

1. Introduction
An organism’s genome is under constant assault. An incessant barrage of endogenous and exogenous genotoxic stress requires continuous activation of DNA damage repair pathways to ensure the integrity of ongoing cellular processes. Normal metabolism produces an abundance of by-products that are reactive toward cellular DNA, for example reactive oxygen species that toxically oxidize nucleotides and β-unsaturated aldehydes that foster interstrand crosslinks [1]. In addition to these endogenous metabolites, outside sources like radiation and chemical mutagens can induce DNA lesions, such as pyrimidine dimers from ultraviolet rays or double strand breaks (DSBs) from γ-irradiation [2, 3].
The faithful replication of cellular DNA from progenitor to progeny and the proper transcription of genes depend on the accurate and timely repair of these nucleic encumbrances. Deficits in repair genes reduce stem cell proliferation and often engender mutations in cell cycle genes, so people harboring repair gene mutations are predisposed to developing hematopoietic deficiencies and cancer [4, 5]. Understanding the idiosyncrasies of repair pathways therefore becomes imperative for developing new medical strategies to prevent deleterious physiological consequences in patients lacking repair functions. However, despite substantial recent progress, the molecular mechanics governing these biochemical processes are far from transparent.
An illustrative cellular program in which to further characterize the various DNA damage repair pathways is meiosis. An absolute requirement for the correct completion of meiosis is the formation of an obligate crossover between each homologous pair [6]. This exchange of DNA between homologues is contingent upon the induction and subsequent repair of DSBs via homologous recombination (HR) [7]. Furthermore, since more DSBs are formed than crossovers made, alternate repair pathways exist to shunt DSBs to non-crossover outcomes and prevent the persistence of these toxic lesions [8]. Although some proteins are unique to meiosis, most of the requisite proteins that mediate DSB repair have been co-opted from the repair pathways used during mitotic proliferation [1, 8, 9]. Due to the high degree of endogenously produced DNA damage, the replisome regularly runs into adducts that thwart its ability to continue DNA synthesis. These impediments must be remodeled for replication to proceed and failure to do so during pre-meiotic S phase can lead to DNA lesions that are carried into meiotic prophase [8]. While nuclei harboring DNA damage—whether from lesions bypassed during replication or from unrepaired meiotically programmed DSBs—are culled by apoptosis, damage can persist in the germlines of animals with repair defects, leading to embryonic lethality in their progeny [8, 10]. Thus, the meiotic germline integrates numerous mechanisms to maintain genome integrity while ensuring the proper segregation of chromosomes into developing progeny and thereby lends itself to the investigation of multiple repair pathways.
C. elegans presents a convenient organism in which to explore meiotic outcomes. Meiotic progression is easily visualized by DAPI-staining the germline, which is organized in a spatio-temporal gradient [11]. Additionally, egg viability is a handy readout of meiotic success. By comparing the embryonic viability of wild type worms after mutagen exposure to that of mutant strains, it is possible to glean the genetic requirements for various DNA damage response pathways. Immunofluorescence of dissected gonads after mutagen exposure with antibodies of known repair pathway constituents, for instance RAD-51, an essential homologous recombination enzyme [12], further augments the capacity to determine the genetic prerequisites for repair.
Numerous studies have implemented mutagen exposure in C. elegans to examine repair pathways [13–22]. In fact, a comprehensive review enumerates the assays available to induce DNA damage and subsequently analyze the effects of the damage through egg viability, immunofluorescence, and apoptosis counts [23]. However, certain details of mutagen exposure lack consistency between studies and have thus far not been addressed, for instance the age of the worm at the initiation of treatment, the doses of each mutagen, and the implications behind the timing of post-exposure assays. This article aims to describe the detailed protocols for the most commonly cited methods of exposure, deliberate on particular parameters for mutagen exposure, discuss specific considerations for various mutagens, and, in general, consolidate useful information for those grappling with these tricky assays.
2. Mutagen Exposure via Liquid Culture
2.1 Materials
Glass pipets
1×M9 buffer: 0.3 g KH2PO4, 0.6 g Na2HPO4 and 0.5 g NaCl dissolved in 100 ml MQ water. Autoclaved for 20 min. Add 0.1 ml 1M MgSO4 once media has cooled to about 60°C.*
Triton X-100
Glass collection tubes (e.g. Kimble disposable centrifuge tubes #7378510 plus lids)
12-well cell culture plate (Cellstar®)
Mutagen
Saturated culture of OP50
Table top shaker
Aluminum foil
1 N Sodium hydroxide solution
Siliconized 1.5 ml Eppendorf tubes
35 mm NGM plates
Red and black markers
*If a precipitate is observed in the M9 buffer or a stock from which it is made, do not use the solution and remake it. M9, as well as other solutions, should be stored in glass due to leaching of chemicals from plastic during long-term storage. We have observed lethality induced by M9 that has been stored in plastic.
2.2 Preparation for Mutagen Exposure
2.2.1 Safety
Mutagens are toxic to humans, as well as worms! Thus, the utmost care must be taken to limit exposure to the user and her labmates. It is essential to prepare all reagents ahead of time, have everything in the open, so drawers and cabinets do not need to be opened with dirty, gloved hands. Wear two layers of gloves and a lab coat. If weighing out mutagen from a powder, wear a face mask. Do all mutagen manipulations in the hood, when possible. If your scale is not in a fume hood, be sure to wipe down the balance with a paper towel soaked in 1N NaOH. Stock solutions of mutagen can be made ahead of time (refer to Table 1), but we recommend only short term storage (less than 2 weeks) as we have found that many mutagens are labile in solution and can degrade into less efficacious substances or even spurious, toxic species after a couple of weeks. All reusable glassware should be neutralized with 1N NaOH for at least 24 hours prior to disposal. In addition, other glass and plastics should be neutralized before disposal with hazardous waste, according to the requirements of each institution.
Table 1.
Summary for each mutagen of the type of lesion incurred, the range of doses commonly used for sensitivity assays (Sections 2.3, 3.2, 3.3, 4.3, 5.2), the dose cited for immunofluorescence (αRAD-51, αRPA-1, αFCD-2) in the mitotic compartment, if available (Section 2.3, Step 15), and mutants strains with published sensitivity to the mutagen.
| Mutagen | Lesion | Dose Range (Sensitivity) | Dose (Immunofluorescence) | Published Sensitive Strains |
|---|---|---|---|---|
| Cisplatin | ICL | 90–360 μM | 180 μM13,17 |
polq-1(tm2026)19 hel-308(tm2134)19 brc-1(tm1145)17 rfs-1(ok1372)17 fcd-2(tm1298)39 |
| Nitrogen Mustard | ICL | 50–200 μM | 200 μM17 |
brc-1(tm1145)17 rfs-1(ok1372)17 rtel-1(tm1866)40 polq-1(tm2026)19 hel-308(tm2134)19 fcd-2(tm1298)13 dog-1(gk10)13 slx-1(tm2644)15 |
| Psoralen | ICL | 10 μg/ml + 50–200 J/m2 UVA | N/A |
rtel-1(tm1866)40 fcd-2(tm1298)13 dog-1(gk10)13 |
| Mitomycin C | ICL | 250–500 μM | N/A |
fan - 1(tm423)16 him-18(tm2181)16 |
| Campothecin | RF stall | 100–1000 nM | N/A |
rtel-1(tm1866)40 him-18(tm2181)14 slx-1(tm2644)15 rfs-1(ok1372)17 brc-1(tm1145)17 |
| γ-Irradiation | DSB | 10–200 Gy | 75 Gy |
him-18(tm2181)14,15 brc-1(tm1145)17 rfs-1(ok1372)17 polq-1(tm2026)19 hel-308(tm2134)19 |
| Hydroxyurea | RF stall | 5–40 mM | 40 mM17 | clk-2(mn159)22 |
2.2.2. Kill curves
Due to differences in stability of mutagens, we strongly encourage each lab to establish kill curves for each mutagen prior to use. Perform the assay with a range of doses on wild-type and a published sensitive strain (Table 1) to decide which doses work best in your lab. Remember that each lab is different. Something as seemingly trivial as the bacterial strain used to feed the worms or mild fluctuations in incubator temperature could impact the observed sensitivities to the same strains across labs. To normalize for variation between trials within your lab, always include wild-type and the positive control used to generate your kill curve in each experiment. These controls will allow one to account for differences across experiments and to ultimately determine the importance of a component in a specific repair pathway.
2.3 Mutagen Exposure
- Synchronize worm cultures. We suggest one of the following methods:
-
1.1Perform a bleach prep as described in Stiernagle 2006 to isolate eggs [24]. Apply ~200 eggs to each 60 mm plate.
-
1.2Allow 10 gravid adults to lay eggs for 4 hours on 60 mm plates. This should result in approximately 150–200 eggs per plate. Incubate plates at 20°C. Young adults should be ready for use approximately 60–70 hours later, but the exact timing will depend on your incubator and its inherent kinetics of heating and cooling [25]. Examine plates prior to use to ensure age of worms.
-
1.3Hand pick L4s from an asynchronous culture. Allow worms to develop overnight at 20°C, and use the resultant young adults for experimentation.
-
1.1
If working with glass collection tubes that do not have the 100 μl line marked on them, do so with a marker by filling one with 100 μl M9, lining this up with the collection tubes to be used, and drawing a relative measurement line.
Use a glass pipet to wash the synchronized young adults off of the plates with 1×M9 + Triton X-100 (100 μl/L). Rinse the plate multiple times with the M9 in order to maximize the number of worms that are in liquid. Transfer the liquid to the glass collection tube.
Allow worms to settle to the bottom of the tubes. There should be a visible bolus of worms at the bottom of the tubes to ensure a hearty sample size for subsequent experimentation.
Remove as much of the liquid as possible without sucking up any worms. Wash with 500 μl M9 + Triton X-100 (100 μl/L). Repeat washes until the liquid has been cleared of the bacteria from the plate.
Once the liquid has become transparent, remove liquid down to the 100 μl mark on the tube. Suspend worms in a volume of M9 (no detergent) that will allow for 100 μl to be available for each dose that will be used. For instance, if the worms are to be treated with 0, 50, and 100 μM of nitrogen mustard, then 200 μl M9 should be added to the tube to create a final volume of 300 μl.
Prepare 12-well plate by adding 150 μl OP50 to each well. Next, dispense 100 μl M9 + worms into each well. Finally add the desired concentration of mutagen diluted in 750 μl M9. The final volume in each well will be 1 ml.
Cover the 12-well plate with foil, and shake at 70 rpm for 19 hours on tabletop shaker at room temperature.
At some point during this incubation, apply approximately 25 μl saturated OP50 culture to the 35 mm NGM plates. We recommend seeding the plates 4–12 hours prior to use. Older plates will have a thicker bacterial lawn that will make counting more difficult.
After the worms have spent 19 hours in the mutagen, use a glass pipet to transfer the volume in each well to pre-labeled, siliconized 1.5 ml Eppendorf tubes. Suck the volume in the well up and down a few times to minimize the number of worms left on the plastic.
Allow worms to settle. Remove as much liquid as possible without disturbing the worms, and wash with 500 μl M9 + Triton X-100 (100 μl/L). Repeat this wash 2 times to minimize any residual mutagen.
Remove as much of the liquid as possible without sucking up any worms. Dispense the remaining volume containing the worms onto pre-labeled plates. It helps to suck up and down with the micropipette once or twice to suspend the worms in the remaining liquid.
Dry plates at room temperature, and then transfer them to 20°C for a 3-hour recovery.
For viability assays, plate 5 worms per plate on the 35 mm plates seeded in Step 9. Incubate at 20°C for 4 hours (this is the most commonly used time window; however, see Section 7.1 for a discussion on timing).
Remove adults. These can either be sacrificed or moved to a new plate if they are destined for DAPI or antibody staining. Refer to Craig et al 2012 for staining protocols [23].
Count the number of eggs on each 35 mm plate. We find it easiest to draw approximately 8 lines across the bottom of the plate with a fine-tip red marker, thereby dividing the plate into regions that can fit into the field of view of a stereo-microscope set on its highest power.
After counting eggs, store plates at 20°C.
Three days later, count viable L4 and adult progeny.
3. Mutagen Exposure via Treated Plates
3.1 Materials
Worm Media Base: 74.6% Agar, 13.2% Tryptone, 8.8% NaCl, 2.4% Tris acid, 1.1% Tris base
Cholesterol, 10 mg/ml, dissolved in absolute ethanol
35 or 60 mm petri dishes
Mutagen
Saturated OP50 culture
3.2 Infusing the Mutagen
Suspend the appropriate mass of Worm Media Base needed for the desired volume of water (for each liter of water, use 22.79 g). Add 500 μl cholesterol for each liter of media. Autoclave.
Remove media from autoclave. Once the media has cooled to around 60°C, add the volume of mutagen to create the proper concentration for the subsequent assay (Table 1). Pour into 35 mm or 60 mm plates.
Allow plates to dry for 1–2 days at room temperature.
Heat kill the saturated OP50: Transfer 10 ml of the saturated OP50 culture in 50 ml Falcon tubes and place at 75°C for 2 hours. Spin tubes at 4000g in a tabletop centrifuge and discard the supernatant. Resuspend in 2 ml of fresh growth media.
Apply 25 μl of heat-killed, concentrated OP50 to the plates. Incubate plates at room temperature for 1 day to allow the bacteria to dry. Use plates within 24 hours. Although saturated cultures of bacteria can be used here, we have found greater reproducibility with dead bacteria.
Pick synchronized young adults to treated plates (See Section 2.3 Step 1). Keep them on the plates for 22 hours at 20°C.
Transfer 5 worms per plate onto freshly seeded NGM plates containing no mutagen (See Step 9 of Section 2.3 for considerations on seeding plates). For each dose being tested on each strain, we recommend 8–10 plates (i.e. 40–50 worms). Incubate at 20°C for 4 hours.
Complete Steps 12–18 in Section 2.3.
3.3 Spreading Mutagen
For each plate needed, create a 250 μl aliquot of the appropriate concentration of mutagen dissolved in 1×M9. Apply each aliquot onto a plate and disperse evenly by gently rotating.
Incubate plates at room temperature overnight to allow chemical to soak into the plate.
Complete Steps 4–8 in Section 3.2.
4. Photoactivation of Trimethyl Psoralen
4.1 Materials
Synchronized Nematode Growth Media plates (as described in Section 2.3. Step 1)
15 ml Falcon tubes
1×M9 (see Section 2.1 for recipe)
4,5′, 8-Trimethylpsoralen (TMP, also called Trioxsalen, Sigma T-6137)
Acetone
Triton X-100
Nutator mixer
Vacuum aspirator
UVA source
Centrifuge
6-well cell culture plates
4.2 Preparation of Trimethylpsoralen
Prepare a 2.5 mg/ml stock by dissolving solid TMP in acetone. TMP is not soluble in water, and even in acetone, it takes time and agitation to fully dissolve.
4.3 Trimethyl Psoralen Exposure
Wash 3–4 synchronized young adults (See Section 2.3, Step 1) of each genotype into 15 ml Falcon tubes using 1×M9 + Triton X-100 (100 μl/L).
Spin worms at 1500 rpm for 2 min. Remove as much buffer as possible.
Add 1×M9 + Triton X-100 to a final volume of 5 ml per tube.
Add 20 μl of 2.5 mg/ml TMP to each tube for a final concentration of 10 μg/ml TMP (See Section 8.1.3 for alternate approach). For the solvent only control, add 20 μl of acetone to a tube of 5 ml.
Place tubes of worms on a Nutator mixer. Incubate in the dark at room temperature for an hour.
Spin worms at 1500 rpm for 2 min. Remove the TMP (or solvent only) solution using a vacuum aspirator. Leave 0.5 ml of worms in liquid in each tube.
Add 1.5 ml 1×M9 + Triton X-100 to each tube for a final volume of 2 ml. Transfer this 2 ml into individual wells of 6-well plates.
Expose worms to UVA at 0, 50, 100, and 150 J/m2 (See Section 8.1.3 for alternate approach).
Transfer worms to plates in the same manner as detailed in Section 2.3, Step 8.
Incubate worms at 20°C overnight.
At some point during this incubation, apply approximately 25 μl saturated OP50 to the 35 mm NGM plates. As stated in Section 2.3, Step 9, we recommend seeding 4–12 hours before use.
At 22 hours after UVA exposure, pick 5 worms per plate to the freshly seeded 35 mm NGM plates. Allow these worms to lay eggs for 4 hours.
Complete Steps 15–18 of Section 2.3.
5. Gamma Radiation Exposure
5.1 Materials
Synchronized Nematode Growth Media plates (as described in Section 2.3, Step 1)
Gamma irradiation source*
*Dose rates are different for every machine and can range from a high of 20 Gy/min to a low of 10 Gy/hour, depending on strength and decay of source. Cobalt sources are also available but show different survival curves [26], so be careful to note the source, model, and dose rate of the machine used.
5.2 Ionizing Radiation (IR)
Irradiate synchronized young adults (See Section 2.3, Step 1) on NGM growth plates. For IR sensitivity assays, doses between 50 and 200 Gy are used [13–15, 17, 19]. Exposure to 0, 75, and 150 Gy should be sufficient to determine whether a gene confers susceptibility.
The time points chosen will vary depending on which germline nuclei you choose to assay (See Section 7.1 for a discussion on timing). We recommend performing a full time course in which worms are transferred individually to freshly seeded 35 mm NGM plates (See Section 2.3, Step 9) every 12 hours for 72 hours, as differences in the embryonic lethality observed in the various 12-hour time windows could be informative. If a less extensive approach is desired, eggs laid from 24–36 hours should assay repair defects in pachytene and those laid from 48–72 hours should identify repair defects in the mitotic compartment (Section 7.1).
Complete Steps 15–18 in Section 2.3 at the end of each time point. Transfer the worms to new plates if later time points are to be assayed.
6. Comparison between Liquid Culture and Treated Plates
6.1 Liquid Culture
One major concern when dealing with liquid culture is ensuring an adequate sample size. In order to achieve statistical significance, experiments should be done at least twice, preferably thrice. Typically, a cohort of 20 worms per dose is sufficient for each trial, as these worms will lay, on average, 100–200 embryos in the 4 hour time window. You may need additional worms if you are assaying a mutant with reduced fecundity. If 4 doses of mutagen are being tested, this requires 80 worms overall per genotype. Thus, it becomes imperative to retain as many worms as possible. The predominant issue with the liquid culture method is that it demands multiple transfers and washes of the worms. There are many ways to reduce the number of worms lost during these steps. As indicated in Step 3 of Section 2.3, the initial wash of the worms from plates into liquid is the most crucial since they will cling to the bacteria. Furthermore, worms tend to stick to plastic surfaces, so using glass materials when possible greatly reduces the number of worms lost from manipulation. If eliminating plastic is not feasible, including a small amount of detergent, such as Triton X-100 or Tween, into the M9 wash buffers decreases the propensity for worms to adhere to plastic. Even so, it is advisable to start with at least 30 to 40 extra worms for each dose within each genotype to be certain that there will be enough animals (and resultant eggs) for one trial.
Another technical hurdle in using liquid culture is the solubility of the compound in a water-based solvent. Individual levels of solubility in water for each mutagen are discussed in Section 8. However, certain compounds are only soluble or active in a non-polar solvent. For instance, when using camptothecin, spiking the liquid culture with dimethyl sulfonate (DMSO) can ameliorate the solubility of the compound to a point where its activity is useful for biological purposes. However, DMSO affects the viability of the resultant embryos (Fig 1). Therefore, for compounds insoluble in aqueous culture, a mock dose is required to establish baseline sensitivity to the solvent in which the compound must be prepared so that any further post-exposure embryonic sub-viability can be confidently attributed to the mutagen being tested.
Figure 1.
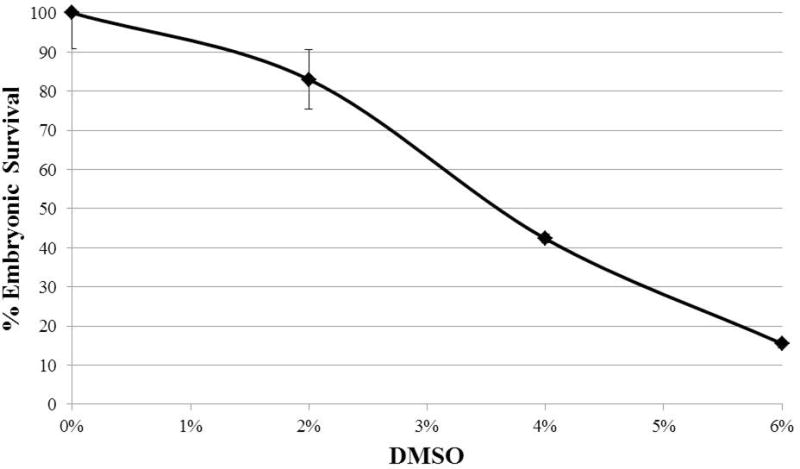
Shows the effect of increasing concentrations of dimethyl sulfide (DMSO) on the survival of the embryos produced by N2 worms. Young adults were soaked for 19 hours in M9 (pH 6.0) and OP50 with the indicated dose of DMSO. The eggs laid from 22–26 hours after the initiation of exposure were scored for survival. Each point corresponds to the average of eggs laid by 50–60 worms plated on 5–6 plates. Error bars represent the standard deviation between plates.
Despite these mild drawbacks, liquid culture has many redeemable features. It requires no long-term storage of the chemicals in a water-based medium, as would be the case for treated plates that are not used immediately. Thus, loss of efficacy of the drug over time is not a concern. Because the worms are shaken while submerged in the liquid, the mutagen concentration is guaranteed to be as uniform as possible.
6.2 Treated Plates
With the exception of hydroxyurea, the more common approach throughout the literature is to expose the worms to mutagens via liquid culture. In one report in which nitrogen mustard (HN2) was soaked into the plates, a higher concentration was required to induce the level of embryonic inviability in wild-type worms observed at much lower concentrations in other studies that used liquid culture [19]. Whereas those groups who opted for liquid exposure reported 30–60% embryonic survival of wild-type worms at 100–150 μM HN2 [14, 17], when plates were used, wild-type embryos exhibited 95% viability at 150 μM HN2 and 65% viability at 300 μM HN2 [19].
The difference in efficacy is likely a consequence of unequal mutagen concentration across the plate. The reasons for such unequal distribution are multifarious. The immobile population of bacteria could metabolize the mutagen into DNA adducts or alkylated proteins that represent no threat to the worm, so as the worm feeds, it may be exposed to a reduced local concentration of the mutagen. Infusion of the chemical into the media necessarily occurs when the media is close to 60°C, a temperature at which compounds might degrade. Some chemicals are not fully soluble in the media and could sequester themselves into pockets. Application of mutagen to plates immediately prior to use can result in uneven spreading across and into the medium. In all of these scenarios, since the worms are not physically shaken in the compound, they, by chance or choice, may spend most of their time on the plate residing in areas of depleted mutagen concentration.
These issues prescribe that higher doses should be used when choosing treated plates over liquid culture, but they also challenge whether consistency can be achieved between separate trials. In order to mitigate inconsistency when exposing worms on treated plates, each plate should be poured with an equal volume of agar. When soaking the mutagen into the media, this will help to ensure a similar concentration gradient is formed by dispersion of the chemical into the plate and a consistent surface concentration of the compound. To mitigate any effects borne by the presence of the bacteria, heat kill the bacteria (Section 3.2, Step 4) and dispense a consistent amount of OP50 onto each plate and spread it evenly. As with antibiotic plates, mutagen-treated plates can be stored at 4°C and maintain their potency. However, this provides an additional confounding factor since the extent of degradation of these compounds in distant trials. Therefore, as stated in the detailed protocols, it is best to use the plates within 24 hours of preparing them. These precautions may not completely abolish the concern of chemical degradation or microenvironments, but they should help protect against unsavory error bars.
Treating the worms on plates does have certain advantages, foremost amongst which is simplicity. Aside from actually making the plates, all that is required is to pick the worms to new plates at various time points. In contrast with preparing worms for liquid culture, one does not have to harvest 1.5 to 2 times as many worms as will eventually be needed. Although excess worms never hurt, especially in dealing with low level HU toxicity [23], much fewer worms must be initially cultivated for this method of exposure.
7. Procedural Parameters
7.1 Timing
In their investigation of the influence of mitotic defects on meiotic outcomes, Jaramillo-Lambert et al 2007 used an experimental system in which a fluorescent nucleotide, Cy3-dUTP, was injected into the mitotic tip of the gonad [27]. By looking at various time points after injection, they could determine how long it takes nuclei that incorporated the fluorescent nucleotide to progress through the various stages of meiosis. In summary, the stage of fluorescently-tagged nuclei at their time points were as follows: 6 hr, pre-meiotic and transition zones; 12 hr, mostly transition zone, some early pachytene; 24 hr, pachytene; 48 hr, late pachytene; 54 hr, some diplotene, mostly diakinesis; 60 hr, either diakinesis or no longer able to be observed. Therefore, a nucleus takes 60 or more hours to traverse the gonad from the mitotic tip to become a fertilized embryo. According to this timeline and as noted in Bailly et al 2010, the embryos that are counted in Steps 11–14 of Section 2.3 would have been in pachytene at the time of mutagen exposure (Fig. 3) [22]. To assay embryos produced from nuclei that were mitotically dividing at the time of exposure, one would have to look 48–60 hours post-exposure. This time frame is corroborated by chromosomal fragmentation in diakinesis nuclei of gen-1 worms exposed to 90 Gy irradiation only at 48 hours post-treatment [22]. As GEN-1 appears to be dispensable meiotically, this fragmentation must be a reflection of unrepaired damage from the mitotic compartment.
Figure 3.
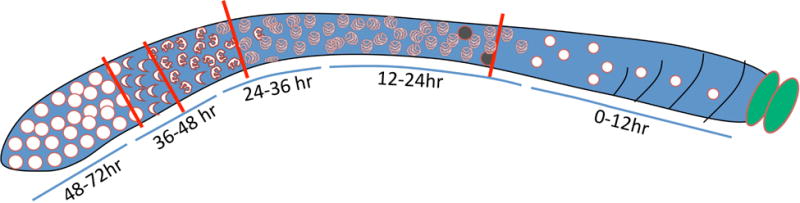
Schematic of the C. elegans germline. The mitotic tip is shown to the left, and stages of meiosis are demarcated by characteristic chromosome morphologies. Using nuclear progression data from Jaramillo-Lambert et al. 2007 [22] and embryonic survival data from our lab and others, we devised a timeline for the analysis of different populations of germline nuclei after mutagen exposure that assumes a negligible effect of mutagen-induced arrest on embryonic output.
Since gen-1 mutants are compromised for DNA-damage-induced mitotic arrest [22], the extrapolation of nuclear progression does not account for the effect of cell cycle arrest on embryonic output. If the mitotic cell divisions provide the driving force behind continued meiotic progression, then mitotic arrest should attenuate the speed with which nuclei travel through the gonad. Mitotic arrest in response to mutagen exposure has been addressed in multiple studies and can be visualized either by immunostaining with a G2 marker, such as anti-phospho-CDK1 antibody (a human epitope that cross reacts with worms [28]) or with DIC microscopy by the appearance of grossly enlarged nuclei [29]. Wild type worms arrest mitotic progression for at least 12 hours after irradiation with 60 and 120 Gy [29]. Treatment of worms with 40 mM hydroxyurea and 200 μM nitrogen mustard also induce mitotic arrest [13]. It should be noted that although mitotic arrest has been demonstrated after incursion of DNA damage, no investigation of the dynamics of the arrest induced by various doses of each mutagen, nor a study of how the induced arrest impacts the movement of nuclei proximally through the germline, exists. Such an understanding would greatly improve the solidity of conclusions drawn from mutagenic viability assays.
One published experiment speaks directly to the relationship of mutagen exposure to resultant embryonic survival. When the embryonic survival of fcd-2 mutants exposed to 180 μM cisplatin was determined over the course of 72 hours, it fell from 82% survival from 24–48 hours post-exposure to 47% survival from 48–72 hours after treatment [20]. As demonstrated by robustly increased α-FCD-2 staining in the mitotic compartment following treatment with interstrand crosslinking agents and hydroxyurea [13], FCD-2 is involved in mounting a response to stalled replication forks in the mitotic compartment. Thus, the reduced survival of embryos laid by cisplatin-treated worms from 48–72 hours post-exposure is likely due to damage induced in mitotically dividing nuclei that remained unrepaired in the resultant fertilized oocyte.
7.2 Developmental Stage
One area of disagreement between protocols from disparate groups is whether worms should be L4s or young adults at the time of exposure. According to the protocols described in Sections 2.3, 3.2, 3.3 and 3.4, the embryos being scored are laid between 22 and 26 hours after the initiation of exposure. During this time window, there is a significant difference in the number of eggs laid by animals that were L4s at the start of exposure and their more developed counterparts that began exposure as young adults. Young adults ultimately lay 1.5- to 2-fold more eggs per worm than the L4s (Fig 2). If performing an experiment in triplicate with the goal of 60 total worms per dose, this could result in a difference of 99 or 126 embryos scored per dose for a water soluble or insoluble drug, respectively. In this context, treating young adults is the better approach because they will produce a more abundant cohort of embryos.
Figure 2.
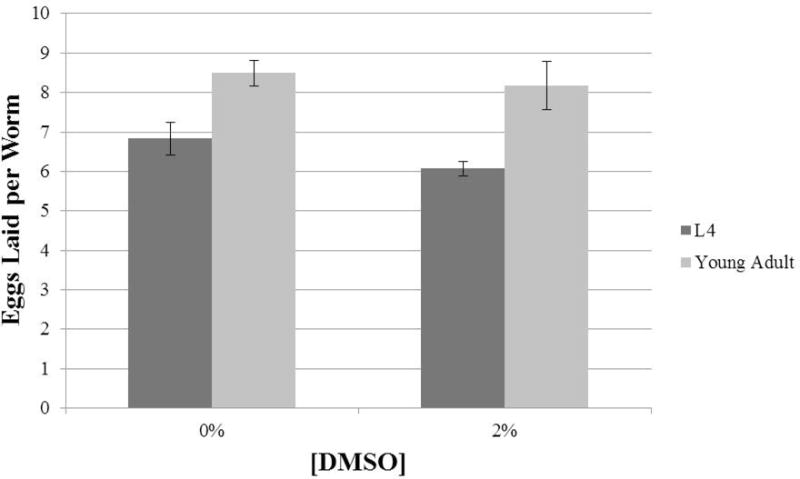
Number of eggs laid per worm 22–26 hours after initiation of liquid culture for N2 worms that were either L4s or young adults at the beginning of treatment. Each data set is comprised of average eggs laid by 50–60 worms with around 10 worms per plate.
However, the number of eggs laid is only one consideration when deciding which age of worm to use. After all, reduced egg output can be overcome by using more worms or expanding the time frame in which eggs are counted. A more pressing concern is whether there is a difference between wild-type L4s and young adults in their sensitivity to mutagenic perturbations. As shown in Fig. 4, the viability of eggs laid by N2 worms exposed to 20 Gy of γ-irradiation as L4s and those exposed as young adults differs significantly. While the young adults do not appear to exhibit any sensitivity from 24–48 hour post-exposure, L4 worms seem to be more sensitive, laying 20% inviable embryos from 24–36 hour post-treatment and 12% from 36–48 hours. The higher sensitivity of L4s might be due to the action of residual spermatogenesis proteins on L4 [30] or perhaps to developmental differences in stress tolerance [25]. Alternatively, the differential sensitivities may reflect differences in the population of nuclei that are monitored in these time windows. Transit of nuclei through the germline is reported to slow down as resided in the mitotic or transitions zone whereas the same time window in adults is likely to arise from pachytene nuclei [27]. Whatever the cause, these data underscore the necessity for careful synchronization of worm populations for maximum consistency between experiments. As detailed in all of protocols detailed herein (Section 2.3, Step 3; Section 3.2, Step 6; Section 4.2, Step 1; Section 5.2, Step 1), we recommend exposing young adults to the mutagens because it circumvents potential confounding physiological processes that are occurring as the worms transition from L4 to adulthood. However, as long as fastidious attention is paid to the staging of the worms each time an experiment is performed, using L4s is perfectly valid.
Figure 4.
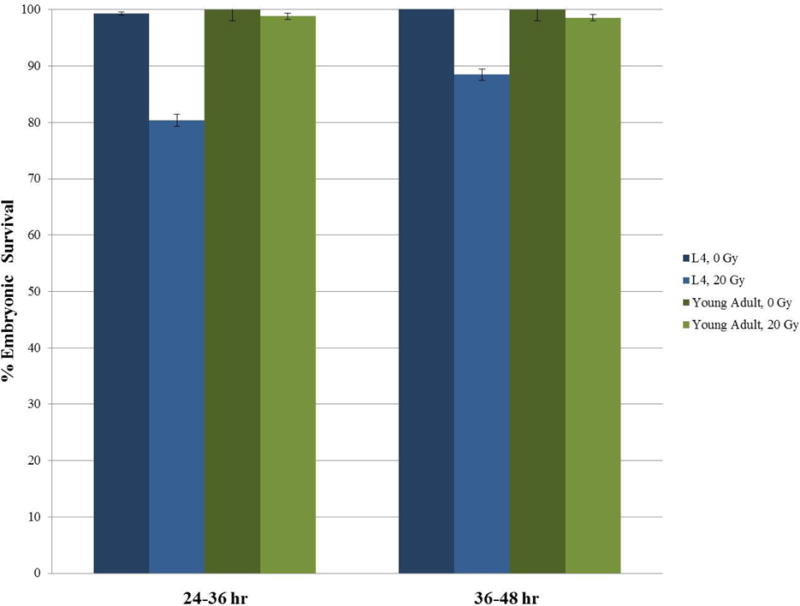
Response of N2 worms exposed to 20 Gy of irradiation as either L4s or young adults. After exposure, 2 worms per plate (0 Gy: 8–10 worms; 20 Gy: 18–20 worms). Eggs laid 24–36 hours and 36–48 hours post-exposure were scored for viability. Error bars represent the standard error between plates.
All of the protocols for mutagen exposure exploit the fact that, as a self-fertilizing hermaphrodite, spermatogenesis is already completed in C. elegans at late L4 or later stages. The effects of mutagen exposure on the pre-made sperm is the hermaphrodite spermatheca have not been studied. Therefore, one should be cautious in ascribing the effects of mutagen exclusively to developing oocytes.
8. Considerations for Individual Compounds
8.1 Interstrand Crosslinking Agents
Interstrand crosslinking agents utilized in C. elegans include the nitrogen mustard mechlorethamine hydrochloride, cisplatin, trimethyl psoralen, and, less commonly, mitomycin C. All compounds that produce interstrand crosslinks possess two reactive centers that potentiate a reaction with two separate nucleotides. For detailed mechanistic insight into the reactions incurred when these compounds are introduced into cells, reference Deans & West 2011 [1]. Included herein are more methodological considerations for each of the compounds.
8.1.1 Nitrogen Mustard
Nitrogen mustard (HN2) is soluble in water. However, aqueous aliquots do not keep well over time. It is best to make fresh aliquots every two weeks or so.
One major concern with HN2 is storage of the solid. It should absolutely be stored in a desiccator. We have noticed a dramatic reduction in survival of embryos produced by exposed wild type worms when HN2 was stored at 4°C without desiccation. This could be due to reactions of mechlorethamine hydrochloride with the moisture in the air, as the compound has been shown to react with water in unbuffered solution to produce toxic species [31].
8.1.2 Cisplatin
Cisplatin (CDDP) is much more soluble in DMSO than water. However, it is inadvisable to dissolve CDDP in DMSO because the two compounds react to form an adduct that has a reduced affinity for double stranded DNA [32]. In aqueous solution, CDDP tends to isomerize to a less active form. Its stability in water is aided by higher NaCl concentrations (0.9%) and disrupted by alkaline solutions [33] (in fact, it demonstrates increased cytotoxicity in solutions with lower pH values [34]). For best consistency, we recommend making CDDP aliquots fresh. If problems with its efficacy are encountered, lower M9 pH and slightly higher NaCl concentrations should be used.
8.1.3 Trimethyl psoralen
Trimethyl psoralen (TMP) must be exposed to UVA in order to become an active interstrand crosslinking agent [1]. This is useful for targeting specific tumors in human patients but presents another methodological complication in the worm. Reference the separate protocol (Section 4) for TMP exposure for considerations about photoactivation.
TMP is insoluble in water. It should be prepared in acetone. However, even in acetone, it does not dissolve quickly. It may require multiple rounds of vortexing before it is fully solubilized. Make sure that it is completely dissolved before usage.
The protocol detailed here for the photoactivation of TMP recommends using a constant concentration of TMP and varying the dose of UVA to which the worms are exposed in order to establish a dose-response curve, but it is equally appropriate to use increasing concentrations of TMP and a steady dose of UVA. When embarking on the latter approach, the TMP exposure is between 0 and 2.0 μM, and the UVA dose is 200 J/m2 [35].
8.1.4 Mitomycin C
Mitomycin C (MMC) undergoes intracellular reduction to become biologically active [1]. Although not often cited in C. elegans literature, MMC has been used successfully with the protocol detailed in Section 2.3 [16] and is water soluble.
8.2. Campothecin
Camptothecin inhibits topoisomerase I by preventing its re-ligation activity, which leads to the formation of a one-sided double strand break when the replication fork encounters the camptothecin-bound enzyme [36]. Camptothecin is insoluble in water and must be dissolved in DMSO. It exists in two forms. Its lactone form actively inhibits topoisomerase, while its carboxylate form does not. The balance between these two forms is dependent upon pH. In a buffer with pH 7, less than 40% of the chemical is in the active lactone form, whereas when the buffer is at pH 6, closer to 85% is in the lactone form [37]. The difference in the activity of CPT in buffers of different pH values is exemplified in Fig 5, where both wild-type and slx-1 worms gave a much more robust response to the camptothecin when M9 with a pH of 6 was used.
Figure 5.
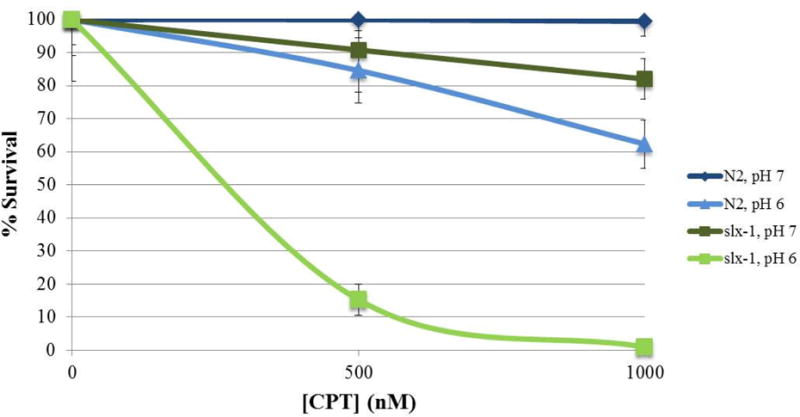
Effect of pH on the efficacy of camptothecin. The pH of the 1×M9 buffer in which the liquid exposure was performed was altered. N2 and slx-1 late L4s were exposed to either 500 or 1000 nM CPT or a mock dose of DMSO. Eggs laid from 22–26 hours after the initiation of exposure were scored for viability. Embryonic survival at each dose was normalized using the survival at the mock dose. Each point corresponds to the average of eggs laid by 30–50 worms plated on 3–4 plates. Error bars represent the standard deviation between plates.
8.3 Hydroxyurea
Hydroxyurea (HU) inhibits ribonucleotide reductase, which depletes the pool of deoxyribonucleotides available for DNA synthesis and causes replication fork stalls [38]. HU is most commonly used in plates, by either the infusion or spreading method. It is freely soluble in water up to 50 mg/ml but should not be kept in aqueous aliquots since it decomposes in the presence of moisture [38]. For this reason, the solid should be stored in a desiccator at 4°C once the seal has been broken.
Highlights.
Differences in exposure protocols confound comparison and normalization.
Numerous parameters affect nematode sensitivity to DNA damaging agents.
Precise staging and handling of nematodes is essential for reproducible results.
Time points post-exposure report on different phases of meiosis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deans AJ, West SC. Nature reviews Cancer. 2011;11:467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mouret S, Baudouin C, Charveron M, Favier A, Cadet J, Douki T. Proceedings of the National Academy of Sciences. 2006;103:13765–13770. doi: 10.1073/pnas.0604213103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ward JF. Radiation Research. 1995;142:362–368. [PubMed] [Google Scholar]
- 4.Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Nature. 2007;447:725–729. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- 5.D’Andrea AD, Grompe M. Blood. 1997;90:1725–1736. [PubMed] [Google Scholar]
- 6.Page SL, Hawley RS. Science (New York, NY) 2003;301:785–789. doi: 10.1126/science.1086605. [DOI] [PubMed] [Google Scholar]
- 7.Keeney S, Neale MJ. Biochemical Society transactions. 2006;34:523–525. doi: 10.1042/BST0340523. [DOI] [PubMed] [Google Scholar]
- 8.Chapman JR, Taylor Martin RG, Boulton Simon J. Molecular cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 9.Kottemann MC, Smogorzewska A. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Derry WB, Putzke AP, Rothman JH. Science (New York, NY) 2001;294:591–595. doi: 10.1126/science.1065486. [DOI] [PubMed] [Google Scholar]
- 11.Colaiácovo MP, MacQueen AJ, Martinez-Perez E, McDonald K, Adamo A, Volpe A La, Villeneuve AM. Developmental cell. 2003;5:463–474. doi: 10.1016/s1534-5807(03)00232-6. [DOI] [PubMed] [Google Scholar]
- 12.Alpi A, Pasierbek P, Gartner A, Loidl J. Chromosoma. 2003;112:6–16. doi: 10.1007/s00412-003-0237-5. [DOI] [PubMed] [Google Scholar]
- 13.Youds JL, Barber LJ, Ward JD, Collis SJ, O’Neil NJ, Boulton SJ, Rose AM. Molecular and cellular biology. 2008;28:1470–1479. doi: 10.1128/MCB.01641-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saito TT, Youds JL, Boulton SJ, Colaiacovo MP. PLoS genetics. 2009;5:e1000735. doi: 10.1371/journal.pgen.1000735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saito TT, Mohideen F, Meyer K, Harper JW, Colaiacovo MP. PLoS genetics. 2012;8:e1002888. doi: 10.1371/journal.pgen.1002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smogorzewska A, Desetty R, Saito TT, Schlabach M, Lach FP, Sowa ME, Clark AB, Kunkel TA, Harper JW, Colaiacovo MP, Elledge SJ. Molecular cell. 2010;39:36–47. doi: 10.1016/j.molcel.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ward JD, Barber LJ, Petalcorin MI, Yanowitz J, Boulton SJ. The EMBO journal. 2007;26:3384–3396. doi: 10.1038/sj.emboj.7601766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Haaften G, Romeijn R, Pothof J, Koole W, Mullenders LH, Pastink A, Plasterk RH, Tijsterman M. Current biology : CB. 2006;16:1344–1350. doi: 10.1016/j.cub.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 19.Muzzini DM, Plevani P, Boulton SJ, Cassata G, Marini F. DNA repair. 2008;7:941–950. doi: 10.1016/j.dnarep.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 20.Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ, La Volpe A. Molecular cell. 2010;39:25–35. doi: 10.1016/j.molcel.2010.06.026. [DOI] [PubMed] [Google Scholar]
- 21.Lee KY, Yang I, Park JE, Baek OR, Chung KY, Koo HS. Biochemical and Biophysical Research Communications. 2007;352:479–485. doi: 10.1016/j.bbrc.2006.11.039. [DOI] [PubMed] [Google Scholar]
- 22.Bailly AP, Freeman A, Hall J, Declais AC, Alpi A, Lilley DM, Ahmed S, Gartner A. PLoS genetics. 2010;6:e1001025. doi: 10.1371/journal.pgen.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Craig AL, Moser SC, Bailly AP, Gartner A. Methods in cell biology. 2012;107:321–352. doi: 10.1016/B978-0-12-394620-1.00011-4. [DOI] [PubMed] [Google Scholar]
- 24.Stiernagle T. WormBook. 2006 doi: 10.1895/wormbook.1.101.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zevian S, Yanowitz J. Methods. 2014 doi: 10.1016/j.ymeth.2014.04.015. In this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartman P, Goldstein P, Algarra M, Hubbard D, Mabery J. Mutation research. 1996;363:201–208. doi: 10.1016/0921-8777(96)00012-2. [DOI] [PubMed] [Google Scholar]
- 27.Jaramillo-Lambert A, Ellefson M, Villeneuve AM, Engebrecht J. Developmental biology. 2007;308:206–221. doi: 10.1016/j.ydbio.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 28.Hachet V, Canard C, Gönczy P. Developmental cell. 2007;12:531–541. doi: 10.1016/j.devcel.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 29.Gartner A, Milstein S, Ahmed S, Hodgkin J, Hengartner MO. Molecular cell. 2000;5:435–443. doi: 10.1016/s1097-2765(00)80438-4. [DOI] [PubMed] [Google Scholar]
- 30.Tang L, Machacek T, Mamnun YM, Penkner A, Gloggnitzer J, Wegrostek C, Konrat R, Jantsch MF, Loidl J, Jantsch V. Molecular biology of the cell. 2010;21:885–896. doi: 10.1091/mbc.E09-09-0811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Golumbic C, Fruton JS, Bergmann M. The Journal of organic chemistry. 1946;11:518–535. [PubMed] [Google Scholar]
- 32.Fischer SJ, Benson LM, Fauq A, Naylor S, Windebank AJ. Neurotoxicology. 2008;29:444–452. doi: 10.1016/j.neuro.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 33.Enzo Life Sciences.
- 34.Atema A, Buurman KJ, Noteboom E, Smets LA. International journal of cancer. Journal international du cancer. 1993;54:166–172. doi: 10.1002/ijc.2910540126. [DOI] [PubMed] [Google Scholar]
- 35.Hsiang YH, Lihou MG, Liu LF. Cancer research. 1989;49:5077–5082. [PubMed] [Google Scholar]
- 36.Saetern AM, Nguyen NB, Bauer-Brandl A, Brandl M. International journal of pharmaceutics. 2004;284:61–68. doi: 10.1016/j.ijpharm.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 37.Koc A, Wheeler LJ, Mathews CK, Merrill GF. The Journal of biological chemistry. 2004;279:223–230. doi: 10.1074/jbc.M303952200. [DOI] [PubMed] [Google Scholar]
- 38.Sigma Aldrich Product Information.