc electron density map contoured at 3σ. Orientation of the molecules and colour code are analogous to Figure 3C. (C) Detailed view of the THF-binding site in the apo- (left) and the cofactor-bound (right) form. The binding site for 5-formyl-THF in DMGO is shown for comparison. (D) Binding of 5-formyl-THF to TrmE determined by isothermal titration calorimetry, producing a Kd of 0.67 μM.
Apart from the C-terminal loop, the only other but less well-conserved parts of TrmE are the tip region of the four-helix bundle and the back of the dimer interface (Figures 3A and 4D).
Tetrahydrofolate-binding site of the N-terminal domain
Using the DALI server to find homologous structures, we find a high similarity between the N-terminal domain and the tetrahydrofolate (THF)-binding domain of N,N-dimethylglycine oxidase (DMGO) (PDB: 1PJ7; z-score: 10.1; r.m.s.d.: 3.1 Å) (Leys et al, 2003). As shown in Figure 5A, the dimer of the N-terminal domains with its local noncrystallographic symmetry axis has the same topology as the THF-binding region in DMGO, but in the case of the latter, the THF-binding domain is present on a single polypeptide, whereas both N-terminal domains of TrmE would be required to form a similar THF-binding site.
The homology to the THF-binding domain of DMGO suggested that TrmE might also bind THF and that some form of THF would be used to transfer a C1 group onto tRNA. To test this hypothesis, crystals of TrmE were soaked with 5-formyl-tetrahydrofolate (5-formyl-THF). The crystals diffracted up to 2.9 Å and additional density for bound 5-formyl-THF was clearly visible (Figure 5B). In DMGO, the two subdomains form a single binding site for THF, which is located at the periphery of the contact area of the subdomains. In TrmE instead, the two N-terminal domains are related to each other by a local two-fold noncrystallographic symmetry. The THF-binding site is located at the periphery of the dimer interface as seen for DMGO and involves residues from both subunits. Due to the two-fold symmetry of the N-terminal domains in TrmE, an additional THF-binding site is formed at the symmetry-related position in the TrmE dimer interface (see Figure 5A and B).
Binding of 5-formyl-THF did not lead to a significant rearrangement of the backbone. The binding site for 5-formyl-THF is very similar to the one of DMGO (Leys et al, 2003) (Figure 5C). The pteridin group is bound by a double hydrogen bond to the totally invariant Glu78 from molecule A (Glu78A), which corresponds to Glu658 in DMGO. This is analogous to the double hydrogen bond between the base of guanine nucleotides and the conserved aspartic acid in the NKxD motif (Vetter and Wittinghofer, 2001). In the apo form, Glu78A stabilizes Arg20A, which contacts the C-terminal FCV/IGK loop. In the 5-formyl-THF-bound form, this interaction of Arg20A and Glu78A is broken up and the residues are pushed away from each other by the ligand. Arg20A directly stabilizes the carbonyl group of the pteridin ring but still maintains the contact to the C-terminal loop. In DMGO, a glutamate instead of an arginine indirectly binds to the carbonyl position via a bridging water molecule. The residue at position 59B (conserved as Glu, Gln, Asp or Asn) could take over the role of Asp552 in DMGO by stabilizing the N10 position of THF. Ile16B (conserved as Ile/Val), Tyr71A (Tyr/Phe) and Val61B (Val, Met, Leu, Ile) form a hydrophobic pocket for the pteridin ring comparable to Tyr651 and Leu508 in DMGO. In cells, the THF cofactor has a variable length (1–8) of glutamate residues. A number of positively charged residues are found close to the THF-binding pocket, which could stabilize a possible poly-Glu tail of THF.
Although it had been suggested that THF might be a one-carbon unit (C1) donor in the modification reaction, the question as to which oxidation state of THF is used still remains. For the mechanism that is becoming apparent, we would favour it to be to the 5-formyl-THF derivative. This hypothesis is supported by the structure. The invariant His82 from the symmetry-related monomer (His82B), stabilized by the highly conserved Asp58B, contacts the oxygen of the 5-formyl group. In the apo form, the position of 5-formyl is occupied by a sulphate ion. Lys450A, which is rather flexible in the apo structure, is in a position to contact the oxygen of the formyl group as well. The contacts to His82B and Lys450A could stabilize the negative charge that develops at the oxygen in the assumed reaction intermediate (see below) between uracil and the formyl group. All of this supports the notion that 5-formyl-THF is the likely substrate of TrmE.
Apart from the structural evidence, we can also demonstrate biochemically that 5-formyl-THF can bind to TrmE. Using TrmE from E. coli (40 μM) in the cell and 5-formyl-THF (400 μM) as injectant, we determined by isothermal titration calorimetry that 5-formyl-tetrahydrofolic acid is bound with an affinity of 0.67 μM (Figure 5D). The binding is due to both a favourable enthalpy (ΔH=−6640 cal/mol) and entropy (T ΔS=1846 cal/mol). Although the stoichiometry obtained is somewhat less than unity, we are confident from the structural analysis that the TrmE dimer binds two molecules of 5-formyl-THF.
Discussion
Taking into account the essential role of Cys447 in the modification reaction in vivo, the position and orientation of the cysteine and the proximity to 5-formyl-THF in the TrmE structure support a catalytic role for this totally conserved residue. We would postulate that Cys447 forms a covalent adduct via the 6 position of the pyrimidine ring of uracil, as also discussed previously (Yim et al, 2003), and that the formyl group is added to the 5 position. Although the distance between Cys447 and the 5-formyl group of THF of approximately 11 Å is large, it is not unreasonable to assume that the binding of tRNA, GidA or nucleotide or a combination of these and other as yet unknown factors should induce a conformational change in TrmE large enough to close the gap between the two focal points of the reaction. Proteolytic digest of TrmE leads to major fragments of approximately 36 and 39 kDa. Based on the domain organization of TrmE reported here and the fragmentation pattern found in the crystal, such fragments could correspond to TrmE molecules, which lack either the complete N-terminal domain (1–118, approximately 37 kDa) or the central core of the N-terminal domain (1–102, approximately 39 kDa). In the presence of GTP, TrmE proteolysis produces additional fragments of 28 and 21 kDa, indicating a GTP-induced conformational change. This is supported by experiments with mutants that have a defect in GTP binding and/or GTP hydrolysis and are much less sensitive to further proteolysis (Yim et al, 2003). The link between the N-terminal domain and the helical part seems to be flexible and accessible for proteases, which is a hint that this region could act as a hinge for the conformational change that brings the Cys and the cofactor THF closer to each other.
Nothing is known about the interaction between TrmE and tRNA. It is not even clear if TrmE alone is sufficient for binding since we have been unable to biochemically demonstrate an interaction between the protein and in vitro-transcribed RNA. We have also not been able to reconstitute the modification reaction of tRNA with purified components. Nevertheless, on the basis of the postulated mechanism (see below), the anticodon loop of tRNA would have to bind near the THF-binding pocket and the catalytic cysteine. Indeed, the region around the invariant Cys447 and Lys449 of the C-terminal motif is the second most highly conserved area of the molecule, and also contains the invariant residues Glu78, His82 and Lys450 (Figure 4D), which we show to be required for 5-formyl-THF binding. Moreover, TrmE contains three additional conserved surface patches. While one of these contains the conserved canonical G-domain residues, the other two patches could mark the binding sites of tRNA, GidA or other as yet unidentified components of the reaction. We could speculate that the distance between Cys447 and the conserved tip of the helical domain (55 Å) approximately matches the distance between the anticodon loop and the TψC arm (approximately 63 Å) or the D arm (approximately 46 Å). In human tRNALys from MERRF patients, a single point mutation in the TψC arm is responsible for hypomodification at U34 and development of the disease; in MELAS disease, a point mutation in tRNALeu present in the D arm leads to hypomodification at U34 (Yasukawa et al, 2000a; 2000b; 2001). For the exact localization of the tRNA-binding site, structural studies with tRNA and protein components of the modification reaction will obviously be required.
Taking all the structural and biochemical results together, we propose the following mechanism of the modification reaction shown in Figure 6. According to the model, TrmE first activates the 5 position of U34 in tRNA by a mechanism analogous to DNA cytosine-5-methyltransferase or other pyrimidine modification systems. Cys447 would be an active thiolate that nucleophilically attacks the 6 position of uracil and creates a carbanion at the 5 position. This could possibly be aided by the totally invariant lysine in the C-terminal loop whose positive charge would electrostatically stabilize and thus activate the nucleophile. Similar mechanisms have been postulated for a conserved arginine found in thymidylate synthase, which performs a methylation reaction at C5 (Finer-Moore et al, 2003), or the conserved arginine in protein-tyrosine phosphatases, where a thiolate attacks and cleaves a phosphotyrosine bond (Stone and Dixon, 1994).
Figure 6.
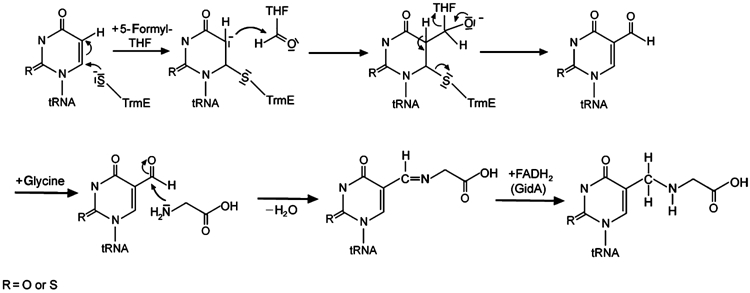
Proposed mechanism for the biosynthetic pathway leading to cmnm5U34, taking into account the results presented here. Cys447 of TrmE activates the 5 position of uracil by covalent bond formation to the 6 position. This enables a C1-group transfer from 5-formyl-THF bound in TrmE near the catalytic cysteine residue. Glycine is incorporated, analogous to the incorporation of taurine in H. sapiens (Suzuki et al, 2002), by a Schiff's base, which is then hydrogenated by GidA, an FAD-containing reduction system, as described in detail in Results. R can be oxygen or sulphur, corresponding to uridine or 2-thiouridine, respectively.
We would further propose that the C1 group being attached to the 5 position is a formyl group supplied by 5-formyl-THF. We have shown here that 5-formyl-THF binds directly to TrmE in a pocket that is very similar to the THF-binding site of the enzyme DMGO and that the binding site would apparently favour the 5-formyl group for the condensation reaction. A nucleophilic attack of the activated base onto the carbonyl of the formyl moiety leads to a transfer of the formyl group onto the 5 position of the uridine base. The developing negative charge on the formyl group could in turn be stabilized by positive charges found in the vicinity of the oxygen of the formyl group (Lys450 and His82). Although in the structure determined here, the catalytic residue Cys447 and the formyl group are 11 Å apart and thus not close enough to act in common on the uridine base, we would expect that binding of tRNA, nucleotide and/or GidA induces a conformational change that brings Cys447 and the formyl group in juxtaposition. It could also be that GTP hydrolysis is necessary to reorganize the active site for formyl transfer (Yim et al, 2003). Since the GTPase active sites are not close to the formyl transfer site, such a GTPase-induced conformational change might be coupled to or mediated by further binding partners such as tRNA itself, the protein GidA or other as yet unknown components of the reaction pathway.
It has been shown that taurine is directly incorporated into human mitochondrial tRNAs in a modification scheme involving the human homologues of TrmE and GidA by an unknown mechanism (Suzuki et al, 2002). In analogy to this, we would postulate that glycine is incorporated into the corresponding E. coli tRNAs after the covalent bond between U34 and TrmE is cleaved. Nucleophilic attack of the amine group of glycine onto the formylated uridine leads to the formation of a Schiff's base, which would have to be reduced subsequently to create the product cmnm5U34. While the enzyme donating glycine and reducing the Schiff's base is not known, a good candidate for this is the protein GidA. We find GidA from E. coli to have FAD bound as a cofactor (data not shown), as demonstrated for the homologous protein from Myxococcus xanthus (White et al, 2001), which could be used for reduction of the Schiff's base in an FADH2-dependent manner. Since GidA is large enough to harbour more than just an FAD-binding domain, it might also be able to bind and deliver the glycine moiety. The activity of GidA in the whole modification reaction could possibly be regulated by TrmE, as it has previously been reported that TrmE and GidA form a complex (Colby et al, 1998).
In summary, the structural and biochemical studies presented here suggest how TrmE, a GTP-binding protein conserved between bacteria and man, most likely directly participates in at least the first step of the cmnm modification reaction in the anticodon loop of certain tRNAs. While we have been unable to reconstitute the reaction in vitro and it is likely that further components of the reaction are still missing, we have nevertheless made an important step towards elucidating the complete reaction pathway.
Materials and methods
Plasmids
TrmE from E. coli was expressed from plasmid pIC933 (pET15b derived). For the expression of TrmE from T. maritima, the encoding DNA fragment was amplified by genomic PCR and cloned into a pET20-derived plasmid using HindIII and NotI cleavage sites. The construct for expression of ΔN-TrmE (G102-K454) from E. coli was amplified by PCR using pIC933 as template and cloned into a pET14b plasmid.
Protein expression and purification
N-terminally His6-tagged TrmE from T. maritima was expressed in E. coli strain Rosetta BL21DE3 in TB medium. At an OD600 of 0.6, expression was induced by addition of 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG). After 4 h induction, cells were harvested by centrifugation and resuspended in lysis buffer (50 mM Tris pH 7.5, 300 mM NaCl, 5 mM MgCl2, 5 mM β-mercaptoethanol (β-ME), 10 mM imidazole). The cells were lysed in a microfluidizer in lysis buffer containing 0.15 mM protease inhibitor phenylmethylsulphonyl fluoride (PMSF). After centrifugation at 35 000 g for 1 h, the supernatant was applied to an Ni-NTA column. The column was washed with lysis buffer containing 20 mM imidazole, followed by elution with 50 mM Tris pH 7.5, 300 mM NaCl, 5 mM MgCl2, 5 mM β-ME and 250 mM imidazole. Peak fractions were concentrated and purified by gel filtration on a Superdex S200 26/60 (50 mM Tris pH 7.5, 100 mM NaCl, 5 mM MgCl2, 5 mM dithioerythritol (DTE)). Fractions containing purified TrmE from T. maritima were concentrated to 10 mg/ml, flash-frozen in liquid nitrogen and stored at −80°C.
Full-length TrmE from E. coli was expressed in BL21DE3 (TrmE−) in TB medium. After induction and harvesting of the cells as described for TrmE from T. maritima, cells were resuspended in EDTA-lysis buffer (50 mM Tris pH 7.5, 100 mM NaCl, 5 mM DTE, 2.5 mM EDTA pH 8.4, 0.15 mM PMSF), lysed by microfluidizer and cell debris was removed by centrifugation at 35 000 g for 1 h. As first purification step, a fractionated ammonium sulphate precipitation was performed. Contaminant protein was removed by precipitation at 30% (w/v) ammonium sulphate and centrifugation at 21 000 g for 1 h. TrmE in the supernatant was precipitated by further increase of ammonium sulphate concentration to 40% (w/v). The ammonium sulphate pellet was resuspended in 250 ml low-salt buffer and the protein solution was applied to a Q-Sepharose ion exchange column equilibrated with low-salt buffer (50 mM Tris pH 7.5, 100 mM NaCl, 5 mM MgCl2, 5 mM DTE). After washing with low-salt buffer, protein was eluted by a salt gradient ranging from 100 to 750 mM NaCl. Fractions containing TrmE were pooled, precipitated with 50% (w/v) ammonium sulphate and stored at −20°C. TrmE was purified by gel filtration on a Superdex S200 26/60 (50 mM Tris pH 7.5, 100 mM NaCl, 5 mM MgCl2, 5 mM DTE). Fractions containing purified TrmE were concentrated to 100 mg/ml, flash-frozen in liquid nitrogen and stored at −80°C.
Overexpression and purification of His6-tagged ΔN-TrmE was analogous to the purification of TrmE from T. maritima.
Crystallography
Crystals of T. maritima TrmE were obtained using the hanging-drop/vapour diffusion method. Prior to crystallization, the protein solution was incubated at 65°C for 20 min and precipitated protein was removed by centrifugation. In all, 1 μl drops of 10 mg/ml TrmE solution were mixed with 1 μl of reservoir solution (1.8 M ammonium sulphate, 100 mM 2-[N-morpholino]ethanesulphonic acid pH 5.5). After 3–5 days, large crystals at 12°C grew to a dimension of 0.25 mm × 0.1 mm × 0.1 mm. For data collection, crystals were cryoprotected in reservoir solution containing 25% glycerol as cryoprotectant. A native data set was collected in Grenoble at ID14-1. The data set for the Se-Met protein was collected at ESRF ID-29. To obtain the 5-formyl-THF-bound structure, crystals were soaked in 2.5 M malonate pH 6.4 containing 10 mM 5-formyl-THF. Malonate at 2.5 M concentration was suitable as cryoprotectant and a data set was collected at ESRF ID14-2.
Collected data were processed with XDS (Kabsch, 1993). Initial heavy atom sites for SAD phasing were identified with SHELXD (Usón and Sheldrick, 1999). Refinement of the initial sites, phase determination and density modification with SHARP (de La Fortelle and Bricogne, 1997) led to an interpretable density map. Phase information was afterwards used to perform a phase extension to native 2.3 Å resolution using CNS (Brünger et al, 1998). The atomic model was built using XtalView/Xfit, and all refinement steps, consisting of bulk solvent correction, simulated annealing and B-factor refinement, were carried out with CNS. Table I summarizes the data collection and refinement statistics. Figures were generated using MolScript (Kraulis, 1991) and Raster3D (Merrit and Murphy, 1994). Molecular surfaces were generated with GRASP (Nicholls et al, 1991).
Table 1.
Data collection and refinement statistics
| Data set | Native | 5-Formyl-THF bound | Se-Met |
|---|---|---|---|
| X-ray source | ESRF ID14-1 | ESRF ID14-2 | ESRF ID-29 |
| Space group | p6(2) | p6(2) | p6(2) |
| Cell parameters | a=128.85 | a=130.03 | a=127.67 |
| b=128.85 | b=130.03 | b=127.67 | |
| c=107.16 | c=113.84 | c=113.87 | |
| Resolution (Å) | 19.7–2.3 | 19.9–2.9 | 19.7–3.7 |
| Wavelength (Å) | 0.934 | 0.934 | 0.9791 |
| Completeness (%) | 99.6 (99.7) | 99.3 (99.8) | 99.2 (98.8) |
| Unique reflections | 44 730 | 24 138 | 22 001 |
| I/σI | 15.5 (4.1) | 25.1 (7.4) | 15.8 (5.8) |
| Refinement | |||
| PDB code | 1XZP | 1XZQ | |
| Rworka (%) | 22.6 | 25.0 | |
| Rfreeb (%) | 25.2 | 30.7 | |
| Reflections (work/free) | 42 493/2237 | 21 747/2391 | |
| R.m.s.d. | |||
| Bond length (Å) | 0.006 | 0.010 | |
| Bond angle (deg) | 1.3 | 1.4 | |
| Ramachandran plot | |||
| Core (%) | 89.0 | 84.2 | |
| Allowed (%) | 9.3 | 15.2 | |
| Generously (%) |
1.7 |
0.6 |
|
| Values in parentheses correspond to the highest resolution shell. | |||
| Root mean square deviations (r.m.s.d.) are given as deviations from ideal values. | |||
| Rwork=∑h∣Fo−Fc∣/∑hFo, where Fo and Fc are the observed and calculated structure factor amplitudes of reflection h. | |||
| Rfree is the same as Rwork, but calculated on the reflections set aside from refinement. | |||
Fluorimetry/equilibrium titration
TrmE protein was titrated against 200 nM mant-nucleotides until saturation was reached. The mant fluorophore was excited at 360 nm, and emission was monitored at 450 nm (Fluoromax 2; Spex Industries). The increase in fluorescence upon addition of protein was integrated over at least 10 min and the determination of equilibrium dissociation constant, Kd, was carried out as described by Herrmann and Nassar (1996) by fitting a quadratic function to the data. Experiments were performed at 20°C in 50 mM Tris pH 7.5, 100 mM KCl, 5 mM MgCl2 and 5 mM DTE.
Stopped-flow kinetics
In stopped-flow experiments, E. coli TrmE in the concentration range of 1–13 μM was mixed with 100 nM mant-GDP, providing conditions for pseudo-first-order binding kinetics. Mant-GDP was excited at 360 nm and change in fluorescence was monitored through a 408 nm cutoff filter (SM-17; Applied Photophysics). For each protein concentration, the data obtained were fitted to a single exponential function, yielding the observed rate constant kobs. The association rate constant (kon) was obtained from the slope of a linear fit plotting kobs versus the protein concentration. The dissociation rate constant (koff) was obtained by mixing a preformed equimolar complex of TrmE and mant-GDP (4 μM) with a 200-fold excess of unlabelled GDP. The Kd values are calculated from the ratio of koff and kon. Experiments were performed at 20°C in 50 mM Tris pH 7.5, 100 mM KCl, 5 mM MgCl2 and 5 mM DTE.
Isothermal titration calorimetry
Binding affinity of 5-formyl-THF to E. coli TrmE was determined by isothermal titration calorimetry. The binding energy upon titration of 5-formyl-THF (400 μM) to TrmE (40 μM) was measured at 20°C in 50 mM Tris pH 7.5, 100 mM KCl, 5 mM MgCl2 and 2 mM β-ME; the data obtained were fitted using the manufacturer's software to obtain the Kd.
Analytical gel filtration
All analytical gel filtration experiments were carried out in 50 mM Tris pH 7.5, 100 mM KCl, 5 mM MgCl2 and 5 mM DTE containing additionally 50 μM GDP. A 1 mg portion of protein was incubated for 30 min on ice with 200 μM GDP prior to application onto the Superdex 200 10/30 column.
Unlabelled nucleotides as well as 5-formyl-THF were purchased from Sigma. Mant-labelled nucleotides were purchased from Jena Biosciences or synthesized in-house. Chemicals for crystallography were from Fluka with highest purity grade available.
Coordinates
Atomic coordinates and structure factors have been deposited at the Protein Data Bank (accession codes 1XZP and 1XZQ for the apo- and the THF-bound form of TrmE, respectively).
Acknowledgments
We gratefully acknowledge the use of beamlines ID14-1, ID14-2 and ID29 at ESRF Grenoble and thank the staff for professional support. We thank Ilme Schlichting, Wulf Blankenfeldt, Roman Fedorov, Axel Scheidig, Dennis Fiegen and Oezkan Yildiz for data collection. We thank Mathias Sprinzl for valuable advice on tRNA modifications. We also thank Astrid Krämer for assistance with biochemical experiments, Dorothee Kühlmann for technical and Rita Schebaum for secretarial assistance. We thank Michael Weyand for data collection and crystallographic assistance. ME Armengod acknowledges the support from the Ministerio Español de Ciencia y Technologia (grants BMC2001-1555 and BFU2004-05819).
References
- Bjork GR, Durand JM, Hagervall TG, Leipuviene R, Lundgren HK, Nilsson K, Chen P, Qian Q, Urbonavicius J. (1999) Transfer RNA modification: influence on translational frameshifting and metabolism. FEBS Lett 452: 47–51 [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F (1990) The GTPase superfamily: a conserved switch for diverse cell functions. Nature 348: 125–132 [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F (1991) The GTPase superfamily: conserved structure and molecular mechanism. Nature 349: 117–127 [DOI] [PubMed] [Google Scholar]
- Brégeon D, Colot V, Radman M, Taddei F (2001) Translational misreading: a tRNA modification counteracts a +2 ribosomal frameshift. Genes Dev 15: 2295–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I, Meredith MR, Bloys AJ, Hagervall TG (1997) Expression of a coronavirus ribosomal frameshift signal in Escherichia coli: influence of tRNA anticodon modification on frameshifting. J Mol Biol 270: 360–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Cabedo H, Macian F, Villarroya M, Escudero JC, Martinez-Vicente M, Knecht E, Armengod ME (1999) The Escherichia coli trmE (mnmE) gene, involved in tRNA modification, codes for an evolutionarily conserved GTPase with unusual biochemical properties. EMBO J 18: 7063–7076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby G, Wu M, Tzagoloff A (1998) MTO1 codes for a mitochondrial protein required for respiration in paromomycin-resistant mutants of Saccharomyces cerevisiae. J Biol Chem 273: 27945–27952 [DOI] [PubMed] [Google Scholar]
- Decoster E, Vassal A, Faye G (1993) MSS1, a nuclear-encoded mitochondrial GTPase involved in the expression of COX1 subunit of cytochrome c oxidase. J Mol Biol 232: 79–88 [DOI] [PubMed] [Google Scholar]
- de La Fortelle E, Bricogne G (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol 276: 472–494 [DOI] [PubMed] [Google Scholar]
- Elseviers D, Petrullo LA, Gallagher P (1984) Novel E. coli mutants deficient in biosynthesis of 5-methylaminomethyl-2-thiouridine. Nucleic Acids Res 12: 3521–3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finer-Moore JS, Santi DV, Stroud RM. (2003) Lessons and conclusions from dissecting the mechanism of a bisubstrate enzyme: thymidylate synthase mutagenesis, function, and structure. Biochemistry 42: 248–256 [DOI] [PubMed] [Google Scholar]
- Hagervall TG, Edmonds CG, McCloskey JA, Björk GR (1987) Transfer RNA (5-methylaminomethyl-2-thiouridine)-methyltransferase from Escherichia coli K-12 has two enzymatic activities. J Biol Chem 262: 8488–8495 [PubMed] [Google Scholar]
- Hagervall TG, Pomerantz SC, McCloskey JA (1998) Reduced misreading of asparagine codons by Escherichia coli tRNALys with hypomodified derivatives of 5-methylaminomethyl-2-thiouridine in the wobble position. J Mol Biol 284: 33–42 [DOI] [PubMed] [Google Scholar]
- Herrmann C, Nassar N (1996) Ras and its effectors. Prog Biophys Mol Biol 66: 1–41 [DOI] [PubMed] [Google Scholar]
- Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22: 2577–2637 [DOI] [PubMed] [Google Scholar]
- Kabsch W (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr 26: 795–800 [Google Scholar]
- Kambampati R, Lauhon CT (2003) MnmA and IscS are required for in vitro 2-thiouridine biosynthesis in Escherichia coli. Biochemistry 42: 1109–1117 [DOI] [PubMed] [Google Scholar]
- Kraulis PJ (1991) MolScript—a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24: 946–950 [Google Scholar]
- Lauhon CT (2002) Requirement for IscS in biosynthesis of all thionucleosides in Escherichia coli. J Bacteriol 184: 6820–6829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzen C, Cool RH, Prinz H, Kuhlmann J, Wittinghofer A (1998) Kinetic analysis by fluorescence of the interaction between Ras and the catalytic domain of the guanine nucleotide exchange factor Cdc25Mm. Biochemistry 19: 7420–7430 [DOI] [PubMed] [Google Scholar]
- Leys D, Basran J, Scrutton NS (2003) Channelling and formation of ‘active' formaldehyde in dimethylglycine oxidase. EMBO J 22: 4038–4048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Guan MX (2002) A human mitochondrial GTP binding protein related to tRNA modification may modulate phenotypic expression of the deafness-associated mitochondrial 12S rRNA mutation. Mol Cell Biol 22: 7701–7711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Li R, Lin X, Guan MX (2002) Isolation and characterization of the putative nuclear modifier gene MTO1 involved in the pathogenesis of deafness-associated mitochondrial 12S rRNA A1555G mutation. J Biol Chem 277: 27256–27264 [DOI] [PubMed] [Google Scholar]
- Merrit EA, Murphy MEP (1994) Raster3D version 20—a program for photorealistic molecular graphics. Acta Crystallogr D 50: 869–873 [DOI] [PubMed] [Google Scholar]
- Nicholls A, Sharp KA, Honig B (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281–296 [DOI] [PubMed] [Google Scholar]
- Saraste M, Sibbald PR, Wittinghofer A (1990) The P-loop—a common motif in ATP- and GTP-binding proteins. Trends Biochem Sci 15: 430–434 [DOI] [PubMed] [Google Scholar]
- Stone RL, Dixon JE (1994) Protein-tyrosine phosphatases. J Biol Chem 269: 31323–31326 [PubMed] [Google Scholar]
- Sullivan MA, Cannon JF, Webb FH, Bock RM (1985) Antisuppressor mutation in Escherichia coli defective in biosynthesis of 5-methylaminomethyl-2-thiouridine. J Bacteriol 161: 368–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Suzuki T, Wada T, Saigo K, Watanabe K (2002) Taurine as a constituent of mitochondrial tRNAs: new insights into the functions of taurine and human mitochondrial diseases. EMBO J 21: 6581–6589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbonavicius J, Qian Q, Durand JMB, Hagervall TG, Björk GR (2001) Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J 20: 4863–4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usón I, Sheldrick GM (1999) Advances in direct methods for protein crystallography. Curr Opin Struct Biol 9: 643–648 [DOI] [PubMed] [Google Scholar]
- Vetter IR, Wittinghofer A (2001) The guanine nucleotide-binding switch in three dimensions. Science 294: 1299–1304 [DOI] [PubMed] [Google Scholar]
- Vilkaitis G, Merkienè E, Serva S, Weinhold E, Klimasauskas S (2001) The mechanism of DNA cytosine-5 methylation. J Biol Chem 276: 20924–20934 [DOI] [PubMed] [Google Scholar]
- White DJ, Merod R, Thomasson B, Hartzell PL (2001) GidA is an FAD-binding protein involved in development of Myxococcus xanthus. Mol Microbiol 42: 503–517 [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Hwang J, Inouye M (2000) Characterization of GTPase activity of TrmE, a member of a novel GTPase superfamily, from Thermotoga maritima. J Bacteriol 182: 7078–7082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa T, Suzuki T, Ishii N, Ohta S, Watanabe K (2001) Wobble modification defect in tRNA disturbs codon–anticodon interaction in a mitochondrial disease. EMBO J 20: 4794–4802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa T, Suzuki T, Ishii N, Ueda T, Ohta S, Watanabe K (2000a) Defect in modification at the anticodon wobble nucleotide of mitochondrial tRNA(Lys) with the MERRF encephalomyopathy pathogenic mutation. FEBS Lett 467: 175–178 [DOI] [PubMed] [Google Scholar]
- Yasukawa T, Suzuki T, Ueda T, Ohta S, Watanabe K (2000b) Modification defect at anticodon wobble nucleotide of mitochondrial tRNAs(Leu)(UUR) with pathogenic mutations of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. J Biol Chem 275: 4251–4257 [DOI] [PubMed] [Google Scholar]
- Yim L, Martinez-Vicente M, Villarroya M, Aguado C, Knecht E, Armengod ME (2003) The GTPase activity and C-terminal cysteine of the Escherichia coli MnmE protein are essential for its tRNA modifying function. J Biol Chem 278: 28378–28387 [DOI] [PubMed] [Google Scholar]
- Yokoyama S, Nishimura S (1995) Modified nucleotides and codon recognition. In tRNA: Structure, Biosynthesis and Function, Söll D, RajBhandary UL (eds) pp 207–223. Washington, DC: American Society for Microbiology [Google Scholar]
- Yokoyama S, Yamaizumi Z, Nishimura S, Miyazawa T (1979) 1H NMR studies on the conformational characteristics of 2-thiopyrimidine nucleotides found in transfer RNAs. Nucleic Acids Res 6: 2611–2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama S, Watanabe T, Murao K, Ishikura H, Yamaizumi Z, Nishimura S, Miyazawa T (1985) Molecular mechanism of codon recognition by tRNA species with modified uridine in the first position of the anticodon. Proc Natl Acad Sci USA 82: 4905–4909 [DOI] [PMC free article] [PubMed] [Google Scholar]