

Abstract
ApoE3 is the major chylomicron apolipoprotein, binding in a specific liver peripheral cell receptor, allowing transport and normal catabolism of triglyceride-rich lipoprotein constituents. Point mutations in ApoE3 have been associated with Alzheimer’s disease, type III hyperlipoproteinemia, atherosclerosis, telomere shortening and impaired cognitive function. Here, we evaluate the impact of missense SNPs in APOE retrieved from dbSNP through 16 computational prediction tools, and further evaluate the structural impact of convergent deleterious changes using 100 ns molecular dynamics simulations. We have found structural changes in four analyzed variants (Pro102Arg, Arg132Ser, Arg176Cys and Trp294Cys), two of them (Pro102Arg and Arg176Cys) being previously associated with human diseases. In all cases, except for Trp294Cys, there was a loss in the number of hydrogen bonds between CT and NT domains that could result in their detachment. In conclusion, data presented here could increase the knowledge of ApoE3 activity and be a starting point for the study of the impact of variations on APOE gene.
Introduction
Apolipoproteins (Apo) compose a family of proteins involved in lipid metabolism, participating in many transport pathways, with major physiological importance. In humans, a large number of apolipoproteins that perform different functions have been described, including ApoA1, 2, ApoB3, 4, ApoC5, ApoD6, and ApoE7, 8. ApoA and ApoD have been described as components of the High Density Lipoprotein (HDL) transport, ApoA being the major component in plasma2, 5, whereas ApoB plays a critical role in the low-density lipoprotein (LDL) transport system3, 4. Meanwhile, ApoC has been described as a component of very low-density lipoprotein (VLDL)5; and ApoE is the major apoliprotein of chylomicrons.
ApoE is capable of binding to a specific liver peripheral cell receptor, allowing transport and normal catabolism of triglyceride-rich lipoprotein constituents7, 8. It is known that ApoE forms oligomers9 and, when bound to heparan sulfate proteoglycans (HSPG) and lipids, it adopts an active conformation that allows binding and transport of the low-density lipoprotein receptor (LDLR)10–12. Currently, three common isoforms of ApoE are known. These isoforms may be generated by polymorphisms in two different positions within coding regions of the APOE gene that lead to amino acid residue changes in positions 130 (site A) and 176 (site B) of the mature ApoE protein: ApoE2 (C130/C176), ApoE3 (C130/R176) and ApoE4 (R130/R176)9, 11, 13. As a result, these differences alter the ApoE function14, 15. ApoE isoforms have been associated with several human disorders, such as Alzheimer’s disease16, 17, type III hyperlipoproteinemia12, 18, atherosclerosis19, telomere shortening20, impaired cognitive function21 and infectious diseases22, 23. Some of these disorders could be associated with specific isoforms, such as type III hyperlipoproteinemia and Alzheimer’s disease, which are associated with ApoE2 and ApoE42, 16, respectively.
In humans, the most common isoform is ApoE3, characterized as the wild type12, and this is the unique isoform with a fully elucidated structure, while the other isoforms have only partial structures (e.g. receptor binding domain). Since ApoE3 forms oligomers, some variations (F257A/W264R/V269A/L279Q/V287E) were needed to be inserted in the C-terminus to allow structure elucidation, making a monomeric ApoE39. The ApoE3 structure can be divided into three structural domains: (i) the NT domain comprises the region between residues 1 and 167, (ii) the hinge domain from residues 168 to 205, and (iii) the CT domain from residues 206 to 2999. It is known that the CT domain undergoes structural changes when ApoE binds to lipids, leading to activation of the molecule9, 12, 15. Nevertheless, residues 140-160 from the NT domain are important for the interaction with LDL24, 25. Furthermore, since the protein is mostly stabilized by hydrogen bond interactions and salt bridges, loss of interactions of this type can cause folding errors or loss of affinity for ligands and they could be involved in disease development9, 26, 27. These interactions are very important for the correct folding of CT and Hinge domains9. In addition, the interaction between NT and CT exposes hydrophobic residues in CT, increasing lipid affinity9.
Several studies have shown the effect of point mutations on the functionality of ApoE3. When examining patients with lipoprotein glomerulopathy, Oikawa et al. (1991) found that the Arg163Pro point mutation could cause a lower affinity for the LDL receptor (LDLR)28. Also, Suehiro et al. (1990) demonstrated that the substitution of the same arginine at position 163 of mature protein by a histidine might lead to a lower receptor interaction, increasing the risk for dysbetalipoproteinemia25, 29. However, despite the fact that several point mutations present in the coding region of APOE have been suggested to be associated with human diseases, the potential impact of missense SNPs described in the dbSNP database has not yet been evaluated.
Currently, computational methods designed to predict the impact of amino acid residue changes in proteins have been widely used in order to assess whether changes are deleterious or not30. Among several existing tools, four different groups can be defined based on their methodology: protein-sequence and structure, sequence homology, supervised-learning, and consensus methods31.
Although there are currently a number of tools used to predict the potential structural and functional impact caused by amino acid changes, these tools are not highly accurate32. However, they can still be used as an initial filter of potentially deleterious changes31. Then, more refined analysis, such as molecular dynamics simulations, can be used in order to evaluate more precisely the structural impact caused by amino acid changes31, 33.
The use of molecular dynamics simulations enables the evaluation of structural changes in molecules over a short time window, also allowing observations of changes in physicochemical properties and interactions in simulated environments34. However, the use of this method requires high computational power, making it difficult to simulate longer periods. Hence, simulations are limited to just hundreds of nanoseconds. Nevertheless, this method has been widely used to evaluate changes in protein structure caused by point mutations and missense SNPs, such as in the study of α- and β-defensins35, p5336, lamin A/C protein37, guanylin31, aldosterone synthase38 and aurora-A kinase33.
Here, we evaluate the impact of APOE missense SNPs from dbSNP by means of a number of computational prediction tools, and further evaluate the structural impact of potentially deleterious changes using molecular dynamics simulations. Our hypothesis is that these variations could cause a significant impact on the protein structure and stability.
Material and Methods
Datasets
The dbSNP database contains SNPs and multiple small-scale variations that include insertions/deletions, microsatellites, and non-polymorphic variants39. Using the dbSNP search engine available from the NCBI, only human validated APOE SNPs and non-polymorphic single nucleotide variants (SNVs) were filtered. The ApoE3 protein sequence (NCBI Accession: NP_000032.1) was retrieved from the NCBI Protein database (http://www.ncbi.nlm.nih.gov/protein), and the protein structure file of ApoE3 (PDB ID: 2L7B) was obtained from the RCSB Protein Data Bank9, 40. The frequency data of missense SNPs found in the APOE gene were obtained from the publicly available 1000 Genomes Project (phase I) (http://www.1000genomes.org)41. The variant format file (phase 1 release v3.20101123) corresponding to chromosome 19 contained the frequencies of all SNPs identified in the genomes of 1,092 individuals from 14 populations obtained through a combination of low-coverage (2–6x) whole-genome sequence data, targeted deep (50–100x) exome sequencing and dense SNP genotype data. The 14 populations studied were grouped by the predominant component of ancestry into four super-populations: African (AFR) (246 samples), East Asian (ASN) (286 samples), European (EUR) (379 samples) and Ad Mixed American (AMR) (181 samples).
SNP Selection
As rare SNPs occur at very low frequencies (<1%), there is great concern to avoid confounding putative SNPs with sequencing errors common in next-generation sequencing technologies. Therefore, initially we selected from dbSNP only the ones that fit at least one of the following conditions: (i) it has been sequenced in the 1000 Genomes Project; (ii) it has frequency or genotype data (minor alleles observed in at least two chromosomes); and (iii) it has multiple, independent submissions to the refSNP cluster. Then, in order to evaluate the potential functional impact of the obtained APOE missense SNPs, we utilized a total of 16 prediction tools, divided into four different methods, as shown below. We filtered all missense SNPs that were classified as deleterious by at least three tools in each of the four groups, and denominated these as convergent deleterious predicted SNPs.
Sequence homology-based methods
The following methods based on sequence homology principles were used to produce missense SNP functional predictions: Sorting Intolerant From Tolerant (SIFT)42, Provean43, Mutation Assessor and Panther44, 45.
Supervised learning methods
Supervised learning algorithms used for missense SNP impact prediction included neural networks (SNAP)46, support vector machines (MutPred and SuSPect) and random forests (EFIN)47–49.
Protein sequence and structure-based methods
The following methods either combine information from protein sequence and structure or use protein structural information alone to analyze missense variants: PolyPhen50, Site Directed Mutator (SDM)51, Fold-X52 and PoPMuSiC53.
Consensus-based methods
In order to obtain a consensus score based on many different SNP impact prediction strategies, the following types of consensus software were used: Condel54, Meta-SNP55, PON-P2 and PredictSNP56, 57.
Evolutionary Conservation Analysis
The ConSurf server is a tool for estimating the evolutionary conservation of amino acid positions in a protein molecule based on the phylogenetic relations between homologous sequences58. Using the ApoE3 protein sequence (NCBI Accession: NP_000032.1)40, 59, ConSurf, in ConSeq mode, a search was carried out for close homologous sequences using CSI-BLAST (3 iterations and 0.0001 e-value cutoff) against the UNIREF-90 protein database60, 61. The maximum number of homologs to collect was set as 150, and the minimal and maximal percentage ID between sequences were set as 35 and 95, respectively. The multiple sequence alignment and calculation methods were left as default (MAFFT-L-INS-i and Bayesian). The sequences were then clustered and highly similar sequences removed using CD-HIT62. Position-specific conservation scores were computed using the empirical Bayesian algorithm63.
Signal Peptide Prediction
In order to verify the impact of convergent deleterious SNPs in the signal peptide, Phobius64 and SignalP 4.065 were used for signal peptide topology prediction.
Molecular Modeling
The structural models containing each missense SNP were separately made by means of MODELLER 9.1466 using the class automodel with default settings. The template used as wild type was the monomeric ApoE3 structure (PDB ID: 2L7B)59. One hundred models were generated for each variant. The best models were selected according to DOPE (Discrete Optimized Protein Structure) score, which indicates the most probable structure. The best models were evaluated by PROSA II67 and PROCHECK68 softwares. PROSA II evaluates the model quality while PROCHECK evaluates the stereochemical quality of the model through Ramachandran plot. Good quality models were selected by more than 90% of residues in most favoured and additional allowed regions. The visualization of the structures was done in PyMOL (http://www.pymol.org).
Molecular dynamics simulation
The molecular dynamics simulations of the wild type and the four variant structures were performed by GROMACS 4 computational package using the GROMOS96 43A1 force field69. Structures are immersed in water cubic boxes with a 12 Å distance between the edge of the box and the protein. The simulations were done under ionic strength conditions (0,2 M NaCl)70. The box was filled using the Single Point Charge water model71. The dynamics used the wild type and variants three-dimensional models as initial structures. Additional chlorine ions were also inserted into the complexes with positive charges in order to neutralize the system charge. Geometry of water molecules was constrained by using the SETTLE algorithm72. Atomic connections were made through LINCS algorithm73. Electrostatic corrections were made by Particle Mesh Ewald algorithm74, with a threshold of 1.4 nm to minimize the computational time. The same cut-off radius was applied for van der Waals interactions. The steepest descent algorithm was applied to minimize system energy for 50,000 steps. After the energy minimization, the temperature (NVT ensemble) and pressure (NPT ensemble) systems were normalized to 300 K and 1 bar, respectively, each per 100 steps. The velocity-rescaling thermostat and the Parrinello-Rahman barostat were used for normalization of temperature and pressure, respectively. Full simulation of the system was made by 100 ns using the leap-frog algorithm as the integrator.
Analyses of molecular dynamics trajectories
Molecular dynamics simulations were analyzed by means of the backbone root mean square deviation (RMSD), radius of gyration (Rg) and solvent accessible surface area (SASA) using the g_rms, g_gyrate and g_sas built in functions of the GROMACS package69, respectively. The essential dynamics was performed using the g_covar and g_anaeig utilities of the GROMACS package. The number of hydrogen bonds between the NT domain (residues 1–167) and the CT domain (residues 206–299) was analyzed using g_hbond, also from the GROMACS package. In addition, we analyzed the interactions between known regions of the protein previously described by Chen et al.9. Rg, SASA and the number of hydrogen bonds were plotted as boxplots, because these allow the visualization of the fluctuation and the range in which at least 50% of the data lies.
Results
Distribution and Frequency of APOE SNPs
Out of 183 validated APOE SNPs, 31 are missense, 21 are synonymous, and two are nonsense variants. There are also 98 intronic, 7 5′ UTR, 6 3′ UTR, 7 downstream, 8 upstream, 1 splice donor and 2 splice acceptor variants. A graphical representation of the distribution of SNPs in the coding and non-coding regions of the gene represented in terms of percentage is shown in Fig. 1. Frequency information was obtained from the 1000 Genomes Project for eight APOE missense SNPs (Table S1). All SNPs retrieved from the 1000 Genomes Project are disposed on Table S1. Five of them are rare SNPs with Global Allele Frequency (GAF) values below 1% and occurring only in one of the four populations studied, while the other three variants (Cys130Arg, Arg163Cys and Arg176Cys) have GAFs ≥1%. These variants represent ApoE4 (Cys130Arg), ApoE2* (Arg163Cys) and ApoE2 (Arg176Cys).
Figure 1.
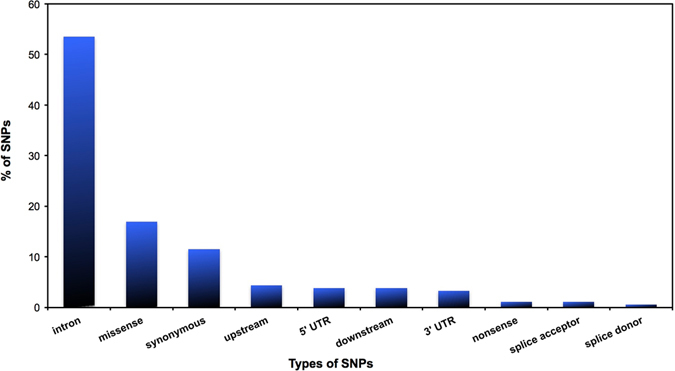
Distribution of SNPs within the APOE gene. The distribution was based on amino acid coding regions (missense, synonymous, nonsense, splice acceptor and splice donor) and on non-coding regions (intronic, upstream and downstream). It can be seen that the majority of the SNPs occur in non-coding regions: 53.6% in introns, 4.4% in upstream regions, 3.8% in 5′ UTR, 3.8% downstream and 3.3% in 3′ UTR. In the coding regions, the majority of the SNPs are missense (16.9%), followed by synonymous (11.5%), nonsense (1.1%), splice acceptor (1.1%) and splice donor (0.6%) variants.
ApoE3 Convergent Deleterious Predicted SNPs
There are currently a wide variety of computational tools used for predicting the effects of missense SNPs on protein function. In general, depending on the strategy, these tools can be classified into four groups: sequence homology, supervised-learning, protein-sequence and structure, and consensus-based methods. We filtered all missense SNPs that were classified as deleterious by at least three tools in each of the four groups (Table S2). A total of four SNPs (Pro102 Arg, Arg132Ser, Arg176Cys and Trp294Cys), which we previously named as convergent deleterious predicted SNPs31 were obtained from this filtration (Table 1). Only three SNPs (Thr11Ala, Ala14Thr and Ala18Thr) occur within the signal peptide region, and all remaining SNPs occur within the mature peptide region (Fig. 2A).
Table 1.
Results of APOE convergent deleterious predicted SNPs analyzed by 16 prediction tools classified in four different groups.
| SNP rs # | Amino Acid Changea | ValidationMethodb | Sequence-Basedc | SLM-Basedc | Consensus-Basedc | Structure-Basedc | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SIFT | Provean | Mutation Assessor | Panther | MutPred | EFIN | SNAP | SuSPect | Condel | MetaSNP | PON-P2 | Predict SNP | PolyPhen | SDM | Fold-X | PoPMuSiC | |||
| rs11083750:C > A | Pro102Arg | Cluster | D | D | D | U | N | D | D | D | D | D | P | D | D | N | DT | DT |
| rs11542041:C > A | Arg132Ser | 1000 G | D | D | D | D | D | D | D | D | N | D | P | D | D | D | DT | DT |
| rs7412:C > T | Arg176Cys | 1000 G, cluster, freq. | D | D | D | D | D | D | D | D | D | D | N | D | D | N | DT | DT |
| rs557715042:G > T | Trp294Cys | 1000 G, freq. | D | D | D | U | D | D | D | N | D | N | P | D | D | D | DT | DT |
aAPOE amino acid positions is relative to GenBank Accession number NP_000032.1.
b1000G: SNP has been sequenced in the 1000 Genomes Project; freq.: Validated by frequency or genotype data: minor alleles observed in at least two chromosomes; cluster: Validated by multiple, independent submissions to the refSNP cluster.
cN: Neutral; D: Deleterious; ST: Stabilizing; DT: Destabilizing; P: Pathogenic; U: Unknown.
Figure 2.
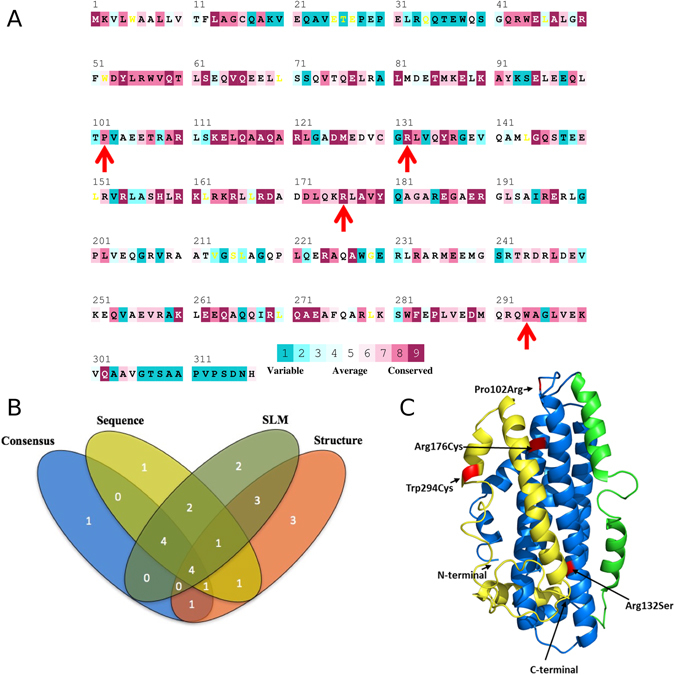
Missense SNPs identified in the APOE gene and Structural domains of native ApoE3. Conservation pattern of amino acid residues within the mature peptide region of ApoE3 obtained from multiple sequence alignment using ConSurf. Color intensity increases with degree of conservation. The amino acids are coloured based on their conservation grades and conservation levels. A grade of 1 indicates rapidly evolving (variable) sites, which are colour-coded in turquoise; 5 indicates sites that are evolving at an average rate, which are coloured white; and 9 indicates slowly evolving (evolutionarily conserved) sites, which are colour-coded in maroon. The four convergent deleterious predicted SNPs are marked below the peptide sequence as red arrows (A). Venn diagram showing the relationships between missense SNPs predicted as deleterious by the four different groups (sequence homology, supervised-learning (SLM), protein-sequence and structure, and consensus-based methods). A total of four convergent deleterious predicted SNPs (classified as deleterious by at least three tools in each of the four different groups) were obtained (B). Structural domains of native ApoE3. In blue is represented the NT domain, CT domain is represented in yellow and hinge region is showed in green. In red are highlighted the different variations analyzed in this work, identified by arrows (C).
In addition, we analyzed the evolutionary conservation of all missense SNPs within the mature region of ApoE3 using ConSurf58, 75. ConSurf exploits evolutionary variation in multiple sequence alignments in order to determine the degrees of conservation. The results from this analysis showed that the majority of the variations (66.7%) occur in sites classified as “conserved” (Fig. 2A), including all four convergent deleterious predicted SNPs (Pro102Arg, Arg132Ser, Arg176Cys and Trp294Cys).
The Thr11Ala, Ala14Thr and Ala18Thr Variants Seem not to Alter the Signal Peptide
In order to evaluate the impact of Thr11Ala, Ala14Thr and Ala18Thr in the signal peptide, two prediction servers were used (Phobius and SignalP 4.0). However, none of them indicated any changes in the signal peptide topology.
The impact of variations on protein structure
ApoE3 native structure is characterized by ten α-helices stabilized by hydrogen bonds, salt bridges and hydrophobic interactions (Fig. 2C). The monomeric ApoE3 (PDB ID: 2L7B) was used to construct the variant structures. Since the ApoE3 monomeric structure has some modifications in the C-terminal, we modeled the native structure by the substitution of respective residues in the C-terminus. Table S3 summarizes the validation assessments. We performed molecular dynamics simulations to evaluate which probable structural changes occur within each modelled ApoE3 structure. The best model for each variant was simulated for 100 ns. The analysis of RMSD was carried out to measure differences in movement between native and variant backbones. The RMSD analysis showed that the native structure had little variation during the simulation time, ranging from 3 to 4 Å (Fig. 3A). Despite that, all analyzed variants presented a higher variation in the backbone of the protein ranging from 3 to 6 Å in Pro102Arg and Arg132Ser and from 3 to 5 Å in Arg176Cys and Trp294 simulations (Fig. 3A).
Figure 3.
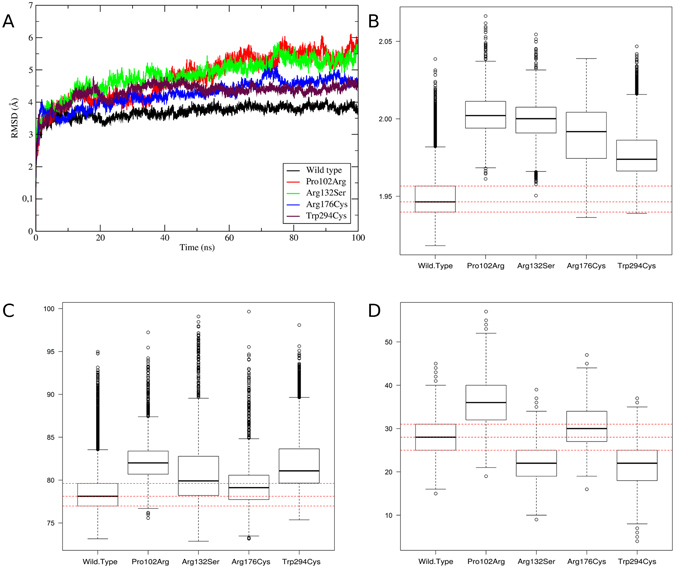
ApoE3 native and variants trajectories analyses. In Backbone RMSD variation the variants are identified in the plots by different colors (A). Radius of gyration (B), solvent accessible surface area (C) and number of hydrogen bonds (D) are plotted in boxplots. On backbone RMSD (A) the variants are identified in the plots by colors: Native structure (black), Pro102Arg (red), Arg132Ser (green), Arg176Cys (blue) and Trp296Cys (turquoise). Only the number of hydrogen bonds between NT and CT domains was computed (D). Dotted red lines on solvent accessible surface area, radius of gyration and number of hydrogen bonds plots indicate the reference values of wild type. The solvent accessible surface and radius of gyration values are in nm² and to RMSD values in Å.
In contrast, analysis of the radius of gyration showed wide differences between variant and wild structures, with an increase for all variants (Fig. 3B). The protein flexibility was also analyzed, by means of essential dynamics, showing that all the variants had a gain in flexibility (Figure S1). Therefore, solvent accessible surface area and radius of gyration were measured in order to evaluate the maintenance of protein packing. The solvent accessible surface area analysis of the variant structures showed little difference between the wild type structure and Arg132Ser and Arg176Cys variants, with little or no increase (Fig. 3C). However, Pro102Arg and Trp294Cys showed a higher increase on this property (Fig. 3C).
Structural changes in CT may reduce the ApoE3 affinity to lipids59. Since NT stabilizes CT, we verified whether some of the convergent deleterious SNPs could affect the number of hydrogen bonds made between CT and NT amino acid residues. There were differences between wild type and variant structures in all cases. While the Arg132Ser and Trp294Cys variants showed a decrease in the number of hydrogen bonds in comparison to the wild type structure (Fig. 3D), the Pro102Arg variant exhibited an increase (Fig. 3D). Moreover, Arg176Cys showed a little increase in the number of hydrogen bonds in comparison to the wild type structure, however, almost the same behavior as the wild type (Fig. 3D). Furthermore, we analyzed differences in the number of interactions between known structural regions in native and variant structures over time. From this, we measured the variant effects on known interactions of native structure. In analyzes with NT and CT domains, almost all variants presented differences when compared to the native structure, with a decrease in Arg132Ser and Trp294Cys variants, an increase in Pro102Arg and the same number of interactions in Arg176Cys (Fig. 3D). However, only Trp294Cys presented a loss of hydrogen bonds between known regions in ApoE3 (Figure S2 and Table S4). Meanwhile, the other three variants presented a great increase in these interactions. However, Pro102Ser presented the greatest impact on the number of hydrogen bonds between the structural domains, with an average gain of about 17 hydrogen bonds in relation to the native structure.
Discussion
ApoE3 presents an helical structure stabilized by hydrogen bonds and salt bridges59. This characteristic confers protein plasticity and capacity of large conformational changes, important for the activity performed by this protein. Here, we used molecular dynamics simulations to assess conformational changes caused by the presence of missense SNPs that lead to amino acid residue changes in the coded protein. We were able to simulate a protein of 299 amino acid residues for 100 ns. For short peptides, it is not difficult to reach this simulation time31, but for proteins greater than 200 amino acids it is common to simulate for less than 10 ns36–38, with few exceptions being simulated for more than 100 ns33.
All of the four variants analyzed here are present in conserved regions of the protein (Fig. 2B). Therefore, the implementation of 16 prediction tools to pre-filter potentially damaging SNPs present in the APOE gene could in fact lead to the discovery of variations that have an impact on protein structure and, consequently, on its function. Furthermore, the use of a consensus of different types of tools (e.g sequence homology-based, supervised learning method and protein sequence and structure-based) to screen potentially damaging SNPs increases their prediction accuracy. Out of the four variants, Pro102Arg presented an increase in all analyses compared to the native protein (Fig. 3D). Interestingly, despite a gain in the number of hydrogen bonds between both CT and NT, as well as between known structural domains, this variant presents the largest differences relative to the wild type structure (Fig. 3).
However, this variant has not been associated with any diseases reported in the literature yet. It is known that ApoE4 is associated with hyperlipidemia2, nevertheless, the double mutant (Cys112Arg/Pro102Arg) has not been described as having this association76, 77. Despite the compensatory effect of Pro102Arg on ApoE4, in ApoE3 it could be deleterious due to the gain in radius of gyration, surface and hydrogen bonds.
On the other hand, the Trp294Cys and Arg132Ser variants presented loss in hydrogen bonds between CT and NT domains (Fig. 3D). This occurs due to the loss of a hydrophobic amino acid in the CT domain. Besides that, the substitution of Trp294 could interfere with lipid interaction mediated by the CT domain, causing loss of affinity59. This step of interaction with lipid was previously associated with activation of the protein, starting the essential structural changes that expose the LDLR binding region in the NT domain59. Moreover, previously, single point changes in CT were used to inhibit the oligomerization of ApoE359. Therefore, it is possible that missense SNPs present in this region could also interpose the normal behavior of the protein. On the other hand, Arg132Ser is important in interdomain interaction, performing two hydrogen bonds with CT domain residues (Gln235 and Glu238)59. Then, loss of hydrogen bonds caused by Arg132Ser could generate the separation of the CT and NT domains, exposing the LDLR interaction domain without the activation by lipids59.
Finally, the Arg176Cys variant presented a more similar behavior compared to the native protein (Fig. 3). Despite this, given the large variation in the radius of gyration analysis of the Arg176Ser variant and the increase of the RMSD, it is possible that this variant generates an opening and closing movement of the Arg176Ser variant, which causes the highest variation on radius of gyration. The Arg176Cys variant characterizes the E2 isoform, which is associated with diseases such as hyperlipoproteinemia III78, 79 and atherosclerosis78. Our analysis showed that this variant could result in a change in affinity between ApoE and LDLR, generating the clinical condition13, 24, 59, 80. The Arg176Cys variant did not show great differences in number of hydrogen bonds (Fig. 3D) or solvent accessible surface analyses (Fig. 3C). Furthermore, this variation has a GAF ≥1%, being the most common variation in this study.
Conclusions
Although many variations have been identified in the APOE gene, the potential structural and functional impact of many of them have not been analyzed yet. However, the four analyzed variants could lead the protein to lose affinity with lipids. The loss of hydrogen bonds between NT and CT domains viewed in variants may be an important factor for research into association between diseases and ApoE variations. Furthermore, the similarity in ApoE2 and other variations could be significant to analyses of impact of these variations and their association with diseases. In conclusion, data presented here could increase the knowledge of ApoE3 activity and be a starting point for the study of impact of variations on the APOE gene.
Electronic supplementary material
Acknowledgements
This work was supported by CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico); CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior); FAPDF (Fundação de Amparo à Pesquisa do Distrito Federal); FUNDECT (Fundação de Apoio ao Desenvolvimento do Ensino, Ciência e Tecnologia do Estado de Mato Grosso do Sul) and UCB (Universidade Católica de Brasília).
Author Contributions
Conceived and designed the experiments: W.P. and S.A. Analyzed the data: A.P., W.P. and S.A. Wrote the main manuscript text: A.P., W.P., O.L.F. and S.A. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01737-w
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Narayanaswami V, Ryan RO. Molecular basis of exchangeable apolipoprotein function. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids. 2000;1483:15–36. doi: 10.1016/S1388-1981(99)00176-6. [DOI] [PubMed] [Google Scholar]
- 2.Breslow JL, et al. Isolation and characterization of cDNA clones for human apolipoprotein A-I. Proc. Natl. Acad. Sci. USA. 1982;79:6861–6865. doi: 10.1073/pnas.79.22.6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lusis AJ, et al. Cloning and expression of apolipoprotein B, the major protein of low and very low density lipoproteins. Proc. Natl. Acad. Sci. USA. 1985;82:4597–4601. doi: 10.1073/pnas.82.14.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Law SW, et al. Human apolipoprotein B-100: cloning, analysis of liver mRNA, and assignment of the gene to chromosome 2. Proc. Natl. Acad. Sci. USA. 1985;82:8340–8344. doi: 10.1073/pnas.82.24.8340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaith P, Assmann G, Uhlenbruck G. Characterization of the oligosaccharide side chain of apolipoprotein C-III from human plasma very low density lipoproteins. Biochim. Biophys. Acta. 1978;541:234–240. doi: 10.1016/0304-4165(78)90396-3. [DOI] [PubMed] [Google Scholar]
- 6.Rassart E, et al. Apolipoprotein D. Biochim. Biophys. Acta - Protein Struct. Mol. Enzymol. 2000;1482:185–198. doi: 10.1016/S0167-4838(00)00162-X. [DOI] [PubMed] [Google Scholar]
- 7.Utermann G, Weber W, Beisiegel U. Different mobility in SDS-polyacrylamide gel electrophoresis of Apolipoprotein E from phenotypes Apo E-N and Apo E-D. FEBS Lett. 1979;101:21–26. doi: 10.1016/0014-5793(79)81286-7. [DOI] [PubMed] [Google Scholar]
- 8.Utermann G, Pruin N, Steinmetz A. Polymorphism of apolipoprotein E. III. Effect of a single polymorphic gene locus on plasma lipid levels in man. Clin. Genet. 1979;15:63–72. doi: 10.1111/j.1399-0004.1979.tb02028.x. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Li Q, Wang J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc. Natl. Acad. Sci. USA. 2011;108:14813–14818. doi: 10.1073/pnas.1106420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: insights into function. Trends Biochem. Sci. 2006;31:445–454. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Weisgraber KH, Apolipoprotein E. structure-function relationships. Adv. Protein Chem. 1994;41:853–72. doi: 10.1016/s0065-3233(08)60642-7. [DOI] [PubMed] [Google Scholar]
- 12.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid Res. 2009;50(Suppl):S183–S188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weisgraber KH, Rall SC, Mahley RW. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J. Biol. Chem. 1981;256:9077–9083. [PubMed] [Google Scholar]
- 14.Mahley RW, Huang Y, Rall SC., Jr. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). Questions, quandaries, and paradoxes. J Lipid Res. 1999;40:1933–1949. [PubMed] [Google Scholar]
- 15.Zuo L, et al. Variation at APOE and STH loci and Alzheimer’s disease. Behav. Brain Funct. 2006;2:13. doi: 10.1186/1744-9081-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 17.Wolk Da, Dickerson BC. Apolipoprotein E (APOE) genotype has dissociable effects on memory and attentional-executive network function in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA. 2010;107:10256–10261. doi: 10.1073/pnas.1001412107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marais AD, Solomon GAE, Blom DJ. Dysbetalipoproteinaemia: A mixed hyperlipidaemia of remnant lipoproteins due to mutations in apolipoprotein E. Crit. Rev. Clin. Lab. Sci. 2014;51:46–62. doi: 10.3109/10408363.2013.870526. [DOI] [PubMed] [Google Scholar]
- 19.McNeill E, Channon KM, Greaves DR. Inflammatory cell recruitment in cardiovascular disease: murine models and potential clinical applications. Clin. Sci. (Lond). 2010;118:641–55. doi: 10.1042/CS20090488. [DOI] [PubMed] [Google Scholar]
- 20.Jacobs, E. G. et al. Accelerated Cell Aging in Female APOE-?? 4 Carriers: Implications for Hormone Therapy Use. PLoS One8, (2013). [DOI] [PMC free article] [PubMed]
- 21.Deary IJ, et al. Cognitive change and the APOE epsilon 4 allele. Nature. 2002;418:932–932. doi: 10.1038/418932a. [DOI] [PubMed] [Google Scholar]
- 22.Burt TD, et al. Apolipoprotein (apo) E4 enhances HIV-1 cell entry in vitro, and the APOE epsilon4/epsilon4 genotype accelerates HIV disease progression. Proc. Natl. Acad. Sci. USA. 2008;105:8718–8723. doi: 10.1073/pnas.0803526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Bont N, et al. Apolipoprotein E knock-out mice are highly susceptible to endotoxemia and Klebsiella pneumoniae infection. J. Lipid Res. 1999;40:680–685. [PubMed] [Google Scholar]
- 24.Innerarity TL, Friedlander EJ, Rall SC, Weisgraber KH, Mahley RW. The receptor-binding domain of human apolipoprotein E. Binding of apolipoprotein E fragments. J. Biol. Chem. 1983;258:12341–12347. [PubMed] [Google Scholar]
- 25.Suehiro, T., Yoshida, K. & Yamano, T. of a NewVariant of Apolipoprotein E (apo E-Kochi). 29, 587–594 (1990). [DOI] [PubMed]
- 26.Weisgraber KH, Mahley RW. Human apolipoprotein E: the Alzheimer’s disease connection. FASEB J. 1996;10:1485–94. doi: 10.1096/fasebj.10.13.8940294. [DOI] [PubMed] [Google Scholar]
- 27.Mahley RW, Huang Y. Apolipoprotein (apo) E4 and Alzheimer’s disease: Unique conformational and biophysical properties of apoE4 can modulate neuropathology. Acta Neurol. Scand. 2006;114:8–14. doi: 10.1111/j.1600-0404.2006.00679.x. [DOI] [PubMed] [Google Scholar]
- 28.Oikawa S, et al. Abnormal lipoprotein and apolipoprotein pattern in lipoprotein glomerulopathy. Am. J. Kidney Dis. 1991;18:553–558. doi: 10.1016/S0272-6386(12)80649-4. [DOI] [PubMed] [Google Scholar]
- 29.Ishigaki Y, et al. Virus-mediated transduction of apolipoprotein E (ApoE)-Sendai develops lipoprotein glomerulopathy in ApoE-deficient mice. J. Biol. Chem. 2000;275:31269–31273. doi: 10.1074/jbc.M005906200. [DOI] [PubMed] [Google Scholar]
- 30.Zhang, Z., Miteva, M. A., Wang, L. & Alexov, E. Analyzing effects of naturally occurring missense mutations. Comput. Math. Methods Med. 2012, (2012). [DOI] [PMC free article] [PubMed]
- 31.Porto, W. F., Franco, O. L. & Alencar, S. a. Computational analyses and prediction of guanylin deleterious SNPs. Peptides 1–11, 10.1016/j.peptides.2015.04.013 (2015). [DOI] [PubMed]
- 32.Rodrigues C, Santos-Silva A, Costa E, Bronze-da-Rocha E. Performance of In Silico Tools for the Evaluation of UGT1A1 Missense Variants. Hum. Mutat. 2015;36:1215–1225. doi: 10.1002/humu.22903. [DOI] [PubMed] [Google Scholar]
- 33.Kumar, A. & Purohit, R. Use of Long Term Molecular Dynamics Simulation in Predicting Cancer Associated SNPs. PLoS Comput. Biol. 10, (2014). [DOI] [PMC free article] [PubMed]
- 34.Hospital A, Goñi JR, Orozco M, Gelpi J. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinforma. Chem. 2015;8:37–47. doi: 10.2147/AABC.S70333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porto, W. F., Nolasco, D. O., Pires, Á. S., Pereira, R. W. & Octávio, L. Prediction of the Impact of Coding Missense and Nonsense Single Nucleotide Polymorphisms on HD5 and HBD1 Antibacterial Activity against Escherichia coli. Biopolym. Pept. Sci. 1–36 (2016). [DOI] [PubMed]
- 36.Chitrala, K. N. & Yeguvapalli, S. Computational screening and molecular dynamic simulation of breast cancer associated deleterious non-synonymous single nucleotide polymorphisms in TP53 gene. PLoS One9, (2014). [DOI] [PMC free article] [PubMed]
- 37.Rajendran V, Purohit R, Sethumadhavan R. In silico investigation of molecular mechanism of laminopathy caused by a point mutation (R482W) in lamin A/C protein. Amino Acids. 2012;43:603–615. doi: 10.1007/s00726-011-1108-7. [DOI] [PubMed] [Google Scholar]
- 38.Jia, M. et al. Computational analysis of functional single nucleotide polymorphisms associated with the CYP11B2 gene. PLoS One9, (2014). [DOI] [PMC free article] [PubMed]
- 39.Sherry ST, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernstein FC, et al. The protein data bank: A computer-based archival file for macromolecular structures. Arch. Biochem. Biophys. 1978;185:584–591. doi: 10.1016/0003-9861(78)90204-7. [DOI] [PubMed] [Google Scholar]
- 41.1000 Genomes Project Consortium, T. 1000 G. P. et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 43.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118–e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mi, H. et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 33 (2005). [DOI] [PMC free article] [PubMed]
- 46.Bromberg Y, Yachdav G, Rost B. SNAP predicts effect of mutations on protein function. Bioinformatics. 2008;24:2397–2398. doi: 10.1093/bioinformatics/btn435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeng S, Yang J, Chung BH-Y, Lau YL, Yang W. EFIN: predicting the functional impact of nonsynonymous single nucleotide polymorphisms in human genome. BMC Genomics. 2014;15:455. doi: 10.1186/1471-2164-15-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flicek, P. et al. Ensembl 2014. Nucleic Acids Res. 42, (2014). [DOI] [PMC free article] [PubMed]
- 49.Li B, et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25:2744–2750. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Worth CL, Preissner R, Blundell TL. SDM - A server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011;39:W215–W222. doi: 10.1093/nar/gkr363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 53.Dehouck Y, Kwasigroch JM, Gilis D, Rooman M. PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics. 2011;12:151. doi: 10.1186/1471-2105-12-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 2011;88:440–449. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Capriotti E, Altman RB, Bromberg Y. Collective judgment predicts disease-associated single nucleotide variants. BMC Genomics. 2013;14(Suppl 3):S2. doi: 10.1186/1471-2164-14-S3-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bendl J, et al. PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLoS Comput. Biol. 2014;10:e1003440. doi: 10.1371/journal.pcbi.1003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Niroula, A., Urolagin, S. & Vihinen, M. PON-P2: Prediction method for fast and reliable identification of harmful variants. PLoS One10, (2015). [DOI] [PMC free article] [PubMed]
- 58.Celniker G, et al. ConSurf: Using evolutionary data to raise testable hypotheses about protein function. Israel Journal of Chemistry. 2013;53:199–206. doi: 10.1002/ijch.201200096. [DOI] [Google Scholar]
- 59.Chen J, et al. Apolipoprotein E and Alzheimer’ s Disease A Role in Amyloid Catabolism. roc. Natl. Acad. Sci. USA. 2010;256:9077–9083. [Google Scholar]
- 60.Angermüller C, Biegert A, Söding J. Discriminative modelling of context-specific amino acid substitution probabilities. Bioinformatics. 2012;28:3240–3247. doi: 10.1093/bioinformatics/bts622. [DOI] [PubMed] [Google Scholar]
- 61.Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007;23:1282–1288. doi: 10.1093/bioinformatics/btm098. [DOI] [PubMed] [Google Scholar]
- 62.Li W, Godzik A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 63.Mayrose I, Graur D, Ben-Tal N, Pupko T. Comparison of site-specific rate-inference methods for protein sequences: Empirical Bayesian methods are superior. Mol. Biol. Evol. 2004;21:1781–1791. doi: 10.1093/molbev/msh194. [DOI] [PubMed] [Google Scholar]
- 64.Käll L, Krogh A, Sonnhammer ELL. Advantages of combined transmembrane topology and signal peptide prediction-the Phobius web server. Nucleic Acids Res. 2007;35:W429–W432. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods. 2011;8:785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 66.Fiser A, Šali A. MODELLER: Generation and Refinement of Homology-Based Protein Structure Models. Methods Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 67.Wiederstein M, Sippl MJ. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 69.Hess B, Kutzner C, Van Der Spoel D, Lindahl E. GRGMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 70.Ibragimova GT, Wade RC. Importance of explicit salt ions for protein stability in molecular dynamics simulation. Biophysical Journal. 1998;74:2906–2911. doi: 10.1016/S0006-3495(98)77997-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. Interaction Models For Water In Relation To Protein Hydration. Intermol. Forces. 1981;31:331–338. doi: 10.1007/978-94-015-7658-1_21. [DOI] [Google Scholar]
- 72.Miyamoto S, Kollman PA. J. Comput. Chem. 1992. SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water models; pp. 952–962. [Google Scholar]
- 73.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 74.Darden T, York D, Pedersen L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 75.Berezin C, et al. ConSeq: The identification of functionally and structurally important residues in protein sequences. Bioinformatics. 2004;20:1322–1324. doi: 10.1093/bioinformatics/bth070. [DOI] [PubMed] [Google Scholar]
- 76.Ordovas JM, Litwack-Klein L, Wilson PW, Schaefer MM, Schaefer EJ. Apolipoprotein E isoform phenotyping methodology and population frequency with identification of apoE1 and apoE5 isoforms. J. Lipid Res. 1987;28:371–380. [PubMed] [Google Scholar]
- 77.Wardell MR, Rall SC, Schaefer EJ, Kane JP, Weisgraber KH. Two apolipoprotein E5 variants illustrate the importance of the position of additional positive charge on receptor-binding activity. Journal of lipid research. 1991;32:521–528. [PubMed] [Google Scholar]
- 78.Sullivan PM, Mezdour H, Quarfordt SH, Maeda N. Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human APOE*2. J. Clin. Invest. 1998;102:130–135. doi: 10.1172/JCI2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rall SC, et al. Type III hyperlipoproteinemia associated with apolipoprotein E phenotype E3/3. Structure and genetics of an apolipoprotein E3 variant. J. Clin. Invest. 1989;83:1095–1101. doi: 10.1172/JCI113988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mahley RW, Rall SC. Apolipoprotein E: far more than a lipid transport protein. Annu. Rev. Genomics Hum. Genet. 2000;1:507–37. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.