

Abstract
AIM
To investigate the mechanism of hepatoprotection conferred by liver fibrosis through evaluating the activation phenotype of kupffer cells.
METHODS
Control and fibrotic mice were challenged with a lethal dose of D-GalN/lipopolysaccharide (LPS), and hepatic damage was assessed by histology, serum alanine transferase (ALT) levels, and hepatic expression of HMGB1, a potent pro-inflammatory mediator. The localization of F4/80 (a surrogate marker of KCs), HMGB1, and type I collagen (Col-1) was determined by immunofluorescence staining. The phenotype of KCs was characterized by real-time PCR. KCs isolated from control or fibrotic mice were challenged with LPS or HMGB1 peptide, and HMGB1 translocation was analyzed.
RESULTS
Liver fibrosis protected mice against D-GalN/LPS challenge, as shown by improved hepatic histology and reduced elevation of ALT compared with the normal mice treated in the same way. This hepatoprotection was also accompanied by inhibition of HMGB1 expression in the liver. Co-localization of F4/80, HMGB1, and Col-1 was found in fibrotic livers, indicating the close relationship between KCs, HMGB1 and liver fibrosis. KCs isolated from fibrotic mice predominantly exhibited an M2-like phenotype. In vitro experiments showed that HMGB1 was localized in the nucleus of the majority of M2-like KCs and that the translocation of HMGB1 was inhibited following stimulation with LPS or HMGB1 peptide, while both LPS and HMGB1 peptide elicited translocation of intranuclear HMGB1 in KCs isolated from the control mice.
CONCLUSION
M2-like Kupffer cells in fibrotic liver may exert a protective effect against acute insult by inhibiting the translocation of HMGB1.
Keywords: Liver fibrosis, Injury resistance, Kupffer cell activation, high-mobility group box 1, Translocation
Core tip: The hepatoprotective effect conferred by liver fibrosis against acute liver injury is an interesting phenomenon, which has not yet been fully characterized. In the present study, we dissected the underlying mechanism of acute injury in the setting of liver fibrosis through investigating the correlation between KC activation and HMGB1 translocation. Our study showed that liver fibrosis protects mice against D-GalN/LPS challenge, and M2-like KCs in the fibrotic liver may exert a protective effect by inhibiting the translocation of HMGB1, a potent pro-inflammatory mediator.
INTRODUCTION
Liver fibrosis is the most clinically relevant consequence of chronic liver diseases. It is characterized by the activation of hepatic stellate cells and the excessive accumulation of extracellular matrix[1]. Advanced fibrosis results in cirrhosis, portal hypertension and liver failure, and often requires liver transplantation[2-5]. On the other hand, liver fibrosis represents the wound healing response to liver injury due to a variety of etiologies[6,7]. Recently, increasing attention has been focused on the homeostatic and beneficial effects originating from fibrotic liver. Convincing evidence has shown that fibrosis results in increased liver resistance to subsequent acute insults, thus protecting hepatocytes against various toxic stimuli[8-10]. However, the mechanisms governing this hepatoprotection are largely unexplored.
Hepatic macrophages have central roles in maintaining homeostasis in the liver as well as in the pathogenesis of acute or chronic liver injury[11]. Macrophages are pleiotropic cells that assume diverse functions. The microenvironmental milieu is a key determinant of macrophage function during tissue inflammation[12]. Macrophages can undergo “classical” M1 activation when exposed to lipopolysaccharide (LPS), and interferon-gamma (IFN-γ) or “alternative” M2 activation when exposed to interleukin (IL)-4/IL-13[13-15]. M1- and M2-like macrophages exhibit distinct signatures and fulfill different functions. Kupffer cells (KCs), the resident macrophages in the liver, play pivotal roles in the progression and resolution of liver fibrosis[16]. Recently, KCs have been reported to exert a protective effect against tumor necrosis factor (TNF)-α-induced hepatocyte apoptosis[9]. Nevertheless, whether and how KC activation is involved in injury resistance in the setting of liver fibrosis is poorly understood.
HMGB1 is a ubiquitous protein present in the nucleus of most mammalian cells. HMGB1 has diverse functions which depend on its cellular localization. In the intracellular compartment, HMGB1 participates in gene transcription, DNA replication, and DNA repair. To function as a damage-associated molecular pattern (DAMP), HMGB1 can translocate from nucleus to cytoplasm and is subsequently released into the extracellular milieu. This process is implemented by two principal means: active secretion by innate immune cells (e.g., macrophages) after hyperacetylation or passive release from necrotic cells. Extracellular HMGB1 activates pro-inflammatory signaling pathways by ligation of pattern recognition receptors including the receptor for advanced glycation end products or Toll-like receptor 4, leading to severe damage in multiple liver diseases[17-19]. As a biomarker of liver injury, HMGB1 is superior to serum alanine transferase (ALT) levels at identifying acetaminophen-induced acute liver injury and elevated HMGB1 in acute liver failure correlates well with poor outcome[20,21].
In the present study, we investigated the roles of KCs, KC activation, and HMGB1 in the pathogenesis of acute injury in the setting of liver fibrosis, and hypothesized that activated KCs in fibrotic liver protected against acute insult by inhibiting the translocation of HMGB1. We present herein evidence that liver fibrosis protects mice against D-GalN/LPS challenge and that M2-like KCs in fibrotic liver may exert a protective effect by inhibiting the translocation of HMGB1, a potent pro-inflammatory mediator.
MATERIALS AND METHODS
Animals and treatments
Male BALB/c mice (6-8-wk-old) were obtained from the Laboratory Animal Center, Academy of Military Medical Sciences, Beijing, China. The animal protocol was designed to minimize pain or discomfort in the mice. Mice were housed in a specific pathogen-free environment at 22-24 °C in a 12-h light-dark cycle. Animals were fed standard laboratory chow with free access to water. All animal care and experimental procedures performed in this study were in accordance with the guidelines for experimental animals approved by the Animal Care and Use Committee of Capital Medical University, China.
Mice were treated as follows: (1) induction of fibrosis: BALB/c mice received an intraperitoneal injection of carbon tetrachloride (CCl4) in mineral oil, twice weekly, for 6 wk. The initial dose of CCl4 was 0.2 μL/g (2%), and the dose was gradually increased up to 3 μL/g (30%); and (2) acute challenge: Control and fibrotic mice received intraperitoneal injection of a lethal dose of hepatic toxins (1 mg/g DGalN + 50 ng/g LPS; SigmaAldrich, St Louis, MO, United States). Sera and liver tissues were harvested 24 h after acute injury for analysis. A portion of the liver was fixed in 10% neutral-buffered formalin for histological analysis and immunostaining. The remaining liver was cut into pieces and snap-frozen for homogenization to extract total liver RNA.
Evaluation of liver injury
Serum ALT levels were measured using a multiparameteric analyzer (AU 5400; Olympus, Tokyo, Japan) according to an automated procedure. Formalin-fixed liver tissues were embedded in paraffin, sectioned and stained with hematoxylin-eosin for light microscopy.
Isolation of hepatic non-parenchymal cells and KCs
Hepatic non-parenchymal cells (NPCs) were isolated from mice by collagenase digestion and differential centrifugation using a previously reported method with some modifications[22]. Briefly, in situ perfusion was applied through the portal vein and superior vena cava with 0.9% NaCl followed by DMEM/F12 (Gibco, Grand Island, NY, United States) containing 0.5% Pronase (Roche Diagnostics GmbH, Mannheim, Germany) and DMEM/F12 containing 0.04% type IV collagenase (Sigma-Aldrich). The liver was then harvested, excised and digested with DMEM/F12 containing 10 μg/mL DNase (Sigma-Aldrich). Digested livers were passed through a 70 μm cell strainer (BD Falcon, Franklin Lakes, NJ, United States). The filtrate was centrifuged and washed. The pellets were re-suspended in DMEM (Hyclone, Logan, UT, United States), and then overlaid onto a Percoll (Amersham Pharmacia Biotechnology, Buckinghamshire, United Kingdom) gradient (40%-70%), and centrifuged at 1100 × g for 20 min. NPCs were collected from the interface for further purification.
To purify KCs, the liver NPC suspension was further overlaid onto the Percoll gradient (25%-50%), and centrifuged at 1800 × g for 30 min. The KC-enriched NPCs in the interface were harvested and washed. The isolated KCs were then cultured in DMEM medium containing 10% fetal bovine serum (Hyclone) and 1% penicillin-streptomycin (Sigma-Aldrich) in a humidified chamber at 37 °C with 5% CO2. Following incubation for 2 h, the unattached cells were gently removed. The remaining adhered cells were further cultured for 24 h, and the phenotype of KCs was characterized by real-time PCR.
Reverse transcription and SYBR Green real-time quantitative PCR
Total RNA was extracted from isolated KCs using TRIzol reagent (Invitrogen, Carlsbad, CA, United States) following the manufacturer’s instructions. Reverse transcription of the purified RNA (2.5 μg) was performed using random primers and the AMV retrotranscriptase system (TakaRa, Dalian, Liaoning, China) according to the manufacturer’s protocol. SYBR Green real-time PCR was carried out using the ABI StepOne Plus (Applied Biosystems, Foster City, CA, United States). All reactions were performed in triplicate. In a final reaction volume of 20 μL, the followings were added: 1× SYBR Green (TakaRa), cDNA, 0.5 mmol/L of each primer, and ROX. The reaction conditions were as follows: 50 °C (2 min), 95 °C (5 min), followed by 40 cycles at 95 °C (15 s) and 60 °C (30 s). The primers used were designed with Primer 3.0 software and are listed in Table 1. The relative expression of target genes was calculated and normalized to the expression of the housekeeping gene GAPDH.
Table 1.
Primer sequences used for reverse transcription-quantitative polymerase chain reaction analysis
| Genes | Sense (5’-3’) | Anti-sense (5’-3’) |
| GAPDH | AACTTTGGCATTGTGGAAGG | ACACATTGGGGGTAGGAACA |
| IL-1β | GCCCATCCTCTGTGACTCAT | AGGCCACAGGTATTTTGT |
| TNF-α | GCCTCTTCTCATTCCTGCTTGT | TTGAGATCCATGCCGTTG |
| CD206 | ATGCCAAGTGGGAAAATCTG | TGTAGCAGTGGCCTGCATAG |
| iNOS | CGGAGCCTTTAGACCTCAACA | CCCTCGAAGGTGAGCTGAAC |
| YM-1 | ATCTATGCCTTTGCTGGAATGC | TGAATGAATATCTGACGGTTCTGAG |
| CCL17 | TGCTTCTGGGGACTTTTCTG | TGGCCTTCTTCACATGTTTG |
Treatment of isolated KCs with LPS or HMGB1 peptide
KCs isolated from control and fibrotic mice were cultured in DMEM medium for 24 h. The KCs were then treated with LPS (10 ng/mL) or HMGB1 peptide (FKDPNAPKRLPSAFFLFCSE) (30 μg/mL; SBS Genetech Co, Ltd, Beijing, China) for another 20 h. Translocation of HMGB1 in KCs was analyzed by immunofluorescence staining.
Immunofluorescence staining
Frozen liver sections (or KCs) were fixed with 4% paraformaldehyde for 30 min at room temperature. Slices (or KCs) were treated with 0.2% Triton X-100 for 5 min. The slices (or KCs) were then incubated with Tris-buffered saline (TBS) containing 5% fetal bovine serum for 30 min. Immunofluorescence staining was performed using the following primary antibodies: rat anti-mouse F4/80 antigen Alexa Fluor® 488 (clone BM8; eBioscience, San Diego, CA, United States), rabbit anti-mouse HMGB1 (Epitomics, Burlingame, CA, United States), and goat anti-mouse type I collagen (Col-1) (SouthernBiotech, Birmingham, AL, United States). For indirect immunofluorescence staining, FITC-conjugated (Santa Cruz Biotechnology, Inc, Dallas, TX, United States) or Cy3-conjugated anti-rabbit IgG (Sigma-Aldrich) for HMGB1, and Cy3-conjugated rabbit anti-goat IgG for Col-1 (Sigma-Aldrich) were used. A Nikon inverted fluorescence microscope ECLIPSE Ti and NIS-Elements F3.0 software (Nikon Corporation, Tokyo, Japan) were used for image capture.
HMGB1 immunohistochemical staining
After deparaffinization and rehydration, the embedded liver sections were treated with 3% H2O2 for 15 min, followed by microwave antigen retrieval for a further 15 min in citrate buffer. The nonspecific proteins were blocked with 10% goat serum for 30 min. For HMGB1 staining, the specimens were incubated with a rabbit anti-mouse HMGB1 monoclonal antibody (Abcam, Cambridge, MA, United States) overnight at 4 °C, followed by 30 min incubation with horseradish-peroxidase-conjugated goat anti-rabbit secondary antibody (Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China). The sections were incubated with diaminobenzidine as a chromogenic substrate and counterstained with hematoxylin, dehydrated, and stabilized with mounting medium. Images were captured using an Olympus Bx51 microscope (Olympus America, Melville, NY, United States) and CellSens standard 1.4.1 software.
Animal care and use statement
All animal care and experimental procedures performed in this study were in accordance with the guidelines for experimental animals approved by the Animal Care and Use Committee of Capital Medical University, China.
Statistical analysis
Results were expressed as mean ± SE of the values obtained. Group comparisons were performed using one-way analysis of variance (ANOVA) followed by Newman-Keuls multiple comparison test. Statistics and graphs were generated using Prism 5.0 software (GraphPad Software Inc, San Diego, CA, United States). P < 0.05 was considered statistically significant. The statistical methods used in this study were reviewed by Dr. Jun-Feng Li from the First Affiliated Hospital of Lanzhou University, Lanzhou, China.
RESULTS
Inhibition of HMGB1 expression is accompanied by injury resistance in the setting of liver fibrosis
We first assessed hepatic injury in control and fibrotic mice with or without acute insult. As shown in Figure 1, the D-GalN/LPS challenge triggered a sharp increase in serum ALT levels in control mice, which corresponded well with the pathological findings. In contrast, fibrotic mice showed marked resistance to the same insult. In particular, hepatic damage was significantly alleviated in fibrotic mice following the D-GalN/LPS challenge compared with control mice treated in the same way, as shown by improved hepatic histology and reduced serum ALT levels (Figure 1A and B). HMGB1, a potent and classic pro-inflammatory mediator, was induced in acutely injured mice. However, the expression of HMGB1 was markedly inhibited in fibrotic mice, even under acute challenge (Figure 1C). These findings suggest that liver fibrosis protects mice against acute insult, which is accompanied by inhibition of HMGB1 expression.
Figure 1.
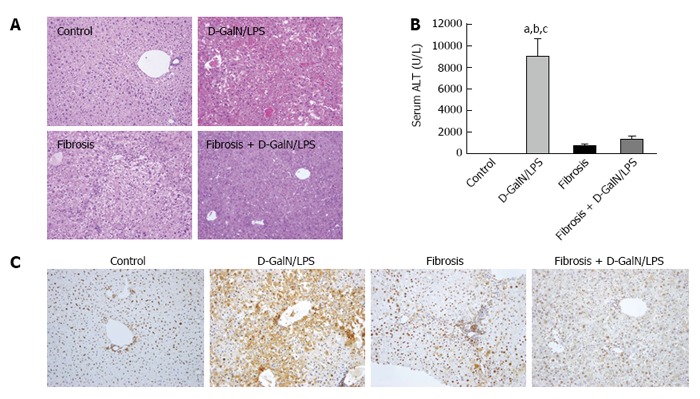
Inhibition of High mobility group box 1 expression is closely associated with the injury resistance in the setting of liver fibrosis. Control and fibrotic mice (treated with CCl4 for 6 wk) were challenged with a lethal dose of D-GalN (1 mg/g)/LPS (50 ng/g), and hepatic damage was assessed by histology (A: HE staining; original magnification, × 200) and serum ALT levels (B). aP < 0.05 vs the control group, bP < 0.05 vs the fibrosis group, cP < 0.05 vs the fibrosis + D-GalN/LPS group. The expression of HMGB1 was determined by immunohistochemical staining (C: original magnification, × 200). Data are expressed as mean ± SEM. CCl4: Carbon tetrachloride; D-GalN: D-galactosamine; HMGB1: High mobility group box 1; LPS: Lipopolysaccharide.
Kupffer cells may be involved in HMGB1-related injury resistance in the setting of liver fibrosis
Kupffer cells are involved in the progression and resolution of liver fibrosis. Recently, KCs have been documented to possess protective effects against hepatocyte apoptosis. In light of this finding, we hypothesized that KCs are involved in HMGB1-related injury resistance in the setting of liver fibrosis. To explore this hypothesis, we analyzed the expression and distribution of KCs and HMGB1 in fibrotic liver by immunofluorescent staining. F4/80, a surrogate marker of KCs, was found to be co-localized with Col-1, supporting the key function of KCs in the pathogenesis of hepatic fibrosis (Figure 2). Interestingly, the co-localization of F4/80, HMGB1 and Col-1 was also found in fibrotic liver (Figure 2), which indicated functional interactions among KCs, the injury mediator HMGB1, and hepatic fibrosis. Thus, KCs may be involved in HMGB1-related injury resistance in the setting of hepatic fibrosis.
Figure 2.
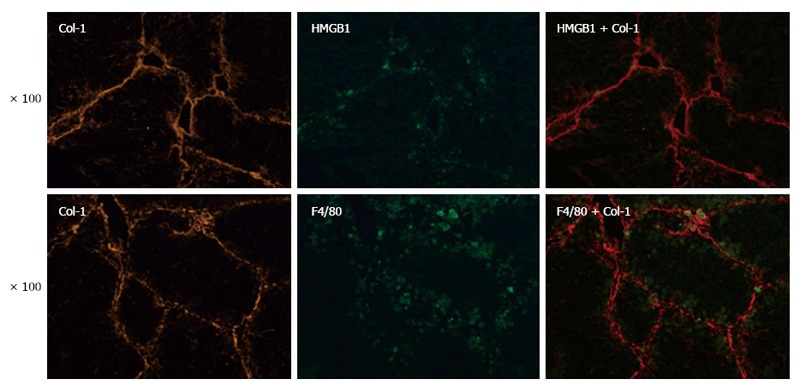
Kupffer cells may be involved in High mobility group box 1-mediated injury resistance. The expression and localization of F4/80 (a surrogate marker of KCs), HMGB1, and Col-1 were determined by immunofluorescence staining (original magnification, × 100). Col-1: Type I collagen; HMGB1: High mobility group box 1; KCs: Kupffer cells.
Kupffer cells in fibrotic liver exhibit a predominant M2-like activation
We next sought to answer the question of how do KCs function in HMGB1-related injury resistance in the setting of liver fibrosis? It has been well established that macrophages are heterogeneous, and their functional plasticity is driven by microenvironmental signals which shape their properties through a wide spectrum of phenotypes[23,24]. Thus, we characterized the phenotype of KCs by their representative markers. KCs were isolated from the livers of normal, acutely injured and fibrotic mice, and M1 and M2 gene signatures were then determined by quantitative real-time PCR. Fibrosis triggered marked up-regulation of M2 markers, including CD206, CCL17 and YM-1. However, macrophages from the acutely injured liver showed no increase or a moderate increase in these M2 signatures (Figure 3). On the other hand, iNOS, a marker of M1 activation, was substantially induced in KCs isolated from the acutely injured liver, but remained unchanged in KCs from the fibrotic liver (Figure 3). Therefore, KCs in fibrotic mice exhibit a predominantly M2-like phenotype.
Figure 3.
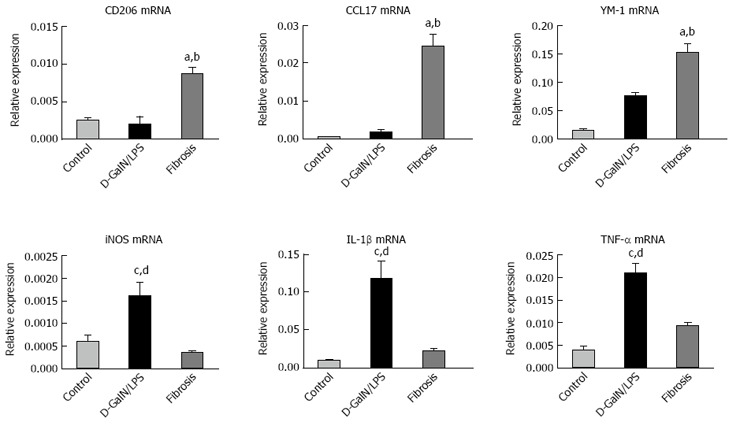
Kupffer cells in the fibrotic liver exhibit predominantly a M2-like activation. KCs were isolated from the livers of normal, acutely injured (D-GalN/LPS) and fibrotic mice, and M1 and M2 gene signatures were then determined by quantitative real-time PCR. Data are expressed as mean ± SEM. aP < 0.05 vs the control group, bP < 0.05 vs the D-GalN/LPS group, cP < 0.05 vs the control group, dP < 0.05 vs the fibrosis group. D-GalN: D-galactosamine; KCs: Kupffer cells; LPS: Lipopolysaccharide.
Translocation of HMGB1 is inhibited in M2-like KCs following LPS or HMGB1 peptide stimuli
How do M2-like KCs and HMGB1 orchestrate injury resistance in the setting of hepatic fibrosis? We speculated that M2-like KCs in fibrotic liver may prevent the translocation of HMGB1 upon acute challenge, thereby hampering its critical function as a DAMP. To confirm our speculation, the translocation of HMGB1 was analyzed in KCs isolated from control and fibrotic mice and treated with LPS or HMGB1 peptide. Both LPS and HMGB1 peptide elicited the translocation of intranuclear HMGB1 in KCs isolated from control mice (Figure 4). Interestingly, HMGB1 was localized in the nucleus of the majority of M2-like KCs isolated from fibrotic liver. Importantly, neither LPS nor HMGB1 peptide elicited the translocation of HMGB1 in M2-like KCs (Figure 4). Collectively, our in vitro data demonstrated that the translocation of HMGB1 was inhibited in M2-like KCs upon acute insult.
Figure 4.
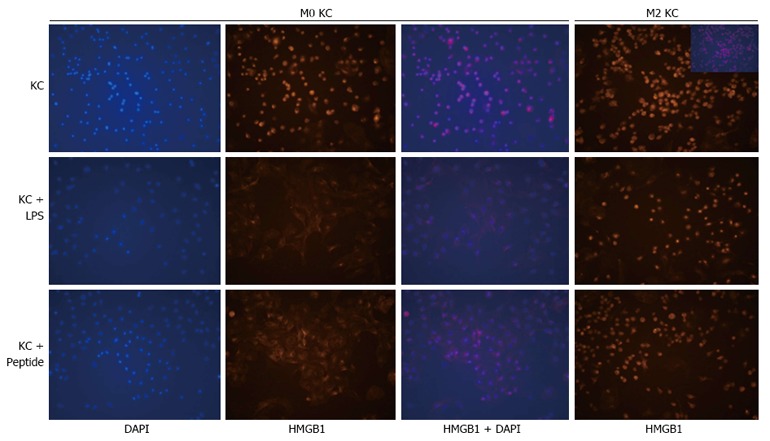
Translocation of High mobility group box 1 triggered by lipopolysaccharide or High mobility group box 1 peptide is inhibited in M2-like Kupffer cells. KCs were isolated from the livers of control and fibrotic mice, and then treated with LPS (10 ng/mL) or HMGB1 peptide (30 μg/mL). The translocation of HMGB1 in KCs was analyzed by immunofluorescence staining (original magnification, × 200). HMGB1: High mobility group box 1; KCs: Kupffer cells; LPS: Lipopolysaccharide.
DISCUSSION
In the present study, we found that liver fibrosis protects mice against D-GalN/LPS challenge, and M2-like KCs in fibrotic liver may exert a protective effect by inhibiting the translocation of HMGB1, a crucial pro-inflammatory mediator. Our findings may provide a potential explanation for the mechanism of injury resistance in the setting of liver fibrosis.
The deleterious effects resulting from liver fibrosis, including cirrhosis, liver failure and hepatocellular carcinoma, are widely accepted. Recent studies have reported the hepatoprotective effects conferred by liver fibrosis. In a mouse model of partial bile duct ligation (PBDL), injured ligated lobes exhibited improved tolerance to TNF-α- and Fas-induced hepatocyte apoptosis, compared with non-ligated lobes[10]. Similarly, thioacetamide-induced fibrotic liver was less vulnerable to acute injury[8]. In the present study, we found that CCl4-induced liver fibrosis conferred significant protection against a lethal challenge with D-GalN/LPS, as shown by improved hepatic histology and reduced serum ALT levels. Importantly, we linked HMGB1 with injury resistance in the setting of liver fibrosis. HMGB1 is an evolutionarily conserved non-histone nuclear protein with abundant expression in most mammalian cells. Recently, HMGB1 was identified as a potent pro-inflammatory mediator. Compared with ALT levels, HMGB1 is more sensitive at identifying acetaminophen-induced acute liver injury[20,21]. In our study, in comparison with acutely injured liver, the expression of HMGB1 was markedly inhibited in fibrotic liver following acute challenge, which corresponded well with a significant improvement in liver histology. These findings provide powerful evidence for injury resistance conferred by liver fibrosis.
Hepatoprotection in the setting of liver fibrosis is an interesting subject and remains to be fully elucidated. Bourbonnais et al[8] attributed a hepatoprotective response to Col-1 produced during liver fibrosis, which significantly protected hepatocytes against toxic stimuli via activation of ERK1 signaling. The study by Osawa et al[9] specifically addressed the role of KCs in cholestatic liver injury using PBDL mice. They found that KCs reduced liver damage, and induced hepatocyte survival and regeneration. These protective and regenerative effects require activation of AKT in hepatocytes and SphK in HSCs[9]. In the present study, KCs were co-localized with HMGB1 in fibrotic liver, indicating the critical role of KCs in HMGB1-related injury resistance in the setting of hepatic fibrosis.
We then explored the hepatoprotective effect of KCs in fibrotic mice from a new perspective, i.e., the activation phenotype of KCs. Our hypothesis was based on the strong association between the function and phenotype of macrophages, as mentioned in the introduction. According to our data, KCs exhibited a predominantly M2-like phenotype in the context of liver fibrosis, which is in line with the latest report on congenital hepatic fibrosis[25]. Finally, we examined the underlying mechanism concerning the potential protective effect of M2-like KCs on HMGB1-related injury resistance in the setting of hepatic fibrosis. As previously noted, nuclear-cytoplasmic translocation is regarded as a key step in HMGB1 functioning as a DAMP, which in turn, triggers inflammation. Accordingly, we speculated that M2-like KCs may inhibit the translocation of HMGB1 induced by acute insult. Hence, we compared HMGB1 translocation in KCs isolated from control and fibrotic mice challenged with LPS or HMGB1 peptide. Both LPS and HMGB1 peptide triggered the translocation of intranuclear HMGB1 in KCs from control mice. However, the translocation of HMGB1 was markedly inhibited in M2-like KCs, even under acute challenge. These results provide strong support for our hypothesis, that is, M2-like KCs in fibrotic liver may protect against acute insult by inhibiting the translocation of HMGB1, thereby suppressing HMGB1-mediated pro-inflammatory responses and attenuating hepatic injury[26].
Post-translational modification seems to be a critical step in regulation of the translocation and secretion of HMGB1 during inflammatory responses. The acetylation of HMGB1 on lysine residues in response to IL-1β, TNF-α, and LPS is regarded as the most feasible and effective way[27]. A recent study has shown that ligand-activated peroxisome proliferator-activated receptor (PPAR)-δ and PPAR-γ modulate LPS-primed release of HMGB1 through SIRT-mediated deacetylation, which in turn, plays a critical role in the cellular response to inflammation[28]. Whether this modulation occurred in our model requires further investigation. Moreover, we cannot exclude the passive release of HMGB1 by injured hepatocytes, as reported by Kao et al[29]. However, active secretion of HMGB1 by macrophages is also important in the context of fibrosis.
In conclusion, although the current findings are preliminary and may be speculative, our findings may shed new light on the pathogenesis of acute hepatic damage in the setting of liver fibrosis. We will elaborate the mechanisms underlying injury resistance by modulating the polarization and function of hepatic macrophages or by blocking HMGB1 signaling. From a clinical perspective, our study findings may pave the way for the treatment of liver failure, particularly acuteonchronic liver failure.
COMMENTS
Background
Convincing evidence has shown that fibrosis confers increased liver resistance to subsequent acute insults, thus protecting hepatocytes against various toxic stimuli. The hepatoprotection conferred by liver fibrosis is intriguing and the mechanism of such protection remains to be fully elucidated.
Research frontiers
Kupffer cells (KCs) have been reported to exert a protective effect against hepatocyte apoptosis in cholestatic liver disease, indicating the potential role of KCs in hepatoprotection. Nevertheless, whether and how the phenotype of KCs is involved in injury resistance in the setting of liver fibrosis is poorly understood.
Innovations and breakthroughs
This study showed that liver fibrosis protects mice against D-GalN/ lipopolysaccharide (LPS) challenge, and M2-like KCs in fibrotic liver may exert a protective effect by inhibiting the translocation of HMGB1.
Applications
The findings from this study may provide a potential explanation for the mechanism of injury resistance in the setting of liver fibrosis. From a clinical perspective, these findings may pave the way for the treatment of liver failure, particularly acuteonchronic liver failure.
Terminology
Macrophages can undergo “classical” M1 activation when exposed to LPS and interferon-γ or “alternative” M2 activation when exposed to interleukin (IL)-4/IL-13. M1- and M2-like macrophages exhibit distinct signatures and fulfill different functions. HMGB1 has diverse functions which depend on its cellular localization. In the intracellular compartment, HMGB1 participates in gene transcription, DNA replication, and DNA repair. To function in a damage-associated molecular pattern, HMGB1 can translocate from the nucleus to the cytoplasm and is subsequently released into the extracellular milieu. Extracellular HMGB1 activates pro-inflammatory signaling pathways by ligation of pattern recognition receptors, leading to severe damage in multiple liver diseases.
Peer-review
This study is well designed, and the results are very interesting. In this study, the authors investigated the mechanism of hepatoprotection conferred by liver fibrosis, evaluated the phenotype of KCs isolated from fibrotic liver. KCs isolated from the fibrotic mice exhibited predominantly an M2-like phenotype. In vitro experiments have shown that HMGB1 was localized in the nucleus of the majority of M2-like KCs and the translocation of HMGB1 was inhibited upon LPS or HMGB1 peptide stimuli, while both LPS and HMGB1 peptide could elicit the conspicuous translocation of intranuclear HMGB1 in KCs isolated from control mice.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: This study was reviewed and approved by the Capital Medical University Institutional Review Board.
Institutional animal care and use committee statement: All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of Capital Medical University (AEEI-2014-071).
Conflict-of-interest statement: The authors declare that they have no conflict of interest related to this study.
Data sharing statement: No additional data are available.
Peer-review started: January 10, 2017
First decision: February 9, 2017
Article in press: March 30, 2017
P- Reviewer: Chadokufa S, Hatta W S- Editor: Gong ZM L- Editor: Filipodia E- Editor: Zhang FF
References
- 1.Friedman SL. Liver fibrosis -- from bench to bedside. J Hepatol. 2003;38 Suppl 1:S38–S53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 2.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramachandran P, Iredale JP. Macrophages: central regulators of hepatic fibrogenesis and fibrosis resolution. J Hepatol. 2012;56:1417–1419. doi: 10.1016/j.jhep.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 4.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7:425–436. doi: 10.1038/nrgastro.2010.97. [DOI] [PubMed] [Google Scholar]
- 6.White ES, Mantovani AR. Inflammation, wound repair, and fibrosis: reassessing the spectrum of tissue injury and resolution. J Pathol. 2013;229:141–144. doi: 10.1002/path.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bourbonnais E, Raymond VA, Ethier C, Nguyen BN, El-Leil MS, Meloche S, Bilodeau M. Liver fibrosis protects mice from acute hepatocellular injury. Gastroenterology. 2012;142:130–139.e4. doi: 10.1053/j.gastro.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 9.Osawa Y, Seki E, Adachi M, Suetsugu A, Ito H, Moriwaki H, Seishima M, Nagaki M. Role of acid sphingomyelinase of Kupffer cells in cholestatic liver injury in mice. Hepatology. 2010;51:237–245. doi: 10.1002/hep.23262. [DOI] [PubMed] [Google Scholar]
- 10.Osawa Y, Hannun YA, Proia RL, Brenner DA. Roles of AKT and sphingosine kinase in the antiapoptotic effects of bile duct ligation in mouse liver. Hepatology. 2005;42:1320–1328. doi: 10.1002/hep.20967. [DOI] [PubMed] [Google Scholar]
- 11.Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell Mol Immunol. 2016;13:316–327. doi: 10.1038/cmi.2015.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Possamai LA, Thursz MR, Wendon JA, Antoniades CG. Modulation of monocyte/macrophage function: a therapeutic strategy in the treatment of acute liver failure. J Hepatol. 2014;61:439–445. doi: 10.1016/j.jhep.2014.03.031. [DOI] [PubMed] [Google Scholar]
- 13.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 14.Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. 2014;59:2034–2042. doi: 10.1002/hep.26754. [DOI] [PubMed] [Google Scholar]
- 15.Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60:1090–1096. doi: 10.1016/j.jhep.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 16.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 18.Dragomir AC, Laskin JD, Laskin DL. Macrophage activation by factors released from acetaminophen-injured hepatocytes: potential role of HMGB1. Toxicol Appl Pharmacol. 2011;253:170–177. doi: 10.1016/j.taap.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang D, Kang R, Van Houten B, Zeh HJ, Billiar TR, Lotze MT. High mobility group box 1 (HMGB1) phenotypic role revealed with stress. Mol Med. 2014;20:359–362. doi: 10.2119/molmed.2014.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antoine DJ, Jenkins RE, Dear JW, Williams DP, McGill MR, Sharpe MR, Craig DG, Simpson KJ, Jaeschke H, Park BK. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J Hepatol. 2012;56:1070–1079. doi: 10.1016/j.jhep.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Antoine DJ, Dear JW, Lewis PS, Platt V, Coyle J, Masson M, Thanacoody RH, Gray AJ, Webb DJ, Moggs JG, et al. Mechanistic biomarkers provide early and sensitive detection of acetaminophen-induced acute liver injury at first presentation to hospital. Hepatology. 2013;58:777–787. doi: 10.1002/hep.26294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung K, Kang M, Park C, Hyun Choi Y, Jeon Y, Park SH, Seo SK, Jin D, Choi I. Protective role of V-set and immunoglobulin domain-containing 4 expressed on kupffer cells during immune-mediated liver injury by inducing tolerance of liver T- and natural killer T-cells. Hepatology. 2012;56:1838–1848. doi: 10.1002/hep.25906. [DOI] [PubMed] [Google Scholar]
- 23.Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, Pecker F, Tran A, Gual P, Mallat A, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. 2014;59:130–142. doi: 10.1002/hep.26607. [DOI] [PubMed] [Google Scholar]
- 24.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Locatelli L, Cadamuro M, Spirlì C, Fiorotto R, Lecchi S, Morell CM, Popov Y, Scirpo R, De Matteis M, Amenduni M, et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis. Hepatology. 2016;63:965–982. doi: 10.1002/hep.28382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kono H, Onda A, Yanagida T. Molecular determinants of sterile inflammation. Curr Opin Immunol. 2014;26:147–156. doi: 10.1016/j.coi.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 27.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hwang JS, Lee WJ, Kang ES, Ham SA, Yoo T, Paek KS, Lim DS, Do JT, Seo HG. Ligand-activated peroxisome proliferator-activated receptor-δ and -γ inhibit lipopolysaccharide-primed release of high mobility group box 1 through upregulation of SIRT1. Cell Death Dis. 2014;5:e1432. doi: 10.1038/cddis.2014.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kao YH, Lin YC, Tsai MS, Sun CK, Yuan SS, Chang CY, Jawan B, Lee PH. Involvement of the nuclear high mobility group B1 peptides released from injured hepatocytes in murine hepatic fibrogenesis. Biochim Biophys Acta. 2014;1842:1720–1732. doi: 10.1016/j.bbadis.2014.06.017. [DOI] [PubMed] [Google Scholar]