

Abstract
Unrestrained growth factor signals can promote carcinogenesis, as well as other hallmarks of cancer such as immune evasion. Our understanding of the function and complex regulation of HER family of receptors has led to the development of targeted therapeutic agents that suppress tumor growth. However, these receptors also mediate escape from recognition by the host immune system. We discuss how HER family of oncogenic receptors downregulate tumor antigen presentation and upregulate suppressive membrane-bound or soluble secreted inhibitory molecules that ultimately lead to impaired cellular immunity mediated by cytotoxic T lymphocyte (CTL) recognition. Implementing this knowledge into new therapeutic strategies to enhance tumor immunogenicity may restore effector cell mediated immune clearance of tumors and clinical efficacy of tumor-targeted immunotherapy against HER receptor overexpression.
Introduction
Growth factor receptors transduce extracellular signals through the activation of intracellular messengers or directly through receptor translocation to the nucleus. Of the receptor tyrosine kinases (RTKs), the HER family, also called ErbB, is one of the most extensively studied for its role in development, physiology, and human cancer (1). The HER family is considered a prototypical oncogenic growth factor receptor, since it activates multiple intracellular signal transduction cascades including the mitogen activated protein kinase (MAPK), phosphatidylinositol-3 kinase (PI3K/AKT), Janus kinase/signal transduced and activator of transcription (JAK/STAT) and protein kinase C (PKC) pathways (2, 3). In turn, these signals induce malignant transformation of tumor cells through increased cell proliferation and survival, resistance to growth inhibition or apoptosis and increased invasion and metastasis, capabilities that are common to the majority of tumors and were recognized as initial “Hallmarks of Cancer” (4).
In addition, recent work has provided evidence for enlarging this list of tumor transforming competences, where cellular metabolism re-conditioning, inflammation-promoting tumor development and evasion of immune destruction have been noted as important additional “Hallmarks of Cancer” (5). In this context, the concept of “tumor immunogenicity” arises, where tumor cells could be more or less immunogenic in regard to expression of molecules that activate or inhibit the host immune system. Three major signals determine a successful immune response, intact antigen processing machinery (APM) and HLA class I mediated antigen presentation (Signal 1), proper co-stimulation (Signal 2) and proinflammatory cytokine stimulation (Signal 3). Tumor cells evade immune recognition by downregulating signal 1 and providing aberrant signals 2 and 3, which in the setting of tumors are represented by increased surface expression of checkpoint receptor ligands, particularly programmed death ligand-1 (PD-L1) and increased secretion of immunosuppressive cytokines and chemokines. In this review, we focus on the molecular mechanisms of how HER family of receptors regulate tumor cell-induced immune escape, not only by downregulating HLA class I antigen presentation but also by upregulating expression of PD-L1 and suppressive cytokines. Ultimately, these oncogenic signals lead to evasion of cellular immunity mediated by cytotoxic T lymphocyte (CTL) and natural killer (NK) cell cytotoxicity, which may be reversed by specific anti-HER monoclonal antibody (mAb) or tyrosine kinase inhibitor (TKI) therapy.
HER family of growth factor receptors: overview and targeted immunotherapy
The HER family of receptors comprises four members: EGFR (ErbB1, HER1), ErbB2 (HER2, neu in rodents), ErbB3 (HER3) and ErbB4 (HER4) (1). Soluble ligand binding to the ectodomain of the receptor promotes homo- and heterodimerization which in turn induces activation of the intracellular tyrosine kinase domain and phosphorylation of the C-terminal tail, intracellular phosphoproteins then bind and activate adaptors that transduce signals that activate MAPK, PI3K, PKC and JAK/STAT pathways (2). Importantly, two members of this growth factor receptor family, the epidermal growth factor receptor (EGFR) and HER2 are the most strongly associated with tumor progression of various human neoplasms, including breast, lung, head and neck squamous cell carcinoma (HNSCC) and glioblastoma (6–8). Furthermore, in the case of the EGFR, in addition to overexpression of the wild type receptor some tumors also exhibit activating mutant forms such as glioblastoma, where a variant called EGFRvIII has been reported (9) or non-small cell lung cancer (NSCLC) where mutations in the EGFR kinase domain (L858R human/T790M murine) have been associated with tumor resistance (10). Given that the mechanism of action of HER receptors depends heavily on the interaction of their extracellular domains with activating ligands, their function can be readily disrupted by targeted receptor-blocking specific mAbs, which prevent ligand binding and possibly induce endocytosis and degradation of the receptor (11). Three humanized mAbs targeting this family of receptors have been approved for clinical use, trastuzumab which targets HER2 and cetuximab or panitumumab which target the EGFR. Moreover, a second strategy to block HER receptor signaling works by inhibiting their tyrosine kinase function with specific small molecule inhibitors. In the setting of cancer treatment, three TKI have been approved for clinical use, gefitinib and erlotinib that are specific for the EGFR and lapatinib that is equally specific for the EGFR and HER2 (12, 13). EGFR blocking mAbs have an additional therapeutic benefit over TKIs, that is the activation of the innate immune system NK cells through interaction of the Fc portion with the FcγRIIIa (CD16) receptor, triggering antibody dependent cellular cytotoxicity (ADCC), activation of dendritic cells (DC) via NK-DC IFNγ-mediated crosstalk and ultimately, inducing upregulation of tumor antigen specific CTL (14).
EGFR and HER2 mediated downregulation of APM components and HLA class I mediated antigen presentation
Tumor antigen presentation is a major pre-requisite for appropriate T cell responses, especially because of the crucial role of this process in the generation of tumor antigen (TA)-specific adaptive immune responses (15, 16). TA-antibody based immunotherapies may rely on this process, where mAbs such as cetuximab or trastuzumab enhance adaptive T cell responses by promoting NK cell activation, Th1 cytokine secretion and DC cross presentation (17, 18). Importantly, impaired HLA class I antigen presentation is associated with reduced CTL recognition, disease progression and metastasis (19, 20). Interestingly, recent studies showed that EGFR signaling regulates the expression of APM components and HLA class I (17, 21). In this regard, EGFR-induced activation of protein phosphatase type 11 (PTNP11, best known as SHP2) dephosphorylates signal transducer and activator of transcription 1 (STAT1), a known transcription factor that mediates expression of HLA class I and APM molecules (Figure 1). Interestingly, treatment with IFNγ and inhibition or depletion of SHP2 in tumor cell lines induced upregulation of phosphorylated STAT1 and restored expression of HLA class I and APM components (17), which ultimately enhanced HLA class I restricted antigen presentation and expansion of EGFR-specific CTL. Furthermore, EGFR-mediated SHP2 activation may not only downregulate HLA class I-peptide presentation (signal 1) but also induce secretion of immunostimulatory cytokines and chemokines (signal 3), since its inhibition upregulated tumor cell secretion of IL-12p70, RANTES (CCL5) and IP-10 (CXCL10). Importantly, a second mechanism of downregulation of HLA class I and APM components downstream the EGFR has been reported, where activated SHP2 dephosphorylates GDP, inducing RAS/MAPK pathway activation (19). Notably, restoration of signal 1 via EGFR inhibition is clinically relevant, since patients from a novel phase II clinical trial who responded to cetuximab single agent therapy showed significant upregulation of tumor HLA class I expression (22), and induction of EGFR specific CTL correlated with better clinical response.
Figure 1. EGFR/HER2 mediated immunoescape.
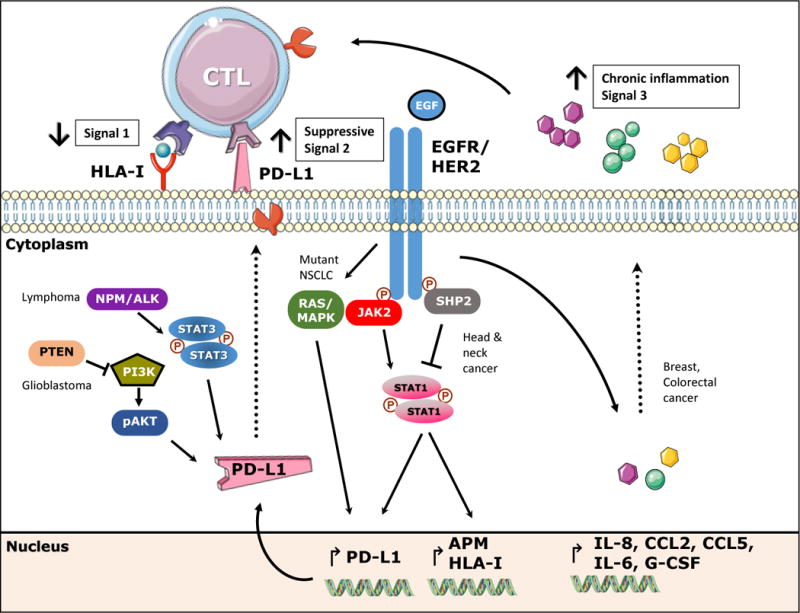
EGFR stimulation induces activation of phosphatase SHP2 which decreases phosphorylation of STAT1 and subsequently expression of HLA class I and APM components. Conversely, EGFR mediated activation of JAK2-STAT1 induces expression of PD-L1. Likewise, mutant EGFR/RAS-MAPK pathway induces expression of PD-L1 as reported in NCSLC. Other pathways involved in PD-L1 expression are depicted on the left, PI3K and NPM/ALK-STAT3 pathways have been reported to upregulate PD-L1 in glioblastoma and T cell Lymphoma, respectively. EGFR/HER2 stimulation induces secretion of IL-8, IL-6, CCL2, CCL3, CCL5, eotaxin, GM-CSF inducing chronic inflammation and escape from T cell recognition. Overall, EGFR mediated downregulation of signal 1 and upregulation of suppressive signals 2 and 3, favoring escape from CTL recognition.
In the setting of HER2, a recent report demonstrated that its oncogenic transformation in murine models was associated with low levels of MHC class I and reduced CTL recognition (23, 24), which could be reversed by IFNγ treatment (25). In humans, HER2 overexpressing tumors showed poor CTL recognition even after IFNγ treatment (26). Furthermore, siRNA mediated silencing of HER2 induced upregulation of HLA class I molecules in several ErbB2+ breast cancer cell lines (27). Likewise, HER2 overexpression in melanoma cells resulted in poor recognition by tyrosinase-specific CTL (28), the mechanism by which HLA class I antigen presentation was affected may be related to HER2-induced mutations within the promoter binding sites of the transcription factor E2F1 in the tapasin locus (29). Moreover, the mechanism by which HER2 downmodulates HLA class I expression may also be associated with MAPK pathway activation since MAPK inhibition with PD98059 resulted in a dose-dependent upregulation of surface HLA class I expression in breast, esophageal and gastric carcinoma cells. Further evidence of the role of the MAPK pathway in the regulation of antigen presentation is the finding of Sapkota et al. which demonstrated that vemurafenib-mediated BRAF inhibition enhanced IFNγ-mediated upregulation of HLA class I expression in melanoma (20). In addition, the view that HER2 orchestrates tumor immune escape is clinically relevant since patients who had high HER2 expression had low HLA class I levels, as determined by immunohistochemistry staining of breast cancer specimens (30). In summary, these findings demonstrate that HER family receptors play an active role mediating immune evasion by downregulating antigen presentation and impairing subsequent CTL-mediated recognition and killing.
EGFR and HER2 mediated upregulation of PD-L1
Recent reports have shown that the PD-L1/PD-1 pathway constitutes a major suppressive mechanism to evade immune activation and tumor clearance by providing an aberrant co-inhibitory signal 2 which downregulates T cell proliferation, survival, cytotoxicity and cytokine release (31, 32). Inhibition of this pathway has proved to be clinically relevant since blocking antibodies against PD-1 or PD-L1 demonstrated substantial clinical activity in patients with metastatic melanoma, renal cell carcinoma (RCC), NSCLC, and HNSCC, where PD-L1 tumor expression enriched for clinical responders (33–35). Mechanisms of PD-L1 upregulation in various cancer types have included IFNγ as well as oncogenic signals through EGFR, PI3K and MAPK/MEK (BRAF V600E+ cancers). Interestingly, HER2 has been implicated in the expression of PD-L1 in gastric carcinoma patients where expression of PD-L1 highly correlated with that of HER2, furthermore, siRNA-mediated silencing of HER2 significantly downregulated PD-L1 expression in gastric carcinoma cell lines (36). In addition, Akbay et al, reported that mutant EGFR/KRAS-MAPK pathway induced the expression of PD-L1 in NSCLC cell lines which could be inhibited in vitro by gefitinib (37), whereas in the setting of HNSCC, overexpressed wild-type EGFR regulated the expression of PD-L1 in a JAK2/STAT1 dependent fashion (Figure 1). Interestingly, inhibiting PD-L1 upregulation via specific JAK2 inhibition enhanced cetuximab-mediated NK cell killing of HNSCC cells (38), supporting the key role of EGFR-PD-L1 axis evading cellular immunity. Curiously, although both cancers upregulate PD-L1 in an EGFR-dependent fashion, the different signaling pathways involved in PD-L1 upregulation could be explained by the unique biology of each cancer type, where NSLSC would rely more on mutant EGFR/KRAS leading to stronger activation of the MAPK pathway (37). In contrast, HNSCC with a much lower EGFR/KRAS mutation rate (2%) (39–41) relies more on an overstimulated wild type EGFR/JAK2 pathway for oncogenic signaling (38). Supporting these results is the recent finding that JAK2 inactivating mutations correlated with resistance to anti-PD-1 therapy in melanoma patients, in this setting we could speculate that tumor cells may evolve new strategies to evade anti-PD-L1/PD-1 pathway blocking therapy by downregulating JAK2-mediated expression of PD-L1 (42). Interestingly, the EGFR-JAK2 mediated PD-L1 upregulation was STAT1 dependent, which constitutes a novel finding, since EGFR is mostly known to upregulate STAT3 oncogenic signaling (21). In this setting, EGFR-STAT1 activation appears to have different kinetics than that of IFNγ, given that the highest peak of activated pSTAT1 that induced PD-L1 upregulation was seen at 24h after EGFR stimulation. Notably, there appears to be a crosstalk between EGFR and IFNγ pathways regarding PD-L1 regulation, since cetuximab-mediated EGFR blockade downregulated the IFNγ-dependent PD-L1 expression at the transcript and protein levels (38). Therefore, EGFR blockade may not only diminish the tumor cell intrinsic PD-L1 upregulation but also the extrinsic IFNγ-mediated signals that have been associated with CD8+ T cell infiltration in the tumor microenvironment (43, 44).
Finally, other oncogenic signals, although not in the context of EGFR stimulation, have been shown to induce PD-L1 expression. In the setting of hematologic malignancies, enhanced activation of chimeric nucleophosmin (NPM)/anaplastic lymphoma kinase (ALK) in T cell lymphoma cells induced an increased STAT3 activation, promoter binding and transcription of PD-L1(45). Likewise, Parsa et al. demonstrated that PTEN loss, and subsequent overactivation of the PI3K pathway increased PD-L1 expression in glioblastoma multiforme (GBM) cell lines (Figure 1) a finding that was further validated in tumor specimens where PTEN loss highly correlated with PD-L1 expression (46).
EGFR and HER2 mediated upregulation of oncogenic cytokines
An optimal T cell activation requires signals from the T cell receptor (TCR) (signal 1), costimulatory receptors (signal 2) and an additional signal mediated by soluble cytokines and chemokines secreted in the milieu (signal 3) (47). In the tumor microenvironment, the presence of suppressive cytokines promotes unresponsiveness of CD8+ T effector immune cell infiltrates and proliferation of suppressive cell subsets such as regulatory T cells (Treg), myeloid derived suppressive cells (MDSC) or tumor-associated macrophages (TAM)(48, 49). In the setting of growth factor receptors inducing secretion of an oncogenic signal 3 a recent report showed that EGFR inhibition in HNSCC cells increased secretion of proinflammatory cytokines such as IL-2, IL-4, IL-6, IL-8, TNF-α, and IFN-γ, which was proposed to induce chronic inflammation, tumor progression and resistance to anti-EGFR therapy (50). Interestingly, this study also reported that erlotinib-mediated EGFR inhibition increased expression of IL-6, a known oncogenic cytokine in HNSCC, and that such upregulation was mediated by NFKB/MAPK pathways. Upregulation of this alternative pathway was postulated as an escape mechanism of erlotinib-resistant cells a view that was further supported by sensitization of these cells to erlotinib by tocilizumab mediated IL-6 receptor inhibition (50). Gelfo et al. showed that activation of the EGFR in colorectal cancer cell lines mediated secretion of proinflammatory cytokines IL-1α, IL-1β and IL-8, which were associated with a cetuximab resistant phenotype in vitro (51). Likewise, HER2 activation in breast cancer cell lines has been reported to induce secretion of proinflammatory cytokines IL-8 and CXCL1 (GRO) that were downregulated by gefitinib treatment, furthermore, circulating levels of IL-8 and CXCL1 in breast cancer patients highly correlated with HER2 expression (52). Additionally, another study reported that HER2+ breast cancer patients had significantly higher levels of CCL2 (MCP-1), eotaxin, CCL5 (RANTES) than healthy controls. Moreover, IL-6, CCL3 (MIP1α) and G-CSF were upregulated in metastatic patients (53), which further links the inflammatory pro-tumorigenic cytokine microenvironment to deregulated oncogenic growth factor signaling. While our understanding of the immunosuppressive tumor microenvironment (TME) has increased notably in recent years, now there is recognition that tumor cell intrinsic signaling through EGFR/HER2 mediates production of suppressive cytokines such as transforming growth factor beta (TGFβ), vascular endothelial growth factor (VEGF) or IL-10, affecting the phenotype and function of tumor infiltrating lymphocytes (TIL).
The effect of EGFR targeted immunotherapy in the tumor microenvironment
Growth factor oncogenic signaling, particularly that of the EGFR/HER2, mediates immunoescape by downregulating signal 1 and providing an immunosuppressive signal 2 and 3 as discussed in previous sections of this review. Therefore, blockade of EGFR/HER2 signals presented as a logical strategy to overcome tumor immunosuppression in the clinical setting. Table 1 lists the major effects of EGFR/HER2 targeted immunotherapy in the different types of cancer for which these agents were FDA approved for. Herein we will focus in more detail describing the effects of the IgG1 anti-EGFR specific mAb cetuximab, which was FDA approved as a monotherapy for recurrent/metastatic HNSCC or in combination with radiation therapy for advanced HNSCC and EGFR+ metastatic colorectal cancer (54). Despite 90% of HNSCC overexpress the EGFR only 10–20% of patients responded to therapy, this observation brought up multiple questions as to what happens in the TME of the responder patients versus the non-responders from an immunology standpoint. Recent reports address these inquiries, in the setting of HNSCC, Srivastava RM et al. recently reported that cetuximab monotherapy induced a significant upregulation of HLA class I in responders compared to non-responders (22). Furthermore, complementing the previous data is the finding of a significant higher frequency of circulating EGFR-specific CD8+ T cells in the same cohort of patients post-cetuximab treatment (14). Moreover, cetuximab monotherapy seems to ameliorate the suppressive phenotype of MDSCs infiltrating the tumor site, since circulating CD11b+CD14+HLA-DRhi monocytes displayed an attenuated M2 polarization in cetuximab responders characterized by decreased IL-10 and CD163 expression after cetuximab treatment (55). On the other hand, the lack of response to EGFR targeted therapy could be explained by increased frequency of circulating CD11b+CD14+HLA-DRlo/neg termed monocytic MDSCs that were observed in patients who did not respond to cetuximab therapy. In addition to this finding is the one where circulating regulatory T cells (Treg) were significantly higher in non-responders to cetuximab monotherapy in the same cohort of patients, interestingly these Tregs were further enriched in the tumor bed and highly expressed the immune checkpoint receptor cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and other known immunosuppressive markers CD39 and TGFβ. These Tregs suppressed NK-cetuximab mediated ADCC in vitro and their increased frequency correlated with poor prognosis of patients (56).
Table 1. Current clinical trials testing the combination of EGFR/HER2 targeted therapy and immunotherapy.
EGFR, Epidermal growth factor receptor; mAb, monoclonal antibody; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; PD-1, programmed death-1; PD-L1, programmed death ligand-1; EGFRmut, EGFR mutant; HNSCC, head and neck squamous cell carcinoma; CRC, colorectal cancer; NSCLC, non-small cell lung cancer.
| EGFR/HER2 targeted mAb + co-stimulatory/checkpoint immunotherapy | |||||
|---|---|---|---|---|---|
|
| |||||
| Targets | Treatments | Phase | Clinical Trial ID | Type of cancer | Status |
| EGFR + CD137 | Cetuximab + Urelumab | Ib | NCT02110082 | HNSCC or CRC | Ongoing |
| EGFR + CTLA-4 | Cetuximab + Ipilimumab + IMRT | Ib | NCT01860430 | HNSCC | Recruiting |
| NCT01935921 | HNSCC | Recruiting | |||
| HER2 + PD-1 | Trastuzumab + Pembrozilumab | Ib/II | NCT02901301 | HER2+ gastric cancer | Not yet recruiting |
|
| |||||
| EGFR/HER2 targeted TKI + checkpoint immunotherapy | |||||
|
| |||||
| Targets | Treatments | Phase | Clinical Trial ID | Type of cancer | Status |
| EGFRmut + PD-L1 | Erlotinib + Atezolizumab | I | NCT02013219 | NSCLC | Recruiting |
| EGFRmut + PD-1 | Erlotinib or Gefitinib + Pembrolizumab | I/II | NCT02039674 | NSCLC | Recruiting |
| EGFRmut + PD-1 | Erlotinib + Nivolumab | I | NCT01454102 | NSCLC | Ongoing |
| EGFRmut + PD-L1 | Erlotinib + Durvalumab | I | NCT02088112 | NSCLC | Recruiting |
| EGFRmut + PD-L1 | Rociletinib + Atezolizumab | I | NCT02630186 | NSCLC | Ongoing |
| EGFRmut + PD-L1 | Osimertinib + Durvalumab | I | NCT02143466 | NSCLC | Recruiting |
| EGFRmut + PD-1 | Afatinib + Pembrolizumab | I | NCT02364609 | NSCLC | Recruiting |
| EGFRmut + PD-L1 | Erlotinib + Atezolizumab | I | NCT02013219 | NSCLC | Recruiting |
Rationale for combination therapy
As discussed in the previous section, cetuximab mediated EGFR targeted therapy is effective only in a small percent of patients (20%). The characterization of circulating and tumor immune cell infiltrates after cetuximab monotherapy in responders versus no-responders has shed light for the onset of combination therapies that will most likely overcome the immunosuppressive strategies that tumors evolve to circumvent cetuximab mediated downregulation of EGFR oncogenic signaling. In this regard, natural killer (NK) cells have been shown to play a crucial role after in vitro activation with cetuximab, mediating ADCC, IFNγ secretion, DC crosstalk that ultimately leads to expansion of TA specific CTLs. Interestingly, in preclinical studies NK cells upregulate the activation marker CD137 after cetuximab exposure (57). In turn, mAb-mediated specific CD137 stimulation enhanced cetuximab ADCC and TA cross-presentation (58). Based on these promising preclinical results a phase Ib clinical trial combining cetuximab and anti-CD137 mAb (urelumab) is currently underway (Table 2). In addition to enhancing NK cytotoxicity, depletion of immunosuppressive cell subsets such as CTLA-4+ Tregs is a logical strategy given the findings shown from cetuximab non-responders. Interestingly, preclinical data showed that the anti-CTLA-4 mAb, ipilimumab, selectively depleted intratumoral Tregs when co-cultured with NK cells ex vivo, based on these results two phase Ib clinical trials were started where the combination of ipilimumab and cetuximab in untreated advanced HNSCC patients (NCT01935921) and ipilimumab, cetuximab and radiation (NCT01860430) are being currently tested (Table 1). Moreover, in light of the preclinical findings of an upregulated PD-L1/PD-1 pathway in HNSCC mediated by the EGFR (21) (discussed in previous sections) the combination of ipilimumab and anti-PD-1 mAb (nivolumab) is being currently tested in a phase III clinical trial in patients with recurrent or metastatic HNSCC (NCT02741570). In this setting, it would be also interesting to test the combination of cetuximab and anti-PD-1 (nivolumab, pembrolizumab) or anti-PD-L1(atezolizumab, avelumab, durvalumab) given the high frequency of PD-L1+ tumor cells in HNSCC specimens and the residual PD-L1 expression seen in vitro when HNSCC cell lines are treated with cetuximab, which hints the possibility of other alternative pathways that could upregulate PD-L1 besides the EGFR, importantly an added benefit of anti-PD-L1 mAb therapy would be to diminish the IFNγ-mediated PD-L1 upregulation in tumors induced via cetuximab-mediated NK cell activation. Finally, given the enhanced expression and signaling of TGFβ in HNSCC (59) and tumor infiltrating Tregs (56), combination of cetuximab and anti-TGFβ/TGFβR1 could potentially enhance cetuximab mediated NK cytotoxicity, decrease generation of Tregs and skew the TME cytokine milieu to a Th1 phenotype. Importantly, some key considerations for the clinical development of targeted therapy plus immunotherapy include optimizing dosing regimens (concurrent vs. sequential) that would allow minimizing treatment-related toxicities and the proper selection of endpoints to assess efficacy.
Table 2. Summary of effects of EGFR/HER2 targeted therapeutic agents on the immune system.
HNSCC, head and neck squamous cell carcinoma; CRC, colorectal carcinoma; NSCLC, non-small cell lung cancer; ADCC, antibody dependent cellular cytotoxicity; DC, dendritic cells; RAF, ERK, AKT, members of the MAPK signaling pathway.
| Examples of effects of EGFR/HER2 targeted therapeutic agents on the immune system | ||
|---|---|---|
|
| ||
| Agents | Indications | Effects |
|
||
| Cetuximab | HNSCC, CRC | |
|
||
| Trastuzumab | Advanced stage breast cancer | |
|
||
| Erlotinib | NSCLC, pancreatic cancer |
|
Concluding remarks
Cumulative data now support the view that tumors evolve intrinsic mechanisms to evade immune recognition and we review those that result from oncogenic signals downstream of the HER family of receptors. Importantly, the better understanding of the mechanisms that tumor cells use to escape immunosurveillance will permit strategies to counteract this deregulation in the TME. Immunotherapy has potential to synergize with current cancer therapies such as tumor-targeted HER-specific therapy. Notably, one of the most important advantages of immunotherapy is the specificity to recognize tumor antigens such as EGFR or HER2, which has shown clinical efficacy in many types of cancer. However, the majority of EGFR/HER2-expressing tumors have a complex genetic background with a significant level of compensatory ‘cross-talk’ among receptors within a signaling network as well as with other pathways regulating cell proliferation, trafficking, and survival. Therefore, combination therapy not only targeting growth factor receptors but also other important molecules that regulate cellular immune responses in the TME such as the PD-L1/PD-1 axis, CTLA-4, TGFβ and other immunosuppressive cytokines should enhance immune responses and improve clinical efficacy. Importantly, combination therapy should always be based on the framework of preclinical studies that critically assess the mechanism of action of these combinational therapeutic approaches.
Highlights.
HER signaling mediates downregulation of HLA class I mediated antigen presentation.
HER signaling regulates expression of PD-L1 in tumor cells.
HER signaling mediates secretion of oncogenic cytokines by tumor cells.
Acknowledgments
Financial support: This work was supported by National Institute of Health grants R01 DE19727, P50 CA097190, CA110249 and University of Pittsburgh Cancer Center Support Grant P30CA047904.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors disclose no potential conflicts of interest.
References
- 1.Linggi B, Carpenter G. ErbB receptors: new insights on mechanisms and biology. Trends Cell Biol. 2006;16:649–56. doi: 10.1016/j.tcb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 2.Marmor MD, Skaria KB, Yarden Y. Signal transduction and oncogenesis by ErbB/HER receptors. Int J Radiat Oncol Biol Phys. 2004;58:903–13. doi: 10.1016/j.ijrobp.2003.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Warren CM, Landgraf R. Signaling through ERBB receptors: multiple layers of diversity and control. Cell Signal. 2006;18:923–33. doi: 10.1016/j.cellsig.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Harari D, Yarden Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene. 2000;19:6102–14. doi: 10.1038/sj.onc.1203973. [DOI] [PubMed] [Google Scholar]
- 7.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–65. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 8.Wada T, Qian XL, Greene MI. Intermolecular association of the p185neu protein and EGF receptor modulates EGF receptor function. Cell. 1990;61:1339–47. doi: 10.1016/0092-8674(90)90697-d. [DOI] [PubMed] [Google Scholar]
- 9.Gan HK, Cvrljevic AN, Johns TG. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J. 2013;280:5350–70. doi: 10.1111/febs.12393. [DOI] [PubMed] [Google Scholar]
- 10.Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. 2013;31:1070–80. doi: 10.1200/JCO.2012.43.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wieduwilt MJ, Moasser MM. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci. 2008;65:1566–84. doi: 10.1007/s00018-008-7440-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–72. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 13.Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–27. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srivastava RM, Lee SC, Andrade Filho PA, Lord CA, Jie HB, Davidson HC, et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res. 2013;19:1858–72. doi: 10.1158/1078-0432.CCR-12-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meissner M, Reichert TE, Kunkel M, Gooding W, Whiteside TL, Ferrone S, et al. Defects in the human leukocyte antigen class I antigen processing machinery in head and neck squamous cell carcinoma: association with clinical outcome. Clin Cancer Res. 2005;11:2552–60. doi: 10.1158/1078-0432.CCR-04-2146. [DOI] [PubMed] [Google Scholar]
- 16.Lopez-Albaitero A, Nayak JV, Ogino T, Machandia A, Gooding W, DeLeo AB, et al. Role of antigen-processing machinery in the in vitro resistance of squamous cell carcinoma of the head and neck cells to recognition by CTL. J Immunol. 2006;176:3402–9. doi: 10.4049/jimmunol.176.6.3402. [DOI] [PubMed] [Google Scholar]
- 17.Leibowitz MS, Srivastava RM, Andrade Filho PA, Egloff AM, Wang L, Seethala RR, et al. SHP2 is overexpressed and inhibits pSTAT1-mediated APM component expression, T-cell attracting chemokine secretion, and CTL recognition in head and neck cancer cells. Clin Cancer Res. 2013;19:798–808. doi: 10.1158/1078-0432.CCR-12-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seliger B, Kiessling R. The two sides of HER2/neu: immune escape versus surveillance. Trends Mol Med. 2013;19:677–84. doi: 10.1016/j.molmed.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Agazie YM, Hayman MJ. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol Cell Biol. 2003;23:7875–86. doi: 10.1128/MCB.23.21.7875-7886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sapkota B, Hill CE, Pollack BP. Vemurafenib enhances MHC induction in BRAFV600E homozygous melanoma cells. Oncoimmunology. 2013;2:e22890. doi: 10.4161/onci.22890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Concha-Benavente F, Srivastava RM, Ferrone S, Ferris RL. EGFR-mediated tumor immunoescape: The imbalance between phosphorylated STAT1 and phosphorylated STAT3. Oncoimmunology. 2013;2:e27215. doi: 10.4161/onci.27215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22*.Srivastava RM, Trivedi S, Concha-Benavente F, Hyun-Bae J, Wang L, Seethala RR, et al. STAT1-Induced HLA Class I Upregulation Enhances Immunogenicity and Clinical Response to Anti-EGFR mAb Cetuximab Therapy in HNC Patients. Cancer Immunol Res. 2015;3:936–45. doi: 10.1158/2326-6066.CIR-15-0053. Authors demostrate that EGFR induces HLA class I downregulation via SHP2-STAT1 suppression, EGFR blockade with cetuximab in receptor, STAT1 activation and upregulation of HLA-B/C alleles more than HLA-A alleles. HLA class I upregulation was significantly associated with clincal response to cetuximab in a single agent neo-adjuvant phase II clinical trial. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seliger B, Maeurer MJ, Ferrone S. Antigen-processing machinery breakdown and tumor growth. Immunol Today. 2000;21:455–64. doi: 10.1016/s0167-5699(00)01692-3. [DOI] [PubMed] [Google Scholar]
- 24.Chang CC, Ogino T, Mullins DW, Oliver JL, Yamshchikov GV, Bandoh N, et al. Defective human leukocyte antigen class I-associated antigen presentation caused by a novel beta2-microglobulin loss-of-function in melanoma cells. J Biol Chem. 2006;281:18763–73. doi: 10.1074/jbc.M511525200. [DOI] [PubMed] [Google Scholar]
- 25.Herrmann F, Lehr HA, Drexler I, Sutter G, Hengstler J, Wollscheid U, et al. HER-2/neu-mediated regulation of components of the MHC class I antigen-processing pathway. Cancer Res. 2004;64:215–20. doi: 10.1158/0008-5472.can-2522-2. [DOI] [PubMed] [Google Scholar]
- 26.Zaks TZ, Rosenberg SA. Immunization with a peptide epitope (p369–377) from HER-2/neu leads to peptide-specific cytotoxic T lymphocytes that fail to recognize HER-2/neu+ tumors. Cancer Res. 1998;58:4902–8. [PubMed] [Google Scholar]
- 27.Choudhury A, Charo J, Parapuram SK, Hunt RC, Hunt DM, Seliger B, et al. Small interfering RNA (siRNA) inhibits the expression of the Her2/neu gene, upregulates HLA class I and induces apoptosis of Her2/neu positive tumor cell lines. Int J Cancer. 2004;108:71–7. doi: 10.1002/ijc.11497. [DOI] [PubMed] [Google Scholar]
- 28.Mimura K, Ando T, Poschke I, Mougiakakos D, Johansson CC, Ichikawa J, et al. T cell recognition of HLA-A2 restricted tumor antigens is impaired by the oncogene HER2. Int J Cancer. 2011;128:390–401. doi: 10.1002/ijc.25613. [DOI] [PubMed] [Google Scholar]
- 29.Bukur J, Herrmann F, Handke D, Recktenwald C, Seliger B. Identification of E2F1 as an important transcription factor for the regulation of tapasin expression. J Biol Chem. 2010;285:30419–26. doi: 10.1074/jbc.M109.094284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inoue M, Mimura K, Izawa S, Shiraishi K, Inoue A, Shiba S, et al. Expression of MHC Class I on breast cancer cells correlates inversely with HER2 expression. Oncoimmunology. 2012;1:1104–10. doi: 10.4161/onci.21056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 32.Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, et al. B7–DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001;193:839–46. doi: 10.1084/jem.193.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferris RL, Blumenschein G, Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016 doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimura Y, Oki E, Yoshida A, Aishima S, Zaitsu Y, Ohtsu H, et al. Significance of accurate human epidermal growth factor receptor-2 (HER2) evaluation as a new biomarker in gastric cancer. Anticancer Res. 2014;34:4207–12. [PubMed] [Google Scholar]
- 37.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–63. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38*.Concha-Benavente F, Srivastava RM, Trivedi S, Lei Y, Chandran U, Seethala RR, et al. Identification of the Cell-Intrinsic and -Extrinsic Pathways Downstream of EGFR and IFNgamma That Induce PD-L1 Expression in Head and Neck Cancer. Cancer Res. 2016;76:1031–43. doi: 10.1158/0008-5472.CAN-15-2001. Authors identify tumor cell extrinsic (IFNα mediated) and intrinsic (EGFR mediated) pathways that induce PD-L1 expression. Importantly, these pathways use JAK2/STAT1 as a common signaling pathway for PD-L1 upregulation. This is the first study that reports the signaling pathway that induces PD-L1 expression downstream wild type EGFR. These findings are clinically relevant since EGFR expression in large cohort of head and neck quamous cell carcinoma specimens (TCGA, n=500) significantly correlated with JAK2 and PD-L1 expression. Specific JAK2 inhibition prevented IFNα and EGFR induced PD-L1 upregulation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McBride SM, Rothenberg SM, Faquin WC, Chan AW, Clark JR, Ellisen LW, et al. Mutation frequency in 15 common cancer genes in high-risk head and neck squamous cell carcinoma. Head Neck. 2014;36:1181–8. doi: 10.1002/hed.23430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–82. doi: 10.1038/nature14129. Characterization of 279 head and neck squamous cell carcinoma specimens identified important mutations in different subsets of patients. HPV-positive patients had PIK3CA, TRAF3 loss-of-function mutations. Smokers had TP53, CDKN2A inactivating mutations. Patients with favourable clinical outcomes had activating mutations of HRAS or PIK3CA coupled with inactivating mutations in CASP8, NOTCH1 and TP53. Importantly, this study identifies altered pathways that are elegible for therapeutic targets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42**.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375:819–29. doi: 10.1056/NEJMoa1604958. Authors describe critical loss-of-function mutations in the IFNα receptor pathway that are associated to resistance to anti-PD-1 therapy in melanoma patients. JAK1, JAK2 and B2M inactivating mutations were reported in specimens from patients who relapsed after anti-PD1 therapy with pembrolizumab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lyford-Pike S, Peng S, Young GD, Taube JM, Westra WH, Akpeng B, et al. Evidence for a role of the PD-1:PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Res. 2013;73:1733–41. doi: 10.1158/0008-5472.CAN-12-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44*.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–61. doi: 10.1016/j.ccell.2015.03.001. Comprehensive review of different checkpoint receptors and ligands expressed in the tumor microenvironment, drugs that have been FDA approved targeting CTLA-4, PD-1, PD-L1, mechanism of tumor resistance to innate and adaptive immunity and candidate combination therapies are discussed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1) Proc Natl Acad Sci U S A. 2008;105:20852–7. doi: 10.1073/pnas.0810958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–8. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 47.Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, et al. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev. 2006;211:81–92. doi: 10.1111/j.0105-2896.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- 48.Burkholder B, Huang RY, Burgess R, Luo S, Jones VS, Zhang W, et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim Biophys Acta. 2014;1845:182–201. doi: 10.1016/j.bbcan.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 49.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fletcher EV, Love-Homan L, Sobhakumari A, Feddersen CR, Koch AT, Goel A, et al. EGFR inhibition induces proinflammatory cytokines via NOX4 in HNSCC. Mol Cancer Res. 2013;11:1574–84. doi: 10.1158/1541-7786.MCR-13-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gelfo V, Rodia MT, Pucci M, Dall’Ora M, Santi S, Solmi R, et al. A module of inflammatory cytokines defines resistance of colorectal cancer to EGFR inhibitors. Oncotarget. 2016 doi: 10.18632/oncotarget.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vazquez-Martin A, Colomer R, Menendez JA. Her-2/neu-induced “cytokine signature” in breast cancer. Adv Exp Med Biol. 2008;617:311–9. doi: 10.1007/978-0-387-69080-3_29. [DOI] [PubMed] [Google Scholar]
- 53.Dehqanzada ZA, Storrer CE, Hueman MT, Foley RJ, Harris KA, Jama YH, et al. Assessing serum cytokine profiles in breast cancer patients receiving a HER2/neu vaccine using Luminex technology. Oncol Rep. 2007;17:687–94. [PubMed] [Google Scholar]
- 54.Allen CT, Clavijo PE, Van Waes C, Chen Z. Anti-Tumor Immunity in Head and Neck Cancer: Understanding the Evidence, How Tumors Escape and Immunotherapeutic Approaches. Cancers (Basel) 2015;7:2397–414. doi: 10.3390/cancers7040900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li J, Srivastava RM, Ettyreddy A, Ferris RL. Cetuximab ameliorates suppressive phenotypes of myeloid antigen presenting cells in head and neck cancer patients. J Immunother Cancer. 2015;3:54. doi: 10.1186/s40425-015-0097-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jie HB, Schuler PJ, Lee SC, Srivastava RM, Argiris A, Ferrone S, et al. CTLA-4(+) Regulatory T Cells Increased in Cetuximab-Treated Head and Neck Cancer Patients Suppress NK Cell Cytotoxicity and Correlate with Poor Prognosis. Cancer Res. 2015;75:2200–10. doi: 10.1158/0008-5472.CAN-14-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kohrt HE, Colevas AD, Houot R, Weiskopf K, Goldstein MJ, Lund P, et al. Targeting CD137 enhances the efficacy of cetuximab. J Clin Invest. 2014;124:2668–82. doi: 10.1172/JCI73014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 58.Srivastava RM, Trivedi S, Concha-Benavente F, Gibson SP, Reeder C, Ferrone S, et al. CD137 Stimulation Enhances Cetuximab-Induced Natural Killer: Dendritic Cell Priming of Antitumor T-Cell Immunity in Patients with Head and Neck Cancer. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.White RA, Malkoski SP, Wang XJ. TGFbeta signaling in head and neck squamous cell carcinoma. Oncogene. 2010;29:5437–46. doi: 10.1038/onc.2010.306. [DOI] [PMC free article] [PubMed] [Google Scholar]