

Abstract
The biosynthesis of prostaglandins and thromboxanes has been a focus of interest in the management of many liver diseases. Cyclooxygenases are the enzymes involved in the first step of the biosynthesis of these lipid mediators and selective inhibitors for these isoenzymes as well as pharmacological analogues of prostaglandins have been developed and are currently applied therapeutically. Here we discuss the implications of these enzymes in the onset of metabolic and lipid disorders in the liver and their potential role in the progression of the diseases towards fibrosis and hepatocellular carcinogenesis.
Keywords: Cyclooxygenase-2, NAS, Prostaglandin, Non-alcoholic steatohepatitis, Hepatocellular carcinoma
Core tip: The assessment of the role of Cyclooxygenase-2 (COX-2) in hepatic diseases, ranging from non-alcoholic steatohepatitis to hepatocellular carcinoma, constitutes a field in which controversy exists probably because of the use of different experimental models. Since potent and selective inhibitors of COX-2 exist, but also stable PGE2 analogues to be used in therapy, unraveling the precise contribution of this enzyme and its products to the prevention of the progress of liver dysfunctions appears to be a useful approach for managing liver diseases.
INTRODUCTION
Prostaglandins and thromboxanes are bioactive lipids that regulate many physiological responses but in some cases are recognized as players in inflammatory and tumor diseases, including colorectal and hepatic cancer[1,2]. Cyclooxygenase (COX or prostaglandin G/H synthase, EC 1.14.99.1) catalyzes the rate-limiting step in the synthesis of prostaglandins (PGs) and thromboxanes using arachidonic acid (AA) as substrate to generate PGH2, which is the precursor for a number of cell specific prostaglandin and thromboxane synthases that generate the biologically active products PGE2, PGF2α, PGD2, PGI2 and thromboxanes among other bioactive lipids[1]. Various phospholipases cleave membrane bound AA that once released, it serves as substrate for three main routes: the cyclooxygenase (producing PGs and thromboxanes), the lipoxygenase (producing lipoxins) and the cytochrome P-450 monooxygenase pathways. The COX pathway has been extensively studied in view of the important effects exerted by PGs in many physiological and pathological processes. Moreover, this pathway is clinically relevant because this enzyme is the main target of nonsteroidal anti-inflammatory drugs, and selective COX-2 inhibitors are efficient in decreasing inflammation, and mitigating pain and fever[3,4]. Prostanoids exit the cells via a carrier-mediated process to activate specific prostanoids-dependent G protein-coupled receptors (GPCRs). There are at least eleven known PG receptors, all of them belonging to the GPCRs superfamily of seven transmembrane spanning proteins. For PGE2, EP1 receptors are coupled to Gq and activate phospholipase C and increase cytoplasmic Ca2+. EP2 and EP4 receptors are coupled to Gs proteins, which activate adenylate cyclase leading to a rise in cAMP and subsequent protein kinase A activation. EP3 receptors are coupled to Gi proteins and activate phosphodiesterases that decrease cAMP, contributing to the resetting of the signaling[5]. In addition to this, PGs may control gene transcription through the activation of nuclear receptors of the peroxisome proliferators-activating receptor family[6].
Two COX isoenzymes have been identified: COX-1, first purified from bovine vesicular glands, is ubiquitous and constitutively expressed in a wide variety of tissues where it accounts for the low and continuous PG synthesis required for tissue homeostasis; COX-2 is undetectable in tissues under normal conditions. Only few tissues have a constitutive COX-2 expression (i.e., placenta, testes, kidney and some neural tissues). However, a variety of extracellular and intracellular stimuli (inflammation, growth factors, hormones, reactive oxygen intermediates and oncogenes) can rapidly induce COX-2 expression in many cell types. Both COX isoenzymes share 61% primary sequence identity and exhibit similar kinetic properties with minimal biochemical differences. Despite the structural and kinetic similarities between COX-1 and COX-2, these close related enzymes carry out very different functions in tissues and organs due to their specific promoters, genes and mRNAs[7,8]. The second key enzyme that couples with COXs for the production of PGE2 is PGE2 synthase (PGES). Several isoforms of PGES have been characterized with specific enzymatic properties, cellular distribution and biochemical roles. Two main classifications for PGES have been defined: cytosolic (cPGES), and microsomal/membrane associated (mPGES). cPGES is identical to the Hsp90-associated protein 23. It is constitutively expressed in most cell types and is predominantly coupled with COX-1. However, the membrane associated mPGES is an inducible enzyme that is upregulated during inflammatory conditions. It has recently been shown that mPGES-1 is closely associated to the pathological expression of COX-2. Indeed, mPGES-1 deficiency reduces PGE2 synthesis and, in turn, attenuates tumor proliferation and invasion of several tumor cells[9].
The levels of PGE2 are the result of the fine tuning between the synthesis by mPGES and cPGES, and the degradation that is mainly due to the 15-hydroxyprostaglandin dehydrogenase (15-PGDH). 15-PGDH catalyzes the NAD-dependent oxidation of PGE2 (at C15) to generate the inactive PG 15-keto-PGE2. Animals lacking 15-PGDH exhibit an increase in tissue levels of PGE2. Moreover, despite a physiological action of 15-PGDH in parturition and in the uterine tract, recent data propose a prominent role for 15-PGDH in oncologic processes[10].
COX-2 IN LIVER PATHOLOGY
Hepatocytes express receptors for most of the stimuli that induce COX-2 transcription in other cell types, including LPS, IL-1 and TNF-α. However, adult, but not fetal or neonatal hepatocytes do not express COX-2 in response to these pro-inflammatory molecules; only Kupffer, stellate and some hepatoma liver cells (not all) exhibit the capacity to express COX-2. In the case of hepatocytes, only under long-term aggression, COX-2 is expressed due to the decrease in C/EBPα levels[11]. However, fetal hepatocytes, which contain negligible levels of C/EBPα compared to the adult counterparts, express COX-2 after pro-inflammatory challenge[12]. Interestingly, specific constitutive expression of COX-2 in liver protects against acute liver insults by combining an inhibition of apoptotic mechanisms in the hepatocytes, and promoting cell cycle progression and proliferation. To prove this, we analyzed several models of liver injury and compared the contribution of a COX-2 transgene (COX-2-Tg) under the control of a specific hepatocyte promoter. In this regard, in the lipopolysaccharide and D-galactosamine treated mice (LPS/D-GalN), in the concanavalin A (ConA)-induced hepatitis, and in the model of hepatocyte proliferation after partial hepatectomy (PH) the hepatic elevation of PGs due to the COX-2-Tg attenuates the injury induced by these stressors and accelerates proliferation after PH. Conversely, inhibition of COX-2 with a selective COXIB ablates these protective effects. Interestingly, constitutive COX-2 expression in the liver results in an elevation of antiapoptotic genes as well as in the activation of proteins involved in cell survival, such as phospho-Akt and phospho-AMP-kinase, after injury. Moreover, in the model of liver regeneration after PH, hepatocyte commitment to start replication is accelerated in COX-2-Tg mice due to the rapid elevation of PCNA, cyclin-D1 and E, all promoting cell cycle progression[13]. However, using a different COX-2-Tg animal model Han et al[14] found that COX-2 expression notably enhanced the injury after LPS challenge. These contradictory results are of interest in order to stress the relevance of the genetic background in animal studies. In fact, the molecular mechanisms described in our case for the protection, but also the opposite observation by the Han’s group can be explained in view of this circumstance (C57BL/6 vs C57BL/6XDBA)[15]. Indeed, the wild-type animals of Han’s model did not showed a significant injury after LPS/D-GalN challenge as occurred in our C57BL/6XDBA animals that displayed an acute apoptotic response, in the line reported by other groups using this injury model[15]. Figure 1 summarizes these data.
Figure 1.
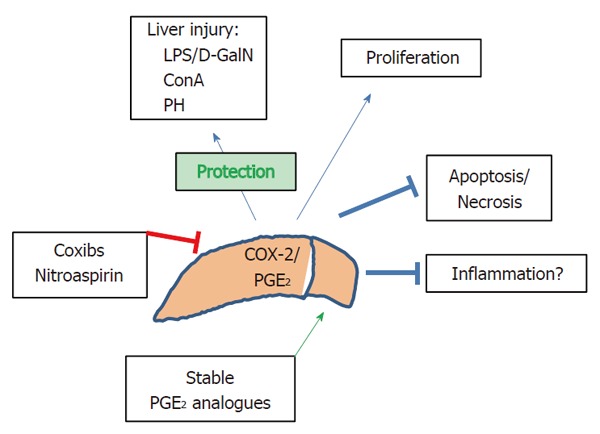
Main regulation and effects of cyclooxygenase-2 in liver. COX-2 and PGE2 exert protection against many liver injuries and promote proliferation of hepatocytes and inhibition of apoptosis and necrosis of hepatic cells. The contribution to inflammation remains controversial depending on the moment of COX-2 expression.
COX-2 AND NAFLD: NAS, NASH AND FIBROSIS
Non-alcoholic fatty liver disease (NAFLD) is defined as a broad clinical pathological entity that appears in the absence of alcohol abuse, but involving fat deposition in the hepatocyte (steatosis, NAS), and worsening to non-alcoholic steatohepatitis (NASH) and fibrosis, all conditions contributing to liver failure and in some cases, to hepatic carcinogenesis. NAFLD is recognized as the hepatic manifestation of metabolic syndrome and constitutes an important health problem that affects one-third of adults and an increasing number of children in developing countries[16]. The pathological definition of metabolic syndrome includes obesity, diabetes, dyslipidemia and hypertension among other symptoms. Around 90% of NAFLD patients have at least one symptom of metabolic syndrome and about 33% have this full canonical profile. Although NAFLD is strongly associated with hyperlipidemia, diabetes mellitus, metabolic syndrome, obesity and insulin resistance (IR), its pathogenesis remains poorly understood and therapeutic options other than lifestyle modification by diet and exercise are limited.
Steatosis is defined as the presence of cytoplasmic TG droplets in more than 5% of the hepatocytes and is the result of an imbalance between the import and/or synthesis of fatty acids by hepatocytes and the rate of usage or export, leading to the formation of the characteristic lipid droplets. Hepatic steatosis (NAS) is the first manifestation of NAFLD, and is identified by the accumulation of triglycerides (TG) as lipid droplets in the cytoplasm of hepatocytes. NAS is often limited and reversible, but it can progress to chronic hepatic inflammation, insulin resistance, liver damage and NASH. A major issue is whether the progression to NAFLD is the cause or the consequence of IR[17]. In this regard, few studies determining hepatic IR at the gene expression level have been performed in NAFLD patients. However, the data available suggest that the insulin signaling pathway, using phospho-Akt and the transcription factor phospho-FoxO1 as read-outs, shows an increase in NASH patients compared to healthy liver[18]. However, recent results from our group demonstrate that hepatic insulin signaling is decreased in NASH patients, and this process is associated with higher apoptotic rates and enhanced collagen deposition. Moreover, IR was not detected in NAS patients. Together, these data suggest that hepatic insulin signaling is preserved in NAS, at the time that point to fact that hepatic lipid overload precedes the impairment of hepatic insulin signaling[17].
The main difference between NAS and NASH is the occurrence of hepatocyte injury, including hepatocyte ballooning and increased cell death, infiltration of circulating inflammatory cells and enhanced parenchymal collagen deposition as morphologic signature of fibrosis. Intralobular inflammation in NASH includes the presence of a small number of lymphocytes, macrophages and neutrophils[18]. Inflammation in NASH involves the contribution of both parenchymal and non-parenchymal cells through the release of bioactive soluble mediators that finally favor the recruitment of lymphoid and myeloid cells in the liver. Activated Kupffer and stellate cells contribute to cytokine expression during steatohepatitis. Among the pro-inflammatory cytokines involved in the progression to NASH, TNF-α and IL-6 seem to be very relevant[19]. Liver cells are also a target for adipose tissue generated factors. More specialized adipokines, such as adiponectin and leptin are also involved. Adiponectin was shown to block TNF-α activation of inflammatory genes, to decrease macrophage recruitment and function and to increase the synthesis of the anti-inflammatory cytokines IL-10 and IL-1Ra, displaying a beneficial effect on NAFLD in mice. Leptin appears to be a key factor in the formation of hepatic droplets due to a polarization in body fat distribution. Beside inflammation, stress signals, including oxidative stress and lipid peroxidation, lead to hepatocyte injury. Apoptotic cell death may also constitute an important component of disease progression. Oxidative stress appears to be a key factor in the pathogenesis of NASH as deduced by a significant increase of oxidative damage markers for lipids, proteins and DNA (MDA, 4-HNE, nitro-tyrosine, and 8-OH-dG), as well as a decrease in the antioxidant capacity, including a reduction in catalase and glutathione reductase and a rise the GSSG/GSH ratio[20]. Despite the high antioxidant capacity of normal liver, hepatocytes sense oxidative stress as reflected by higher rates of cell death both by necrosis and apoptosis. Our results indicated that NASH, but not NAS hepatic samples, have lesser mRNA levels of Mcl-1 and Bcl-2. As a consequence of this, the active content of caspase 3 and apoptosis were higher in NASH than in normal liver and in NAS patients[17]. NASH, in turn, can progress to fibrosis and cirrhosis. Hepatic stellate cells (HSCs) are the main contributors to extracellular matrix deposition and fibrosis as result of its activation in conditions of liver injury. Again, our data demonstrated an increase in the hepatic mRNA levels of COL1A in NASH and no changes in steatosis vs normal liver. In cirrhosis, HSCs are responsible of the type I collagen scar that replaces dying hepatocytes. Cirrhosis can ultimately progress to liver cancer; 4%-27% of individuals with NASH-induced cirrhosis develop hepatocellular carcinoma (HCC).
Regarding COX-2 on these lipid and metabolic disorders, whereas some studies indicate that PGs may favor fat accumulation in hepatocytes and hence the progression from NAS to NASH, others provide evidence where PGE2 suppresses de novo lipogenesis. Therefore, the impact of PGE2 on the insulin-dependent changes in hepatic metabolism is controversial: Hsieh et al[21] reported that rats fed a fructose or high fat diet (HFD) and treated with COX-2 inhibitors improved muscle and fat IR[22]; however, in a different context Coll et al[23] reported that COX-2 inhibition exacerbates palmitate-induced inflammation and IR in skeletal muscle. There are also reports using murine models of NASH induced by high fat or methionine and choline-deficient (MCD) diet describing an increase in COX-2 expression and the beneficial effects after celecoxib or nitro-aspirin treatment[24]. The interaction between PGE2, IL-6 and the IR in hepatocytes has been studied by Henkel et al[25,26] reporting that PGE2 enhanced fat accumulation and interrupted the intracellular signaling of insulin in hepatocytes through serine phosphorylation of IRS via EP3-receptor-dependent ERK1/2 activation. However, using COX-2-Tg mice we observed a protective role for COX-2 in liver injury induced by hyperglycemia in a streptozotocin diabetic mouse involving an increase in PI3K/Akt/AMPK survival pathway[27]. Recently, COX-2 was linked to cold-induced thermogenesis and this COX-2 activity in adipose tissue well correlates with the expression of specific differentiation markers of brown adipose tissue. Furthermore, COX-2 expression in white adipose tissue increased systemic energy expenditure and protected mice against HFD induced obesity[28]. Also, mice carrying a COX-2-Tg in hepatocytes are protected vs IR, adipose tissue proliferation/differentiation and low-grade inflammation when fed HFD. In these COX-2-Tg mice, PGs are associated with increased systemic energy expenditure, higher thermogenesis, and enhanced fatty acid oxidation in the liver, favoring lipid clearance in the hepatocyte[29]. In addition to this, COX-2 represses specific miRNAs. One of them is miR-183, present in liver cells and that is repressed after COX-2 expression and/or in COX-2-Tg hepatocytes. Our group has demonstrated that this decrease in miR-183 is important in the preservation, and even potentiation of the insulin signaling pathway after different hepatic challenges[30]. Accordingly, COX-2 appears as an unexpected potential therapeutic strategy against obesity-associated metabolic dysfunction. In this regard, stable analogs of PGE2, such as 16,16dmPGE2, could be considered as therapeutic alternatives to prevent steatosis progression and/or IR. In fact, PGE2 and analogs are clinically used to ameliorate mild hypercholesterolemia among other liver pathologies[31,32].
The metabolic signals governing the transition from NAS to NASH are not well defined; however, the increase of lipid content in the hepatocyte contributes to an enhancement of oxidative stress, cell death and to create a low-grade pro-inflammatory ambience that sustains the initial lipid stress[33]. Again, the impact of PGE2 on this sequence from NAS to the establishment of NASH and the subsequent hepatic fibrosis remains controversial: in vivo studies in mice using selective COX-2 inhibitors showed a prevention in the progression to NASH, clearly pointing to COX-2-dependent PGs as mediators of the progress[34,35]. However, the opposite approach, that is administration of PGE2, has been show to prevent HSC-dependent fibrogenesis and steatohepatitis[22,36,37]. Furthermore, Cheng et al[38] by using a liver COX-2-Tg mice (again in a genetic background distinct from C57BL/6), failed to show any contribution of these locally produced PGs to NASH progression and steatohepatitis. In this regard, it is relevant to mention that PGE2 impairs the expression of pro-fibrogenic genes in human fat explants from obese individuals as well as antagonizes the TGF-β-dependent fibrogenic activity in adipocytes[39]. In line with this, COX-2-Tg mice fed MCD diet are protected against NASH when compared with the Wt counterparts, essentially through a mechanism that involves a lesser recruitment of circulating immune cells and, as a consequence, a minor presence of pro-inflammatory factors in the liver, resulting in a minor activation of the pro-fibrogenic cells present in the tissue, such as HSCs. Furthermore, these COX-2-Tg mice treated with a classic pro-fibrogenic insult as is CCl4 exhibited a significant protection against fibrosis progression as reflected by lower synthesis of hepatic collagen and accumulation of hydroxyproline when compared with the Wt controls[40]. Figure 2 summarizes the main findings observed in the animal models carrying a COX-2 transgene.
Figure 2.
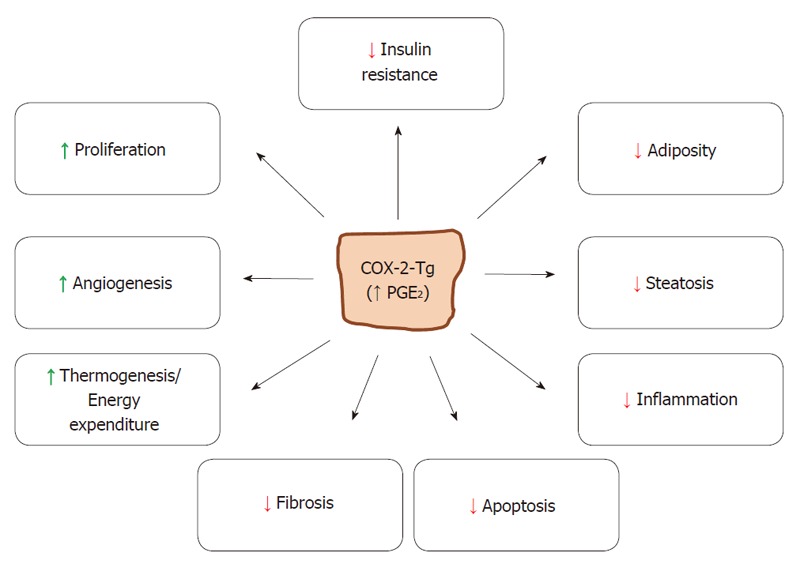
Mice carrying a COX-2 transgene in hepatocytes have elevated PGE2 and are protected against hepatic insults. PGE2 produced by hepatic COX-2 has specific effects in liver against inflammation, steatosis, fibrosis and apoptosis. At the systemic level protection against insulin resistance and adiposity is observed, whereas angiogenesis and thermogenesis are enhanced.
COX-2 AND HCC
Hepatocellular carcinoma (HCC) is one of the most common solid cancers (30% of morbidity) and is very prevalent in cirrhotic patients. The incidence of HCC is increasing worldwide associated to hepatitis C virus (HCV) infection[41,42]. Moreover, hepatitis B virus (HBV) infection, and ambient risk factors (i.e., aflatoxin, alcoholic cirrhosis, etc.) contribute to HCC initiation and progression. Clinical diagnosis of HCC is difficult since no reliable serum markers have been clearly identified, and the therapeutic options for HCC are limited (i.e., sorafenib, cis-Pt, etc.). Despite the recent introduction of potent preventive chemotherapeutic protocols the study of the molecular mechanisms leading to human hepatocarcinogenesis remains an area of intense research in order to understand the progression from NASH and cirrhosis to HCC. Gene expression profiling and proteomic approaches have contributed to identify specific signatures that can be used for the identification of proteins that are differentially expressed between normal and liver tumors in an attempt to define new and more selective biomarkers for HCC. Integrative transcriptome analysis reveals three molecular HCC subclasses, each correlated with different clinical parameters and serum markers[42]. Like in other cancers, the key event driving liver carcinogenesis is the development of simultaneous deregulated proliferation and reduced cell death. Work by different groups have characterized molecular signatures implicated in tumorigenesis: (1) receptor tyrosine kinase pathways; (2) Wnt/β-catenin signaling pathway; (3) ubiquitin/proteasome degradation pathway; (4) epigenetic DNA methylation and histone deacetylation pathways; (5) the PI3K/Akt/mTOR pathway; (6) angiogenic pathways; and (7) telomerase activity[43].
Regarding the involvement of COX-2-derived PGs in hepatocarcinogenesis it should be mentioned that selective COX-2 inhibitors have proved to inhibit HCC cell growth in vitro and in xenograft animal models. As previously mentioned, COX-2 expression in liver appears to be restricted to very special conditions. In fact, COX-2 is transiently expressed in regenerating liver after PH or after challenge with potent hepatotoxic molecules, such as thioacetamide among other[44,45]. In addition to this, COX-2 expression has been observed in animal models of cirrhosis, in human and several (but not all) mouse hepatoma cell lines, after HBV or HCV infection and in human HCC[46-50]. However, although COX-2 expression is detected in early phases of HCC, contradictory observations have been published regarding the nature of the cells expressing the enzyme (both in normal hepatocytes and in hepatoma cells), the role of this expression pattern and the molecular mechanisms by which COX-2-dependent PGs contribute and/or induce tumorigenesis. Interestingly, work from our group[51] has shown that COX-2 expression is not sufficient to exacerbate malignant transformation after administration of chemical hepatocarcinogens. Even more, progression of liver oncogenesis in a well-established model of HCC (the c-myc and TGF-α double transgenic mice), is not affected by COX-2 expression[52]. However, COX-2 expression in this model facilitated the development of preneoplastic foci but failed to promote malignant transformation, probably as result of the contribution of COX-2-derived PGs to inhibit apoptosis and to provide an anti-inflammatory environment, these conditions opposing the initiation of the early phases of HCC. Using COX-2-Tg hepatocyte cell lines we also showed elevated oxidative stress and ROS accumulation after chemical hepatocarcinogenesis, together with an important decrease in the levels of GSH and higher levels of 8-OHdG. Moreover, a moderate activation of JNK, Erk and p38 was detected in COX-2-Tg cells, and COX-2 favored the growth of cell implants in nude mice, probably through the sustained activation of Akt and JNK-c-Jun survival pathways. Recently, it has been shown that PGE2 is able to increase c-Myc levels through the activation of the EP4R/GS/AC/cAMP/PKA/CREB cascade that favors HCC growth and invasion of these cells, contributing PGE2 in this way to hepatocellular carcinogenesis[53].
Genetic deletion of 15-PGDH, the enzyme which degrades PGE2 to an inactive 15-keto- PGE2, leads to increased tissue levels of PGE2. We have shown that 15-PGDH is repressed in human HCC cell lines, together with elevation of COX-2, in chemical and genetic murine models of HCC and in human HCC biopsies. Moreover, transfection of HCC cells with 15-PGDH induces apoptosis and attenuates the growth of these cells when implanted in nude mice; at the same time, transfection with siRNA specific for 15-PGDH promoted tumor growth indicating that the balance between COX-2 and 15-PGDH activities are relevant in the hepatocarcinogenic process[54]. Interestingly, 15-PGDH not only plays a protective role by decreasing PGE2 levels, but also the 15-keto-PGE2 generated has been shown to activate PPARγ. Indeed, activated PPARγ by 15-keto-PGE2 favors the interaction with the p21(WAF1/Cip1) promoter, which in turn results in p21 expression and association with cyclin-dependent kinase 2 (CDK2), CDK4 and PCNA[55]. Altogether, the results of higher pulmonary metastatic incidence of HCC in COX-2-Tg mice and the promotion and migration of HCC cells induced by PGE2[48] suggest that COX-2 might be involved in the expansion and metastatic phase of HCC. COX-2 exerts pro-metastatic effects on cancer stem cells mediated partly through regulation of PDCD4 and PTEN expression[56]. Moreover, PGE2 could upregulate the expression level of Snail, an inducer of epithelial-mesenchymal transition a key player in HCC invasion and metastasis, through the EP2/Src/EGFR/Akt/mTOR pathway[57].
Besides the murine models, we have investigated whether a correlation exists between COX-2 expression and different grades of methylation at the 5’region of the COX-2 gene in hepatoma cell lines and in HCC. We also analyzed the acetylation signatures of the COX-2 promoter and the effects of inhibitors of histone deacetylase (HDAC) on COX-2 expression. Our results indicate that the low COX-2 expression in some hepatoma cell lines and HCC is associated with promoter hypermethylation of COX-2. Histone deacetylation and treatment with demethylating agents or HDAC inhibitors restored the expression of COX-2[41]. Interestingly enough, COX-2 mRNA levels were higher in the non-tumoral liver tissue than in HCC; moreover, inverse correlations between COX-2 levels and the differentiation grade of HCC were observed. Clinical studies showed a reduction of survival in patients when COX-2 expression decreased due to promoter hypermethylation and histone H3 hypoacetylation. Indeed, Giannitrapani et al[47] reported a dispersed range of COX-2 expression in human HCC, from absence of expression in undifferentiated areas to a robust expression in well differentiated tissue. Moreover, COX-2 expression was significantly lower in HCC than in NASH. However, a recent meta-analysis study revealed that the presence of COX-2 in HCC is associated with stages of decreased overall and disease-free survival and represents a worse prognosis[57]. Recent studies in different cancer cells point out to a COX-dependent tumor growth through evasion of immunity and tumor-promoting inflammation. Pre-clinical data indicate that inhibition of COX-2 in breast or colorectal cancer cells favors the activation of immune mechanisms against cancer cells, reinforcing the immunosuppressive role of PGs[58]. Moreover, since HSCs are important mediators of immunosuppression and in the progress of HCC, it is suggested that they contribute to HCC through the recruitment of immunosuppressive cells, mainly myeloid-derived suppressor cells and regulatory T cells, through a mechanism involving the COX-2-PGE2-EP4 pathway[59]. As a summary of these data, controversy exists in the literature regarding the precise role of COX-2 and PGE2 in the development of HCC.
CONCLUSION
The assessment of the role of COX-2 in hepatic diseases, ranging from NASH to HCC, constitutes a field in which controversy exists probably because of the use of different experimental models and specific temporary guidelines of the time-dependent contribution of COX-2-derived metabolites to the onset of these hepatic dysfunctions. Since potent and selective inhibitors of COX-2 exist, but also stable PGE2 analogues to be used in therapy, unraveling the precise contribution of this enzyme and its products is crucial for the prevention of the progression of liver dysfunctions and it appears to be a useful approach for managing the patients. Efforts to identify biomarkers providing indications for the correct use of these therapeutic tools are essential to underline new intervention protocols based on COX-2 and prostaglandin targeting.
ACKNOWLEDGMENTS
The authors thank Mr Adrian Povo-Retana for critical review and editing of the text.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Spain
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: The authors declare no conflict of interests.
Peer-review started: February 1, 2017
First decision: February 23, 2017
Article in press: April 12, 2017
P- Reviewer: Koch TR, Mattner J, Tziomalos K S- Editor: Qi Y L- Editor: A E- Editor: Wang CH
References
- 1.Cha YI, DuBois RN. NSAIDs and cancer prevention: targets downstream of COX-2. Annu Rev Med. 2007;58:239–252. doi: 10.1146/annurev.med.57.121304.131253. [DOI] [PubMed] [Google Scholar]
- 2.Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55:115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 4.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 5.Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- 6.Na HK, Surh YJ. Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands as bifunctional regulators of cell proliferation. Biochem Pharmacol. 2003;66:1381–1391. doi: 10.1016/s0006-2952(03)00488-x. [DOI] [PubMed] [Google Scholar]
- 7.Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donnini S, Finetti F, Terzuoli E, Giachetti A, Iñiguez MA, Hanaka H, Fresno M, Rådmark O, Ziche M. EGFR signaling upregulates expression of microsomal prostaglandin E synthase-1 in cancer cells leading to enhanced tumorigenicity. Oncogene. 2012;31:3457–3466. doi: 10.1038/onc.2011.503. [DOI] [PubMed] [Google Scholar]
- 10.Yang L, Amann JM, Kikuchi T, Porta R, Guix M, Gonzalez A, Park KH, Billheimer D, Arteaga CL, Tai HH, et al. Inhibition of epidermal growth factor receptor signaling elevates 15-hydroxyprostaglandin dehydrogenase in non-small-cell lung cancer. Cancer Res. 2007;67:5587–5593. doi: 10.1158/0008-5472.CAN-06-2287. [DOI] [PubMed] [Google Scholar]
- 11.Callejas NA, Boscá L, Williams CS, DuBOIS RN, Martín-Sanz P. Regulation of cyclooxygenase 2 expression in hepatocytes by CCAAT/enhancer-binding proteins. Gastroenterology. 2000;119:493–501. doi: 10.1053/gast.2000.9374. [DOI] [PubMed] [Google Scholar]
- 12.Martín-Sanz P, Callejas NA, Casado M, Díaz-Guerra MJ, Boscá L. Expression of cyclooxygenase-2 in foetal rat hepatocytes stimulated with lipopolysaccharide and pro-inflammatory cytokines. Br J Pharmacol. 1998;125:1313–1319. doi: 10.1038/sj.bjp.0702196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayoral R, Mollá B, Flores JM, Boscá L, Casado M, Martín-Sanz P. Constitutive expression of cyclo-oxygenase 2 transgene in hepatocytes protects against liver injury. Biochem J. 2008;416:337–346. doi: 10.1042/BJ20081224. [DOI] [PubMed] [Google Scholar]
- 14.Han C, Li G, Lim K, DeFrances MC, Gandhi CR, Wu T. Transgenic expression of cyclooxygenase-2 in hepatocytes accelerates endotoxin-induced acute liver failure. J Immunol. 2008;181:8027–8035. doi: 10.4049/jimmunol.181.11.8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martín-Sanz P, Mayoral R, Casado M, Boscá L. COX-2 in liver, from regeneration to hepatocarcinogenesis: what we have learned from animal models? World J Gastroenterol. 2010;16:1430–1435. doi: 10.3748/wjg.v16.i12.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.García-Monzón C, Lo Iacono O, Mayoral R, González-Rodríguez A, Miquilena-Colina ME, Lozano-Rodríguez T, García-Pozo L, Vargas-Castrillón J, Casado M, Boscá L, et al. Hepatic insulin resistance is associated with increased apoptosis and fibrogenesis in nonalcoholic steatohepatitis and chronic hepatitis C. J Hepatol. 2011;54:142–152. doi: 10.1016/j.jhep.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 18.Valenti L, Rametta R, Dongiovanni P, Maggioni M, Fracanzani AL, Zappa M, Lattuada E, Roviaro G, Fargion S. Increased expression and activity of the transcription factor FOXO1 in nonalcoholic steatohepatitis. Diabetes. 2008;57:1355–1362. doi: 10.2337/db07-0714. [DOI] [PubMed] [Google Scholar]
- 19.Asrih M, Jornayvaz FR. Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance. J Endocrinol. 2013;218:R25–R36. doi: 10.1530/JOE-13-0201. [DOI] [PubMed] [Google Scholar]
- 20.Sumida Y, Niki E, Naito Y, Yoshikawa T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic Res. 2013;47:869–880. doi: 10.3109/10715762.2013.837577. [DOI] [PubMed] [Google Scholar]
- 21.Hsieh PS, Jin JS, Chiang CF, Chan PC, Chen CH, Shih KC. COX-2-mediated inflammation in fat is crucial for obesity-linked insulin resistance and fatty liver. Obesity (Silver Spring) 2009;17:1150–1157. doi: 10.1038/oby.2008.674. [DOI] [PubMed] [Google Scholar]
- 22.Hui AY, Dannenberg AJ, Sung JJ, Subbaramaiah K, Du B, Olinga P, Friedman SL. Prostaglandin E2 inhibits transforming growth factor beta 1-mediated induction of collagen alpha 1(I) in hepatic stellate cells. J Hepatol. 2004;41:251–258. doi: 10.1016/j.jhep.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 23.Coll T, Palomer X, Blanco-Vaca F, Escolà-Gil JC, Sánchez RM, Laguna JC, Vázquez-Carrera M. Cyclooxygenase 2 inhibition exacerbates palmitate-induced inflammation and insulin resistance in skeletal muscle cells. Endocrinology. 2010;151:537–548. doi: 10.1210/en.2009-0874. [DOI] [PubMed] [Google Scholar]
- 24.Chen J, Liu D, Bai Q, Song J, Guan J, Gao J, Liu B, Ma X, Du Y. Celecoxib attenuates liver steatosis and inflammation in non-alcoholic steatohepatitis induced by high-fat diet in rats. Mol Med Rep. 2011;4:811–816. doi: 10.3892/mmr.2011.501. [DOI] [PubMed] [Google Scholar]
- 25.Henkel J, Gärtner D, Dorn C, Hellerbrand C, Schanze N, Elz SR, Püschel GP. Oncostatin M produced in Kupffer cells in response to PGE2: possible contributor to hepatic insulin resistance and steatosis. Lab Invest. 2011;91:1107–1117. doi: 10.1038/labinvest.2011.47. [DOI] [PubMed] [Google Scholar]
- 26.Henkel J, Frede K, Schanze N, Vogel H, Schürmann A, Spruss A, Bergheim I, Püschel GP. Stimulation of fat accumulation in hepatocytes by PGE2-dependent repression of hepatic lipolysis, β-oxidation and VLDL-synthesis. Lab Invest. 2012;92:1597–1606. doi: 10.1038/labinvest.2012.128. [DOI] [PubMed] [Google Scholar]
- 27.Francés DE, Ingaramo PI, Mayoral R, Través P, Casado M, Valverde ÁM, Martín-Sanz P, Carnovale CE. Cyclooxygenase-2 over-expression inhibits liver apoptosis induced by hyperglycemia. J Cell Biochem. 2013;114:669–680. doi: 10.1002/jcb.24409. [DOI] [PubMed] [Google Scholar]
- 28.Vegiopoulos A, Müller-Decker K, Strzoda D, Schmitt I, Chichelnitskiy E, Ostertag A, Berriel Diaz M, Rozman J, Hrabe de Angelis M, Nüsing RM, et al. Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science. 2010;328:1158–1161. doi: 10.1126/science.1186034. [DOI] [PubMed] [Google Scholar]
- 29.Francés DE, Motiño O, Agrá N, González-Rodríguez Á, Fernández-Álvarez A, Cucarella C, Mayoral R, Castro-Sánchez L, García-Casarrubios E, Boscá L, Carnovale CE, Casado M, Valverde ÁM, Martín-Sanz P. Hepatic cyclooxygenase-2 expression protects against diet-induced steatosis, obesity, and insulin resistance. Diabetes. 2015;64:1522–1531. doi: 10.2337/db14-0979. [DOI] [PubMed] [Google Scholar]
- 30.Motiño O, Francés DE, Mayoral R, Castro-Sánchez L, Fernández-Velasco M, Boscá L, García-Monzón C, Brea R, Casado M, Agra N, et al. Regulation of MicroRNA 183 by Cyclooxygenase 2 in Liver Is DEAD-Box Helicase p68 (DDX5) Dependent: Role in Insulin Signaling. Mol Cell Biol. 2015;35:2554–2567. doi: 10.1128/MCB.00198-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madsen L, Pedersen LM, Lillefosse HH, Fjaere E, Bronstad I, Hao Q, Petersen RK, Hallenborg P, Ma T, De Matteis R, et al. UCP1 induction during recruitment of brown adipocytes in white adipose tissue is dependent on cyclooxygenase activity. PLoS One. 2010;5:e11391. doi: 10.1371/journal.pone.0011391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korhonen T, Savolainen MJ, Jääskeläinen T, Kesäniemi YA. Effect of a synthetic prostaglandin E2 analogue, RS-86505-007, on plasma lipids and lipoproteins in patients with moderate hypercholesterolaemia: efficacy and tolerance of treatment and response in different apolipoprotein polymorphism groups. Eur J Clin Pharmacol. 1995;48:97–102. doi: 10.1007/BF00192732. [DOI] [PubMed] [Google Scholar]
- 33.Marra F, Gastaldelli A, Svegliati Baroni G, Tell G, Tiribelli C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol Med. 2008;14:72–81. doi: 10.1016/j.molmed.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 34.Kim SM, Park KC, Kim HG, Han SJ. Effect of selective cyclooxygenase-2 inhibitor meloxicam on liver fibrosis in rats with ligated common bile ducts. Hepatol Res. 2008;38:800–809. doi: 10.1111/j.1872-034X.2008.00339.x. [DOI] [PubMed] [Google Scholar]
- 35.Paik YH, Kim JK, Lee JI, Kang SH, Kim DY, An SH, Lee SJ, Lee DK, Han KH, Chon CY, et al. Celecoxib induces hepatic stellate cell apoptosis through inhibition of Akt activation and suppresses hepatic fibrosis in rats. Gut. 2009;58:1517–1527. doi: 10.1136/gut.2008.157420. [DOI] [PubMed] [Google Scholar]
- 36.Hui AY, Leung WK, Chan HL, Chan FK, Go MY, Chan KK, Tang BD, Chu ES, Sung JJ. Effect of celecoxib on experimental liver fibrosis in rat. Liver Int. 2006;26:125–136. doi: 10.1111/j.1478-3231.2005.01202.x. [DOI] [PubMed] [Google Scholar]
- 37.Kamada Y, Mori K, Matsumoto H, Kiso S, Yoshida Y, Shinzaki S, Hiramatsu N, Ishii M, Moriwaki K, Kawada N, et al. N-Acetylglucosaminyltransferase V regulates TGF-β response in hepatic stellate cells and the progression of steatohepatitis. Glycobiology. 2012;22:778–787. doi: 10.1093/glycob/cws012. [DOI] [PubMed] [Google Scholar]
- 38.Cheng AS, Yu J, Lai PB, Chan HL, Sung JJ. COX-2 mediates hepatitis B virus X protein abrogation of p53-induced apoptosis. Biochem Biophys Res Commun. 2008;374:175–180. doi: 10.1016/j.bbrc.2008.06.098. [DOI] [PubMed] [Google Scholar]
- 39.García-Alonso V, Titos E, Alcaraz-Quiles J, Rius B, Lopategi A, López-Vicario C, Jakobsson PJ, Delgado S, Lozano J, Clària J. Prostaglandin E2 Exerts Multiple Regulatory Actions on Human Obese Adipose Tissue Remodeling, Inflammation, Adaptive Thermogenesis and Lipolysis. PLoS One. 2016;11:e0153751. doi: 10.1371/journal.pone.0153751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Motiño O, Agra N, Brea Contreras R, Domínguez-Moreno M, García-Monzón C, Vargas-Castrillón J, Carnovale CE, Boscá L, Casado M, Mayoral R, et al. Cyclooxygenase-2 expression in hepatocytes attenuates non-alcoholic steatohepatitis and liver fibrosis in mice. Biochim Biophys Acta. 2016;1862:1710–1723. doi: 10.1016/j.bbadis.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 41.Fernández-Alvarez A, Llorente-Izquierdo C, Mayoral R, Agra N, Boscá L, Casado M, Martín-Sanz P. Evaluation of epigenetic modulation of cyclooxygenase-2 as a prognostic marker for hepatocellular carcinoma. Oncogenesis. 2012;1:e23. doi: 10.1038/oncsis.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K, Hashimoto M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385–7392. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Avila MA, Berasain C, Sangro B, Prieto J. New therapies for hepatocellular carcinoma. Oncogene. 2006;25:3866–3884. doi: 10.1038/sj.onc.1209550. [DOI] [PubMed] [Google Scholar]
- 44.Casado M, Callejas NA, Rodrigo J, Zhao X, Dey SK, Boscá L, Martín-Sanz P. Contribution of cyclooxygenase 2 to liver regeneration after partial hepatectomy. FASEB J. 2001;15:2016–2018. doi: 10.1096/fj.01-0158fje. [DOI] [PubMed] [Google Scholar]
- 45.Fernández-Martínez A, Callejas NA, Casado M, Boscá L, Martín-Sanz P. Thioacetamide-induced liver regeneration involves the expression of cyclooxygenase 2 and nitric oxide synthase 2 in hepatocytes. J Hepatol. 2004;40:963–970. doi: 10.1016/j.jhep.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 46.Cusimano A, Foderà D, Lampiasi N, Azzolina A, Notarbartolo M, Giannitrapani L, D’Alessandro N, Montalto G, Cervello M. Prostaglandin E2 receptors and COX enzymes in human hepatocellular carcinoma: role in the regulation of cell growth. Ann N Y Acad Sci. 2009;1155:300–308. doi: 10.1111/j.1749-6632.2009.03701.x. [DOI] [PubMed] [Google Scholar]
- 47.Giannitrapani L, Ingrao S, Soresi M, Florena AM, La Spada E, Sandonato L, D’Alessandro N, Cervello M, Montalto G. Cyclooxygenase-2 expression in chronic liver diseases and hepatocellular carcinoma: an immunohistochemical study. Ann N Y Acad Sci. 2009;1155:293–299. doi: 10.1111/j.1749-6632.2009.03698.x. [DOI] [PubMed] [Google Scholar]
- 48.Mayoral R, Fernández-Martínez A, Boscá L, Martín-Sanz P. Prostaglandin E2 promotes migration and adhesion in hepatocellular carcinoma cells. Carcinogenesis. 2005;26:753–761. doi: 10.1093/carcin/bgi022. [DOI] [PubMed] [Google Scholar]
- 49.Núñez O, Fernández-Martínez A, Majano PL, Apolinario A, Gómez-Gonzalo M, Benedicto I, López-Cabrera M, Boscá L, Clemente G, García-Monzón C, et al. Increased intrahepatic cyclooxygenase 2, matrix metalloproteinase 2, and matrix metalloproteinase 9 expression is associated with progressive liver disease in chronic hepatitis C virus infection: role of viral core and NS5A proteins. Gut. 2004;53:1665–1672. doi: 10.1136/gut.2003.038364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu T. Cyclooxygenase-2 in hepatocellular carcinoma. Cancer Treat Rev. 2006;32:28–44. doi: 10.1016/j.ctrv.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 51.Llorente Izquierdo C, Mayoral R, Flores JM, García-Palencia P, Cucarella C, Boscá L, Casado M, Martín-Sanz P. Transgenic mice expressing cyclooxygenase-2 in hepatocytes reveal a minor contribution of this enzyme to chemical hepatocarcinogenesis. Am J Pathol. 2011;178:1361–1373. doi: 10.1016/j.ajpath.2010.11.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Llorente-Izquierdo C, Mayoral R, Cucarella C, Grau C, Alvarez MS, Flores JM, García-Palencia P, Agra N, Castro-Sánchez L, Boscá L, et al. Progression of liver oncogenesis in the double transgenic mice c-myc/TGF α is not enhanced by cyclooxygenase-2 expression. Prostaglandins Other Lipid Mediat. 2013;106:106–115. doi: 10.1016/j.prostaglandins.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 53.Xia S, Ma J, Bai X, Zhang H, Cheng S, Zhang M, Zhang L, Du M, Wang Y, Li H, et al. Prostaglandin E2 promotes the cell growth and invasive ability of hepatocellular carcinoma cells by upregulating c-Myc expression via EP4 receptor and the PKA signaling pathway. Oncol Rep. 2014;32:1521–1530. doi: 10.3892/or.2014.3393. [DOI] [PubMed] [Google Scholar]
- 54.Castro-Sánchez L, Agra N, Llorente Izquierdo C, Motiño O, Casado M, Boscá L, Martín-Sanz P. Regulation of 15-hydroxyprostaglandin dehydrogenase expression in hepatocellular carcinoma. Int J Biochem Cell Biol. 2013;45:2501–2511. doi: 10.1016/j.biocel.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 55.Lu D, Han C, Wu T. 15-PGDH inhibits hepatocellular carcinoma growth through 15-keto-PGE2/PPARγ-mediated activation of p21WAF1/Cip1. Oncogene. 2014;33:1101–1112. doi: 10.1038/onc.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo Z, Jiang JH, Zhang J, Yang HJ, Yang FQ, Qi YP, Zhong YP, Su J, Yang RR, Li LQ, et al. COX-2 Promotes Migration and Invasion by the Side Population of Cancer Stem Cell-Like Hepatocellular Carcinoma Cells. Medicine (Baltimore) 2015;94:e1806. doi: 10.1097/MD.0000000000001806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen G, Li X, Yang J, Li J, Wang X, He J, Huang Z. Prognostic significance of cyclooxygenase-2 expression in patients with hepatocellular carcinoma: a meta-analysis. Arch Med Sci. 2016;12:1110–1117. doi: 10.5114/aoms.2016.61916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell. 2015;162:1257–1270. doi: 10.1016/j.cell.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu Y, Zhao W, Xu J, Li J, Hong Z, Yin Z, Wang X. Activated hepatic stellate cells promote liver cancer by induction of myeloid-derived suppressor cells through cyclooxygenase-2. Oncotarget. 2016;7:8866–8878. doi: 10.18632/oncotarget.6839. [DOI] [PMC free article] [PubMed] [Google Scholar]