

Abstract
The germinal center (GC) reaction is an adaptive immune response to select B cells bearing high-affinity B cell receptors (BCRs) to undergo further differentiation into antibody-producing cells or memory B cells. To drive affinity maturation, (GC) B cells undergo rounds of hypermutation and rapid proliferation, which can enhance susceptibility to malignant transformation. Lymphomas frequently originate from GC B cells, but the etiology for most lymphoma subtypes is unknown. Work in the past decade has more fully documented the mutational landscape in lymphomas, but the impact of these genomic lesions is often difficult to ascertain. In addition, while mutations affecting BCR signaling are well studied, the impact of extrinsic microenvironmental factors has not been widely addressed. Murine models are useful tools to study lymphomagenesis and disease progression, as well as potential treatment in a pre-clinical setting. Herein we discuss advances in murine models of lymphoma and how they inform on key characteristics of human lymphomas.
Introduction
B cell non-Hodgkin lymphoma (B-NHL) is the most common lymphoma and mainly derives from (GC) B cells. Despite intense research efforts in the field, tools to demonstrate causal relationships are scarce. Mice are the most utilized animal model and a key research tool for human biology due to their genetic similarity, availability of inbred strains, short generation time, cost efficiency and ease of genetic manipulation. Utilization of genetically modified mice can provide insight into the genetic dependencies and microenvironmental influences of human B-NHL, potentially allowing for the clonal tracing of pre-malignant cells to full disease presentation. Here, we review recent advances and opportunities in the use of genetically modified mouse models to represent GC-derived human B-NHL.
Germinal center B cells as the origin of the majority of lymphomas
The life cycle of a B cell is distinguished by the increased risk of genomic lesions associated with immunoglobulin (Ig) gene rearrangement and hypermutation and massive cell proliferation driving clonal expansion. In humans and mice, B cells develop in the bone marrow (BM) and egress upon maturation to populate B cell follicles in secondary lymphoid organs such as the spleen and lymph nodes. To initiate an immune response, B cells bind cognate antigen via the BCR and present derived MHC-II-bound peptides to T cells, resulting in the receipt of critical cytokine support. A cohort of T cell-activated B cells undergo further differentiation in the GC, a distinct microenvironment wherein somatic hypermutation (SHM; Ig point mutations directed to V(D)J rearrangements) and class-switch recombination (CSR; orchestrated double-strand DNA breaks in the Ig heavy chain locus resulting in isotype switch and the acquisition of distinct antibody effector properties) occur. Both SHM and CSR are mediated by the enzyme activation-induced cytidine deaminase (AID).
The GC is functionally organized into a dark zone (DZ) and a light zone (LZ) (Figure 1). In the LZ, B cells with superior BCR affinity are positively selected to undergo cell division in the DZ, which is a dynamic and iterative process [1]. In addition to BCR stimulation, many cytokines impact B cell differentiation and survival in the GC, including CD154 (CD40L), IL-4, IL-21, and BAFF. While most GC B cells undergo apoptosis, the small fraction of positively selected B cells in the LZ induce PI3K signaling and the expression of MYC, a proto-oncogene that is absent in rapidly cycling DZ B cells but obligatory for GC passage [2–5]. These cells then either undergo differentiation into plasma or memory cells, or re-enter the DZ to undergo further rounds of cell division. While PI3K signaling is important for BCR signaling (GC initiation and LZ selection), the canonical (RELA/p50 and c-REL/p50) and non-canonical (RELB/p52) NF-κB pathways also have distinct roles in the GC response (e.g. antigenic stimulation and CD40 signaling) [6]. Interestingly, the transcriptional profile of the majority of B-NHL is consistent with that of a LZ B cell origin [7]. B-NHLs that derive from the GC can be identified by immunophenotype, morphology, gene expression profile and distinct chromosomal translocations or gene mutations mediated by aberrant AID activity. Genetic evidence of SHM is indicative of GC origin, and clonality is readily determined by V(D)J sequencing.
Figure 1.
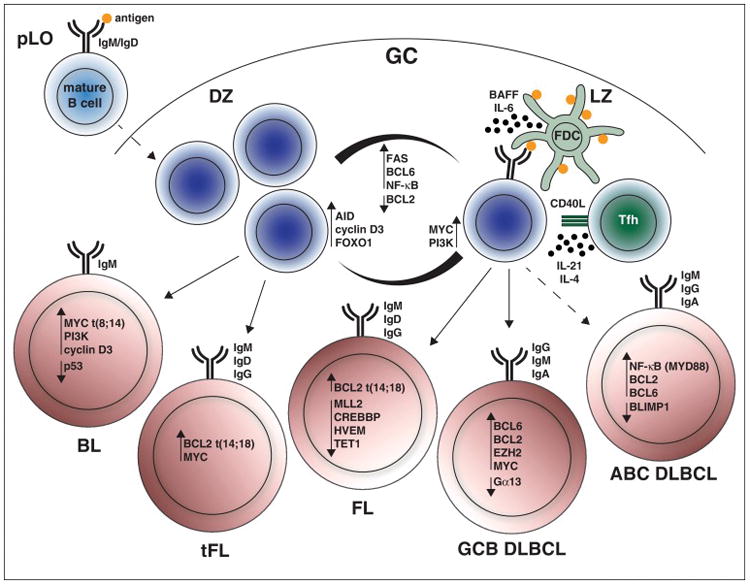
The germinal center reaction and cell of origin in germinal center-derived B lymphomas. In the germinal center (GC), GC B cells undergo clonal expansion in the dark zone (DZ) and are positively selected in the light zone (LZ). In the LZ, follicular dendritic cells (FDC) retain antigen and cooperate with follicular helper T (Tfh) cells to support competitive GC B cells for survival and either differentiation or iterative rounds of cell division in the DZ. Several pathways are differentially regulated in DZ and LZ GC B cells. Mutations in these pathways manifest to different degrees in human B cell non-Hodgkin lymphoma (B-NHL) derived from GC B cells. The oncogenic potential of the highlighted pathways has been mechanistically assessed in murine models of B-NHL.
Regulators of lymphomagenesis in murine in vivo models of B-NHL
Cancer driver genes can be categorized into twelve core signaling pathways that affect cell fate, survival and genome integrity [8]. In GC-derived B-NHL, critical signaling pathways and gene expression signatures have been associated with some lymphoma subtypes, in addition to hallmark translocations (i.e. BCL2 or MYC). In this review, we will mainly focus on recently generated genetically-modified mouse models of lymphoma in which B cells incur genomic lesions at distinct developmental stages (summarized in Table 1). The Cre-loxP system has greatly aided this approach by enabling stage-specific gene modifications in pro-B cells (Cd19-Cre [9] and Mb1-Cre [10]), mature B cells (Cd23-Cre [11] and hCD20-Cre-ERT2 [12]), and GC B cells (Aid-Cre [13] and Cγ1-Cre [14]). The introduction of a specific mutation may have tumorigenic effects at different cell stages, but the nature, impact and relevance may differ.
Table 1.
Regulators of lymphomagenesis in mouse models of B-NHL
| Gene | Mutational type | Functional effect | Implications in B-NHL | References |
|---|---|---|---|---|
| MYC | Gain (CAG; Em) | Growth, Proliferation | BL, tFL, DLBCL | [18] |
| Pik3ca | Gain (P110CA) | Growth, Proliferation, Survival | BL | [18] |
| BCL2 | Gain (VavP; Em) | Survival | FL, tFL, DLBCL | [21••,24•,25•,26,31,47•] |
| Kmt2d | Loss | Proliferation | FL, tFL, DLBCL | [24•,25•] |
| Crebbp | Loss | Differentiation Block, Proliferation | FL, tFL, DLBCL | [26,48] |
| Ezh2 | Gain (Ezh2Y641F) | Proliferation, Differentiation Block | FL, tFL, GCB DLBCL | [27,34] |
| Tet1 | Loss | DNA Damage | FL, DLBCL | [28] |
| Blimp1 | Loss | Differentiation Block | ABC DLBCL | [35,36] |
| Myd88 | Gain (Myd88L252P) | Proliferation, Survival | ABC DLBCL | [38] |
| Bcl6 | Gain (Im) | DNA Damage, Differentiation Block | DLBCL, FL, tFL | [34,39] |
| Gna13 | Loss | Migration, Survival | GCB DLBCL, BL | [46••] |
| Hvem | Loss | Proliferation? | FL, tFL, DLBCL | [47•] |
Burkitt lymphoma
Burkitt lymphoma (BL) is an aggressive lymphoma of IgM+ transformed DZ B cells, that manifests predominantly with translocations between the IGH or IGL locus and MYC. Chronic infections with one or multiple pathogens including, Epstein-Barr virus (EBV), human immunodeficiency virus (HIV) or Plasmodium occur in nearly all three clinical BL variants (endemic BL, sporadic BL and immunodeficiency-associated BL; with the lowest incidence in sporadic BL). In mice lacking p53 in the B lineage via Cd19Cre-mediated deletion, chronic challenge with Plasmodium chabaudi-infected murine erythrocytes shifted the lymphoma phenotype from a mature to GC origin [15••]. Lymphomagenesis is dependent on AID in this model, as B cell hyperplasia is observed with the additional loss of Aid but not frank lymphoma.
The PI3K signaling axis is absent in normal DZ B cells, but functions as a dominant signaling pathway in BL [16] and is inhibitory to CSR via silencing of FoxO1 [17]. In Cγ1-Cre driven induction of hyperactive MYC and P110α (the catalytic subunit of PI3K), a single challenge with sheep red blood cells (SRBCs) induces aggressive tumors bearing GC markers (IgM+ GL7+ Fas+ Bcl6+) within two months [18]. While deregulated MYC and PI3K are sufficient for lymphomagenesis, some tumors presented with mutations in Ccnd3 (encoding cyclin D3) [18]. Interestingly, the same mutations in CCND3 as well as mutations in the upstream transcription factor, E2A (encoding TCF3) or its negative regulator ID3, are common in human BL [16,19]. However, deletion of Id3 (with Cd19-Cre or Cγ1-Cre) in SRBC-challenged mice does not drive malignant transformation, most likely due to redundancy with Id2 [20].
Follicular lymphoma
Follicular lymphoma (FL) is a common and incurable B cell lymphoma. The oncogenic hallmark of FL is BCL2 translocation t(14;18)(q32;q21), which places BCL2 under the IGH regulatory elements. Strikingly, this translocation is also present at low frequency in ~70% of all healthy individuals [21••]. BCL2 is normally not expressed in GC B cells where survival signaling is mediated by the BCL2 family members, BCL-XL, MCL1 and A1 [22]. Forced expression of BCL2 in transgenic mice is insufficient to drive neoplastic growth, but several challenges with SRBCs results in re-entry of memory B cells into the GC reaction. Here, constitutive BCL2 expression confers a survival advantage and an opportunity to acquire secondary lesions by AID, promoting clonal evolution toward malignant FL [21••].
Mutations in histone modifiers MLL2 (encoded by KMT2D), CREBBP, and EZH2 are frequently found in FL patients [23]. Interestingly, Kmt2d deletion with Cγ1-Cre is not sufficient for transformation under continuous SRBC challenge (8-week intervals), but cooperates with BCL2 in transgenic mice (VavP-BCL2) to promote lymphomagenesis [24•]. Additionally, knockdown of Kmt2d in VavP-BCL2 hematopoietic progenitor cells transplanted into recipient mice reduced the latency period of lymphoma relative to VavP-BCL2 alone [25•]. Likewise, knockdown of Crebbp (in VavP-BCL2 cells) accelerates lymphoma onset [26]. Consistent with this phenotype, Cg1-Cre-mediated monoallelic deletion of Crebbp in VavP-BCL2 mice drastically increased FL penetrance [48]. This synergism of BCL2 with histone modifiers is also seen in other B cell lymphomas. For example, introduction of activating Ezh2 mutations (Ezh2Y641F/+) with Cd19-Cre induces B lymphoma (B220+ CD19+ IgM+ CD43+ CD5+ Mac1+), and the added provision of retroviral BCL2 expression accelerates lymphoma progression [27]. Other epigenetic modulators involved in DNA methylation are also commonly found to be dysregulated in B lymphomas. The methyl-cytosine dioxygenase TET1 is transcriptionally silenced in FL, and loss of Tet1 in mice drives B lymphoma (B220+ CD19+ IgM+ IgD+ CD43−) with late onset after one to two years [28].
If left untreated (‘watch and wait’ protocol), FL follows a clinical course with an initial indolent phase followed by an accelerated histologic transformed FL (tFL; ~45% incidence) that often resembles aggressive lymphomas of diffuse large B cell lymphoma (DLBCL) and/or BL [29]. The molecular basis of the indolent to tFL stage is still largely unknown. However, the majority of patients with tFL acquire secondary mutations or deletions in MYC, CDKN2A/B, CCND3 and/or TP53 genes that affect cell cycle regulation and are believed to accelerate disease progression [30•]. Recipient mice transplanted with VavP-BCL2 hematopoietic progenitor cells transduced with MYC develop aggressive large cell lymphomas resembling tFL, whereas ectopic expression of molecules enforcing G1 cell cycle entry (e.g. cyclin D3) produces aggressive tumors that are phenotypically distinct from tFL and feature small (PNA+) cells [31].
Diffuse large B cell lymphoma
Transcriptional profiling defines two major DLBCL subtypes, including germinal center B cell (GCB)-like and activated B cell (ABC)-like DLBCL [32]. The tumors of DLBCL are diverse and not genetically identified by hallmark translocations. However, there are some distinct pathways that are deregulated due to acquired mutations or epigenetic events in one or more subtype(s) [33]. Oncogenic mutations in EZH2 have been shown to potentially cooperate with BCL6 since mice chronically challenged with SRBCs constitutively expressing Ezh2 (IμBcl6;Ezh2Y641F/+;Cγ1-Cre) develop lymphomas but require co-expression of Bcl6 [34]. In GC B cells, Blimp1 (aka Prdm1) promotes plasma cell differentiation and mutations affecting BLIMP1 are specific for ABC DLBCL. In mice, inactivation of Blimp1 with Cγ1-Cre confers a developmental arrest to GC B cells, and chronic SRBC challenge drives lymphomagenesis [35,36].
Constitutive NF-κB signaling occurs frequently in ABC DLBCL via mutations in genes encoding the signaling subunits of the BCR, A20 and other intermediates in the NF-κB pathway, including the TLR adaptor protein MYD88 [37]. In mice, activating mutations in Myd88 (Myd88L252P) induced by Cd19-Cre or Aid-Cre develop tumors (CD138− Bcl6− Irf4+) sharing characteristics with human ABC DLBCL [38]. Further, non-canonical NF-κB signaling during lymphomagenesis is highlighted in hyper-immunized mice overexpressing Nik and Bcl6 (IμBcl6;Nik;Cγ1-Cre) [39]. The necessity for Bcl6 in lymphomagenesis is shared by a large fraction of ABC and GCB lymphomas. Cγ1-Cre-mediated inactivation of Fbxo11, a regulator of Bcl6 proteasomal degradation, in hyper-immunized mice induces lymphoproliferative disease [40], but requires additional factors to drive lymphoma. Analogous to BL, chronic infection with EBV is associated with DLBCL. This infection requires the expression of LMP1, a functional homolog of constitutively active CD40, which drives DLBCL lymphomagenesis following induction with Cd19-Cre in T cell deficient mice [41].
The germinal center microenvironment during pathogenesis
The influence of the GC microenvironment in B cell differentiation and predisposition to transformation is poorly understood, including the differential dependence on tumor stromal elements [42]. Thus, while much attention has been justifiably focused on the role of the BCR in B lymphomagenesis and progression, B cell extrinsic factors in the GC microenvironment need to be considered. Interestingly, it was recently demonstrated that the GC, particularly the LZ, is a hypoxic region that impacts AID expression and promotes glycolysis [43]. This increased glycolytic demand is regulated by GSK3 to prevent metabolic collapse in GC B cells, especially under limited glucose supply [49]. Notably, BCR-dependent DLBCL rely on glycolysis for energy production, whereas the OxPhos-subtype of DLBCL exhibits elevated mitochondrial activity and an independence from BCR signaling [44], which illustrates a need to identify metabolic factors that are of relevance to the transformation and therapeutic targeting of lymphoma.
Recent studies on the impact of GNA13 and HVEM mutations highlight the impact of the GC microenvironment. The guanine nucleotide binding protein Ga13 (encoded by Gna13), a signaling mediator of sphingosine-1-phosphate receptor-2 (S1PR2), represses B cell growth in the context of the GC and is inactivated in a subset of GCB DLBCL. This deficiency promotes the transformation of B cells bearing a GC phenotype (GL7+ CD138 Bcl6+ Irf4 ) following deletion with Mb1-Cre in naı ¨ve mice [46••]. Confinement of these cells to the GC is lost, resulting in their dispersal into circulation and possible seeding of secondary sites. HVEM (encoded by TNFRSF14) is a receptor for the inhibitory ligand BTLA and is frequently mutated in tFL and FL. Interestingly, knockdown of Hvem in VavP-BCL2 hematopoietic progenitor cells contributes to lymphomagenesis in mice and immunotherapeutic delivery of a soluble HVEM receptor kills tumor cells [47•]. These findings illustrate the complexity of cis/trans relationships among receptor/ligand combinations within the GC. Orthotopically engrafted patient-derived xenografts (PDXs) may recapitulate the crosstalk between malignant cells and the microenvironment of the patient [45]. These studies, in conjunction with genetically-modified mouse models, have the potential to elucidate the cytokine dependency and metabolic requirements of human B-NHL.
Concluding remarks
Murine models have significantly advanced our understanding of the etiology of B lymphoma, yet there is much to learn in terms of clonal evolution, metastasis, immune evasion and the tumor microenvironment. Advances in genomics have provided a large and growing resource of genetic information documenting gene mutations, gene expression and epigenetic regulation in B-NHL subtypes. Coupled with the rapid adoption and advancement of CRISPR technology, we have entered a new era of genome engineering in mice that will allow for the modeling of complex genetic diseases such as cancer. However, even faithful mouse models cannot entirely mimic human disease in full scope. Thus, identification of shared and distinct features of murine and human lymphomas needs to be carefully considered. PDXs may help to address these potential discrepancies and fill the gap as a hybrid model. In the age of personalized medicine, tailored mouse models that accurately reflect the origin and progression of lymphoma will allow for better targeted treatments.
Acknowledgments
We thank all authors who contributed insightful scientific work but could not be featured due to limited length and references of this review. We wish to thank all members of the Rickert laboratory for critical feedback in reading the manuscript. This work was supported by National Institutes of Health Grants R01AI041649 and R21CA195431 (to R.C.R.) and a Postdoctoral Fellowship from the Cancer Centers Council (C3) (to P.R-R.).
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Mesin L, Ersching J, Victora GD. Germinal center B cell dynamics. Immunity. 2016;45:471–482. doi: 10.1016/j.immuni.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, Dalla-Favera R. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol. 2012;13:1083–1091. doi: 10.1038/ni.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calado DP, Sasaki Y, Godinho SA, Pellerin A, Kochert K, Sleckman BP, de Alboran IM, Janz M, Rodig S, Rajewsky K. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat Immunol. 2012;13:1092–1100. doi: 10.1038/ni.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sander S, Chu VT, Yasuda T, Franklin A, Graf R, Calado DP, Li S, Imami K, Selbach M, Di Virgilio M, et al. PI3 kinase and FOXO1 transcription factor activity differentially control B cells in the germinal center light and dark zones. Immunity. 2015;43:1075–1086. doi: 10.1016/j.immuni.2015.10.021. [DOI] [PubMed] [Google Scholar]
- 5.Dominguez-Sola D, Kung J, Holmes AB, Wells VA, Mo T, Basso K, Dalla-Favera R. The FOXO1 transcription factor instructs the germinal center dark zone program. Immunity. 2015;43:1064–1074. doi: 10.1016/j.immuni.2015.10.015. [DOI] [PubMed] [Google Scholar]
- 6.De Silva NS, Anderson MM, Carette A, Silva K, Heise N, Bhagat G, Klein U. Transcription factors of the alternative NF-kappaB pathway are required for germinal center B-cell development. Proc Natl Acad Sci USA. 2016;113:9063–9068. doi: 10.1073/pnas.1602728113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, Nussenzweig MC. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood. 2012;120:2240–2248. doi: 10.1182/blood-2012-03-415380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, Reth M. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci USA. 2006;103:13789–13794. doi: 10.1073/pnas.0605944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwon K, Hutter C, Sun Q, Bilic I, Cobaleda C, Malin S, Busslinger M. Instructive role of the transcription factor E2A in early B lymphopoiesis and germinal center B cell development. Immunity. 2008;28:751–762. doi: 10.1016/j.immuni.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 12.Khalil AM, Cambier JC, Shlomchik MJ. B cell receptor signal transduction in the GC is short-circuited by high phosphatase activity. Science. 2012;336:1178–1181. doi: 10.1126/science.1213368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, Bolland DJ, Chen HT, Corcoran AE, Nussenzweig A, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135:1028–1038. doi: 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casola S, Cattoretti G, Uyttersprot N, Koralov SB, Seagal J, Hao Z, Waisman A, Egert A, Ghitza D, Rajewsky K. Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proc Natl Acad Sci USA. 2006;103:7396–7401. doi: 10.1073/pnas.0602353103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15••.Robbiani DF, Deroubaix S, Feldhahn N, Oliveira TY, Callen E, Wang Q, Jankovic M, Silva IT, Rommel PC, Bosque D, et al. Plasmodium infection promotes genomic instability and AID-dependent B cell lymphoma. Cell. 2015;162:727–737. doi: 10.1016/j.cell.2015.07.019. In mice, chronic infections and thereby robust GC responses are often mimicked by constant interval challenges with SRBCs. In this work, the authors used the relevant BL-associated pathogen Plasmodium to investigate lymphomagenesis. These chronically infected mice experienced AID-dependent mutagenesis, including translocations, that led to B lymphomas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, Wright G, Shaffer AL, Hodson DJ, Buras E, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490:116–120. doi: 10.1038/nature11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Omori SA, Cato MH, Anzelon-Mills A, Puri KD, Shapiro-Shelef M, Calame K, Rickert RC. Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity. 2006;25:545–557. doi: 10.1016/j.immuni.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 18.Sander S, Calado DP, Srinivasan L, Kochert K, Zhang B, Rosolowski M, Rodig SJ, Holzmann K, Stilgenbauer S, Siebert R, et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell. 2012;22:167–179. doi: 10.1016/j.ccr.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Love C, Sun Z, Jima D, Li G, Zhang J, Miles R, Richards KL, Dunphy CH, Choi WW, Srivastava G, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44:1321–1325. doi: 10.1038/ng.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen S, Miyazaki M, Chandra V, Fisch KM, Chang AN, Murre C. Id3 orchestrates germinal center B cell development. Mol Cell Biol. 2016;36:2543–2552. doi: 10.1128/MCB.00150-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21••.Sungalee S, Mamessier E, Morgado E, Gregoire E, Brohawn PZ, Morehouse CA, Jouve N, Monvoisin C, Menard C, Debroas G, et al. Germinal center reentries of BCL2-overexpressing B cells drive follicular lymphoma progression. J Clin Invest. 2014;124:5337–5351. doi: 10.1172/JCI72415. In this work, the authors recapitulated the sporadic occurrence of BCL2 translocation of healthy individuals in a mouse model. Chronic antigen challenges were required for multiple re-entries of memory B cell clones carrying BCL2 to accumulate in lymphoid organs. The increased mutation frequency in these cells poses a risk to drive malignant transformation toward FL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peperzak V, Vikstrom IB, Tarlinton DM. Through a glass less darkly: apoptosis and the germinal center response to antigen. Immunol Rev. 2012;247:93–106. doi: 10.1111/j.1600-065X.2012.01123.x. [DOI] [PubMed] [Google Scholar]
- 23.Okosun J, Bodor C, Wang J, Araf S, Yang CY, Pan C, Boller S, Cittaro D, Bozek M, Iqbal S, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46:176–181. doi: 10.1038/ng.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24•.Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015;21:1190–1198. doi: 10.1038/nm.3940. This study and [25•] investigated the frequent loss-of-function mutation in KMT2D of B-NHL in murine models of FL. Both groups identified the tumor suppressive function of KMT2D in mouse models. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25•.Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21:1199–1208. doi: 10.1038/nm.3943. Please see the annotation of [24•] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang Y, Ortega-Molina A, Geng H, Ying HY, Hatzi K, Parsa S, McNally D, Wang L, Doane AS, Agirre Ena X, et al. CREBBP inactivation promotes the development of HDAC3 dependent lymphomas. Cancer Discov. 2016 doi: 10.1158/2159-8290.CD-16-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Souroullas GP, Jeck WR, Parker JS, Simon JM, Liu JY, Paulk J, Xiong J, Clark KS, Fedoriw Y, Qi J, et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat Med. 2016;22:632–640. doi: 10.1038/nm.4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, Yu Y, Bhattacharyya S, Shaknovich R, Geng H, Lobry C, et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol. 2015;16:653–662. doi: 10.1038/ni.3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casulo C, Burack WR, Friedberg JW. Transformed follicular non-Hodgkin lymphoma. Blood. 2015;125:40–47. doi: 10.1182/blood-2014-04-516815. [DOI] [PubMed] [Google Scholar]
- 30•.Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, Ouillette P, Trifonov V, Rossi D, Tabbo F, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6:130–140. doi: 10.1016/j.celrep.2013.12.027. Genetic analysis of sequential FL biopsies showed that progression to tFL mainly occurs through divergent evolution from a common progenitor cell. Mutations in MYC, CDKN2A/B, CCND3 and/or TP53 were specifically enriched in tFL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oricchio E, Ciriello G, Jiang M, Boice MH, Schatz JH, Heguy A, Viale A, de Stanchina E, Teruya-Feldstein J, Bouska A, et al. Frequent disruption of the RB pathway in indolent follicular lymphoma suggests a new combination therapy. J Exp Med. 2014;211:1379–1391. doi: 10.1084/jem.20132120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 33.Pasqualucci L, Dalla-Favera R. SnapShot: diffuse large B cell lymphoma. Cancer Cell. 2014;25:132–132.e131. doi: 10.1016/j.ccr.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 34.Beguelin W, Teater M, Gearhart MD, Calvo Fernandez MT, Goldstein RL, Cardenas MG, Hatzi K, Rosen M, Shen H, Corcoran CM, et al. EZH2 and BCL6 cooperate to assemble CBX8-BCOR complex to repress bivalent promoters, mediate germinal center formation and lymphomagenesis. Cancer Cell. 2016;30:197–213. doi: 10.1016/j.ccell.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mandelbaum J, Bhagat G, Tang H, Mo T, Brahmachary M, Shen Q, Chadburn A, Rajewsky K, Tarakhovsky A, Pasqualucci L, et al. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell. 2010;18:568–579. doi: 10.1016/j.ccr.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calado DP, Zhang B, Srinivasan L, Sasaki Y, Seagal J, Unitt C, Rodig S, Kutok J, Tarakhovsky A, Schmidt-Supprian M, et al. Constitutive canonical NF-kappaB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell. 2010;18:580–589. doi: 10.1016/j.ccr.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pasqualucci L, Zhang B. Genetic drivers of NF-kappaB deregulation in diffuse large B-cell lymphoma. Semin Cancer Biol. 2016;39:26–31. doi: 10.1016/j.semcancer.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 38.Knittel G, Liedgens P, Korovkina D, Seeger JM, Al-Baldawi Y, Al-Maarri M, Fritz C, Vlantis K, Bezhanova S, Scheel AH, et al. B-cell-specific conditional expression of Myd88p. L252P leads to the development of diffuse large B-cell lymphoma in mice. Blood. 2016;127:2732–2741. doi: 10.1182/blood-2015-11-684183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang B, Calado DP, Wang Z, Frohler S, Kochert K, Qian Y, Koralov SB, Schmidt-Supprian M, Sasaki Y, Unitt C, et al. An oncogenic role for alternative NF-kappaB signaling in DLBCL revealed upon deregulated BCL6 expression. Cell Rep. 2015;11:715–726. doi: 10.1016/j.celrep.2015.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneider C, Kon N, Amadori L, Shen Q, Schwartz FH, Tischler B, Bossennec M, Dominguez-Sola D, Bhagat G, Gu W, et al. FBXO11 inactivation leads to abnormal germinal-center formation and lymphoproliferative disease. Blood. 2016;128:660–666. doi: 10.1182/blood-2015-11-684357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang B, Kracker S, Yasuda T, Casola S, Vanneman M, Homig-Holzel C, Wang Z, Derudder E, Li S, Chakraborty T, et al. Immune surveillance and therapy of lymphomas driven by Epstein-Barr virus protein LMP1 in a mouse model. Cell. 2012;148:739–751. doi: 10.1016/j.cell.2011.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott DW, Gascoyne RD. The tumour microenvironment in B cell lymphomas. Nat Rev Cancer. 2014;14:517–534. doi: 10.1038/nrc3774. [DOI] [PubMed] [Google Scholar]
- 43.Cho SH, Raybuck AL, Stengel K, Wei M, Beck TC, Volanakis E, Thomas JW, Hiebert S, Haase VH, Boothby MR. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature. 2016;537:234–238. doi: 10.1038/nature19334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, Polak K, Tondera D, Gounarides J, Yin H, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell. 2012;22:547–560. doi: 10.1016/j.ccr.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Townsend EC, Murakami MA, Christodoulou A, Christie AL, Koster J, DeSouza TA, Morgan EA, Kallgren SP, Liu H, Wu SC, et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice. Cancer Cell. 2016;30:183. doi: 10.1016/j.ccell.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 46••.Muppidi JR, Schmitz R, Green JA, Xiao W, Larsen AB, Braun SE, An J, Xu Y, Rosenwald A, Ott G, et al. Loss of signalling via Galpha13 in germinal centre B-cell-derived lymphoma. Nature. 2014;516:254–258. doi: 10.1038/nature13765. The authors of this work showed that GC confinement is lost in the absence of Ga13. Ga13-dependent G-protein-coupled receptors, S1PR2 or human P2YR8, are involved in GC B regulation and the associated defective signaling led to the development of murine B lymphomas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47•.Boice M, Salloum D, Mourcin F, Sanghvi V, Amin R, Oricchio E, Jiang M, Mottok A, Denis-Lagache N, Ciriello G, et al. Loss of the HVEM tumor suppressor in lymphoma and restoration by modified CAR-T cells. Cell. 2016 doi: 10.1016/j.cell.2016.08.032. In this study, the authors identified that loss of HVEM in cooperation with enhanced BCL2 expression drove FL transformation in murine models. HVEM can bind in cis or trans to BTLA, which results in inhibitory signaling. Immunotherapeutic delivery of soluble HVEM efficiently decreased lymphoma progression in mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J, Vlasevska S, Wells VA, Nataraj S, Holmes AB, Duval R, Meyer SN, Mo T, Basso K, Brindle PK, et al. The Crebbp acetyltransferase is a haploinsufficient tumor suppressor in B cell lymphoma. Cancer Discov. 2017 doi: 10.1158/2159-8290.CD-16-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jellusova J, Cato MH, Apgar JR, Ramezani-Rad P, Leung C, Chen C, Richardson AD, Conner EM, Benschop RJ, Woodgett JR, Rickert RC, et al. Glycogen synthase kinase 3 is a metabolic checkpoint regulator in B cells. Nat Immunol. 2017 doi: 10.1038/ni.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]