


Abstract
Chromatin three-dimensional (3D) structure plays critical roles in gene expression regulation by influencing locus interactions and accessibility of chromatin regions. Here we propose a Markov process model to derive a chromosomal equilibrium distribution of randomly-moving molecules as a functional consequence of spatially organized genome 3D structures. The model calculates steady-state distributions (SSD) from Hi-C data as quantitative measures of each chromatin region's dynamic accessibility for transcription factors and histone modification enzymes. Different from other Hi-C derived features such as compartment A/B and interaction hubs, or traditional methods measuring chromatin accessibility such as DNase-seq and FAIRE-seq, SSD considers both chromatin–chromatin and protein–chromatin interactions. Through our model, we find that SSD could capture the chromosomal equilibrium distributions of activation histone modifications and transcription factors. Compared with compartment A/B, SSD has higher correlations with the binding of these histone modifications and transcription factors. In addition, we find that genes located in high SSD regions tend to be expressed at higher level. Furthermore, we track the change of genome organization during stem cell differentiation, and propose a two-stage model to explain the dynamic change of SSD and gene expression during differentiation, where chromatin organization genes first gain chromatin accessibility and are expressed before lineage-specific genes do. We conclude that SSD is a novel and better measure of dynamic chromatin activity and accessibility.
INTRODUCTION
Gene expression levels are dynamically regulated by transcription factors, epigenetic modifications and spatial genome architecture (1,2). The spatial regulation of gene expression has been evidenced by that genes belonging to a chromosomal domain are often co-regulated (3), and that long range interactions between enhancers and promoters through chromosome loops activate gene expression (4). To study the genome architecture and its functional roles, genome-wide chromosome conformation capture methods, such as ChIA-PET, 5C and Hi-C, have been developed to systematically capture inter- and intra-interactions among chromatin regions (5–7). Among these methods, Hi-C is the most widely used one which combines crosslinking and high-throughput sequencing to measure whole genome interactions at a high resolution (7,8). Initial analyses of Hi-C data find that chromosomes are divided into two compartments, A and B, associated with open and closed chromatins, respectively (7,9). Further analyses with higher resolution Hi-C data reveal topologically associated domains (TAD) which are conserved across cell types and species (10). Chromosome regions within a TAD interact at higher frequencies and genes in the same TAD tend to be co-regulated (10). Furthermore, chromatin loops within topological domains promote long-range interactions between transcriptional regulatory elements and gene promoters (8).
Hi-C derived A/B compartments, TADs and chromatin loops form a hierarchy of genome structures. All the three levels of structures are found to be associated with gene expression regulation. Genes located in compartment A are expressed at higher levels than those in compartment B (7). The expression levels of many genes change after CCCTC-binding factor (CTCF) knock-down, due to that CTCF is critical for maintaining TAD boundaries (11). In addition, pre-existing promoter–enhancer loops facilitate response to external signals (4). These findings support a strong relationship between genome structure and gene expression.
A key mediator between genome structure and transcription is chromatin accessibility. Three-dimensional (3D) chromatin structures not only determine the interactions among DNA elements, but also affect the accessibility of chromatin regions (12,13), which in turn influences the binding of epigenetic modification enzymes, transcription factors and RNA polymerases to DNA (14,15). In supporting this, genome structures are associated with epigenetic modifications that mark different chromatin accessibility (7). Recent studies find that chromatin's epigenetic states are associated with their compactness (16), chromatin hubs have characteristic histone modification patterns (17) and A/B compartments can be reconstructed by epigenetic information (18). However, traditional methods measuring chromatin accessibility such as DNase-seq (19) and FAIRE-seq (20) do not provide information about genome 3D structure, and to our knowledge, there is no method to extract chromatin accessibility information from 3D genome data. Therefore, a method to quantify chromatin's accessibility using 3D genome information will help better understand the relationship between 3D genome organization and gene regulation.
Here we propose a Markov process model to derive a chromosomal equilibrium distribution of randomly-moving molecules as a functional consequence of spatially organized genome structures. The model calculates steady-state distributions (SSD) as quantitative measures of each chromatin region's accessibility for transcription factors and histone modification enzymes. We show that SSD is highly correlated with the distributions of activation histone modifications and transcription factors. In addition, most differentially expressed genes between cell types are transcribed from regions with differential SSD, and chromatin organization genes acquire high SSD before cell type-specific genes do during stem cell differentiation.
MATERIALS AND METHODS
Data sources
The Hi-C data for GM12878 and Normal Human Epidermal Keratinocytes (NHEK) were obtained from Rao et al. (8), which is available at Gene Expression Omnibus (GEO) with accession number GSE63525. The Hi-C data for mouse neuron differentiation was obtained from Fraser et al. (21), which is available at GEO with accession number GSE59027. The Hi-C raw data in FASTQ format for Drosophila melanogaster embryonic nuclei was obtained from Sexton et al. (22), which is available at GEO with accession number GSE34453. The DNase-seq raw data in FASTQ format for D. melanogaster was obtained from Encyclopedia of DNA Elements (ENCODE). RNA-seq data for neuron differentiation was obtained from Linares et al. (23) and could be accessed in GEO with accession number GSE71179. GM12878 and NHEK's RNA-seq, ChIP-seq, FAIRE-seq and DNase-seq data were all obtained from ENCODE (24). We used 19 GM12878's and 15 NHEK ChIP-seq datasets. For Hi-C and RNA-seq data, we downloaded raw sequencing files in Sequence Read Archive (SRA) or FASTQ format. For ChIP-seq data, FAIRE-seq data and DNase-seq data, we downloaded BAM files. The R code to compute SSD from Hi-C could be accessed at https://github.com/ChengLiLab/markov3d.
Hi-C analysis
Hi-C raw data were firstly mapped to hg19 (Human) or dm3 (Drosophila) reference genome and binned into contact matrix by HiC-Pro version 2.7.2b (25). The ICE normalization was performed by HiTC version 1.14.0 (26). To filter out the noise from repetitive regions in the chromosome, we removed regions from each centromere's upstream 500 to downstream 500 kb.
Computing steady-state distribution
For ICE normalized Hi-C matrix, the Floyd–Warshall algorithm was applied to the reciprocal of the matrix to compute the 3D distances between chromosome bins (27). The Floyd–Warshall algorithm computed the shortest path between each pair of points in a distance matrix. For example, there are three points A, B, C and the distance between A and B is 3, between A and C is 5 and between B and C is 10. The shortest path from B to C is 8 (B to A to C) rather than 10 (Figure 1B). After shortest-path correction, we applied the Markov Chain Model to the corrected matrix. For the bins in a chromosome, we treated them as the states of a finite state Markov chain, indicating the possible location of a protein molecule on the chromatin.
Figure 1.

Overview of Markov process model and steady-state distribution (SSD). (A) We use a public Hi-C dataset, the GM12878 cell line from Rao et al. (8) to illustrate the procedure. The input of model is Hi-C raw contact matrix. In the pre-processing step, the raw matrix is normalized and transformed to a distance matrix. Low coverage bins are removed after normalization. The distance matrix estimates relative spatial distances between two chromatin bins in the nucleus, accounting for physical distances captured by Hi-C cross-linking. Then the transition matrix is estimated and SSD is computed. (B) Advantage of shortest-path algorithm. Hi-C crosslinking could anchor region A and C, region B and C but not region A and B. As a result, although region A and B's spatial distance is close, the number of detected Hi-C interactions between A and B is underestimated and needs to be corrected by a shortest-path algorithm. (C) Density and histogram plot of GM12878 whole genome's SSD. (D) A 3D visualization of GM12878's chromosome 1 and SSD, both inferred from Hi-C data.
We denoted D = {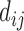 } as the distance matrix between chromosome bins. We modeled the diffusion process of the molecules as a Brownian motion (28), which was used to model random collisions between DNA (29). Therefore, the relationship between two bins’ distance and transition probability was:
} as the distance matrix between chromosome bins. We modeled the diffusion process of the molecules as a Brownian motion (28), which was used to model random collisions between DNA (29). Therefore, the relationship between two bins’ distance and transition probability was:
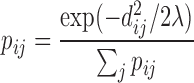 |
The numerator means that transition probability decays when the distance between two bins increases, and the denominator is the sum of all entries across the row of distance matrix, which is used for normalization. Being a classical model for diffusion processes, the advantage of Brownian motion is that we do not use the signal along the diagonal of a Hi-C matrix in transition probability estimation and prevent the possible bias in the Hi-C diagonal signal (4). Since the probability contacts in a Hi-C matrix follow the power law and the exponent 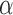 reflects the property of a chromosome (30), for each chromosome we chose the parameter
reflects the property of a chromosome (30), for each chromosome we chose the parameter 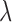 as a function of the chromosome's exponent
as a function of the chromosome's exponent 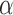 :
:
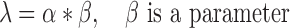 |
To find the optimal parameter 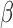 , we conducted a grid search from 1e-8 to 10 on the NHEK cell line and picked the
, we conducted a grid search from 1e-8 to 10 on the NHEK cell line and picked the 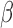 having the largest Spearman correlation with the median ChIP-seq signals across 14 datasets (Supplementary Figure S4). The generality of
having the largest Spearman correlation with the median ChIP-seq signals across 14 datasets (Supplementary Figure S4). The generality of 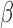 was tested on the GM12878 cell line (Figure 2).
was tested on the GM12878 cell line (Figure 2).
Figure 2.

The distributions of histone modifications and DNA-binding proteins follow SSD. (A) ChIP-seq reads’ distribution along GM12878's chromosome 1. The chromosome is cut into 50 kb non-overlapping bins and RPM (reads per million) of H3K4me1 is computed for each bin to represent the mark's concentration. Black points represent SSD and are scaled for comparison. Only 150–200 Mb regions of chromosome 1 is shown. (B) Spearman correlation between ChIP-seq signals and SSD in 50 kb resolution for GM12878 using whole genome data. Bar plot shows Spearman correlation between SSD and ChIP-seq signals. (C) Hexbin plot showing the correlation between SSD and H3K4me1 signal on GM12878's chromosome 1. The color represents the density of points within each region. Rank ratio is defined as: (rank–min (rank))/(max (rank)–min (rank)). (D) Similar figure as Figure 2C using GM12878 chromosome 16, SSD and CTCF data.
After getting the transition matrix 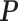 , the SSD was computed, which was the eigenvector corresponding to the largest eigenvalue of an eigenvalue decomposition of
, the SSD was computed, which was the eigenvector corresponding to the largest eigenvalue of an eigenvalue decomposition of 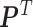 .
.
When considering the situation that biological molecules could leave a chromosome, an additional parameter 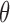 can be added to model the probability that a molecule leaves the chromosome independent of its chromatin location. However,
can be added to model the probability that a molecule leaves the chromosome independent of its chromatin location. However, 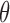 simply reduces SSD to (1–
simply reduces SSD to (1–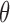 ) * SSD, which does not affect the correlations between SSD and histone modifications, DNA-binding proteins and gene expression as discussed in the ‘Results’ section.
) * SSD, which does not affect the correlations between SSD and histone modifications, DNA-binding proteins and gene expression as discussed in the ‘Results’ section.
RNA-seq analysis
Reads were mapped to hg19 reference genome and gene expression was quantified by RSEM version 1.2.25 (31). Differential expression analysis was conducted by EBSeq version 1.10.0 (32). Differentially expressed genes were defined as having posterior probability of differential expression equal to 1. Gene ontology (GO) analysis was conducted by BiNGO version 3.0.3 (33).
ChIP-seq analysis
For each chromosome, the upstream 500 to downstream 500 kb of centromere was removed to filter out noise coming from repetitive regions. Then each chromosome was cut into bins of the same size as Hi-C. RPM (reads mapped) was computed for each bin. For each histone mark, the ratio between the mark's RPM and control's RPM was calculated as ChIP-seq signals to remove bias.
FAIRE-seq and DNase-seq analysis
For each chromosome, the upstream 500 to downstream 500 kb of centromere was removed to filter out noise coming from repetitive regions. Then each chromosome was cut into bins of the same size as Hi-C. RPM (reads mapped) was computed for each bin.
Comparison with compartment score, hub, directionality index and insulation score
Because compartment A/B was a qualitative description of genome, we also used compartment score (first or second principal component of Hi-C matrix (7)) to represent compartment A/B. Genome was divided into 50 kb non-overlapping bins and then compartment score and directionality index (10) were computed by HiTC version 1.14.0 (26). Hubs and median bins were defined as in Huang et al. (17). Whole genome's SSD were ranked and the top 10 and 45% quantiles were taken as the thresholds for SSD hubs and SSD median, respectively. Histone mark signatures were computed around the center of each hub or median and the computation of normalized signals is the same as Huang et al. (17). Insulation score was defined as in Crane et al. (34). We computed using custom scripts which are available at https://github.com/ChengLiLab/markov3d.
For compartment score, the difference between GM12878 and NHEK cell lines were computed. Here we used difference instead of fold change because compartment score contained negative values. The thresholds of high and low differential features were defined as 5 and 95% quantiles of the fold change or difference. We also labeled each bin as compartment A or B and tracked the change of compartment when correlating it with differential expression data.
RESULTS
Markov process model for chromatin–molecule interactions
We hypothesized that DNA-interacting molecules such as transcription factors and histone modifying enzymes associate and disassociate with chromatin regions and transfer among them constantly, and the concentration of molecules at a chromatin location is partially determined by this dynamic process (35). We first used Hi-C data to model the probabilities of randomly-moving molecules transferring among chromatin regions. Specifically, the raw Hi-C data matrix of a chromosome were firstly normalized by ICE to remove systematic bias (36). In the ICE normalization, the read counts of low coverage regions were normalized to zero and we removed these regions in subsequent analysis. The normalized matrix was then converted to a spatial distance matrix between chromatin bins using a shortest-path algorithm (Figure 1A), which accounts for physical distances beyond the capture limit of Hi-C experiment (27) (Figure 1B). Next, we estimate the transition probability matrix of a finite state Markov chain from the distance matrix by a diffusion model based on Brownian motion (28) (Figure 1A, ‘Materials and Methods’ section). The transition matrix has the following property: the smaller the spatial distance between two chromatin bins is, the higher the likelihood that a molecule transfers from one bin to the other after a given time interval.
Based on this transition matrix, we computed the SSD of the Markov chain, estimating the probability that a randomly-moving biological molecule locates or interacts with individual chromatin bins in a dynamic equilibrium (Figure 1A). SSD follows a long tailed distribution (Figure 1C). A visualization of SSD on the Hi-C based 3D model of GM12878 cell line's chromosome 1 shows that chromatin regions with high SSD locate at both spatially more accessible and less accessible regions, a result that SSD considers both spatial genome structure and dynamic equilibrium of molecule movements (Figure 1D).
SSD correlates with the distribution of histone modification marks and DNA-binding proteins
To evaluate whether SSD can explain observed biological molecules’ chromosomal distribution, we obtained distribution information from DNA binding and histone modification ChIP-seq datasets of GM12878 cell line (24), and calculated ChIP-seq signals for the same chromatin bins used in the Hi-C analysis. SSD and ChIP-seq signals show a high correlation (Figure 2A, Supplementary Figures S1 and 2). Correlation analysis of GM12878's whole genome ChIP-seq signal and SSD shows that the distribution of most histone modification marks is significantly correlated with SSD (Figure 2B). Marks associated with gene activation show the highest correlation with SSD, including H3K27ac, H3K4me1/2/3 and H3K79me2 (Figure 2B and C; Spearman correlation ranges from 0.30 to 0.50, all correlation test P-values < 2.2e-16).
Next we asked whether SSD can explain observed protein–DNA interaction patterns. We used ChIP-seq data of GM12878 for six DNA-binding proteins (CTCF, EZH2, H2AFZ, MYC, STAT1 and STAT3) and two RNA polymerase subunits (POLR2A and POLR2AphosphoS2) (24). We found that the Spearman correlations between these protein factors and SSD range from 0.10 to 0.48 (Figure 2B and D; 50 kb bin, all correlation test P-values < 0.000022). We obtained similar correlation results using a different bin size (Supplementary Figure S3) and another cell line NHEK (Supplementary Figure S4). In total, among 18 GM12878 ChIP-seq datasets of histone modification marks and DNA binding proteins, 16 have significant positive correlation with SSD. These results suggest that ChIP-seq signals of histone modification marks and DNA-binding proteins contain common patterns that can be explained by SSD, in addition to mark-specific or protein-specific patterns.
SSD correlates with gene expression levels and changes
Since SSD is correlated with the levels of activation histone marks and transcriptional machinery, we asked whether SSD is also associated with transcriptional activity. We found that the expression levels of genes locating in high SSD regions are significantly higher than those in low SSD regions (Figure 3A, all t-test P-values < 2.2e-16). High SSD regions also contain more genes than low SSD regions do (Figure 3B, paired t-test P-value < 2.2e-16). Therefore, high SSD regions not only contain more genes but also express these genes at higher levels compared to low SSD regions. We also compared the change of gene expression and the change of SSD genome-wide between two cell lines, GM12878 and NHEK. We found that the change of gene expression levels is positively correlated with the change of corresponding regions’ SSD (Figure 3C and D, Cuzick trend test P-value < 2.2e-16). Next, we compared the change of histone modification marks between GM12878 and NHEK. Similar to transcriptional activity, we found that the change of ChIP-seq signals are positively correlated with the change of corresponding regions’ SSD (Figure 3E, Supplementary Figure S5, all Cuzick trend test P-values < 2.2e-16).
Figure 3.

SSD influences chromatin regions’ accessibility and transcriptional activity. (A) The comparison of expression levels of genes located in high and low SSD regions for each chromosome. For each chromosome, high SSD regions are defined as bins with SSD higher than the median SSD. Low SSD regions are the ones with SSD lower than the median SSD. (B) The number of genes located in high and low SSD regions. (C) Comparing SSD and gene expression levels between two cell lines, GM12878 and NHEK. Points colors represent the log2 fold change of GM12878 and NHEK's gene expression (FPKM). Points above the diagonal are the regions with higher SSD in GM12878 and under the diagonal are those with higher SSD in NHEK. (D) Relationship between SSD fold change and gene expression fold change comparing two cell lines. (E) Relationship between SSD fold change and H3K4me3 ChIP-seq signal fold change comparing two cell lines. (F) Model for how dynamic chromatin accessibility affects gene expression. Regions with high SSD are more accessible for histone modification enzymes and transcriptional factors, leading to higher transcriptional activity in these regions.
Based on these findings, we propose a model of dynamic chromatin accessibility, where the equilibrium distribution of randomly-moving protein molecules on chromatin follows a Markov process. The resultant dynamic chromatin accessibility arises from physical chromatin structures and influences transcriptional activities (Figure 3F).
Comparison between SSD and other Hi-C derived chromatin features
We next compared SSD with other four Hi-C derived features of chromatin structure: A/B compartment (7), chromatin hub (17), directionality index (10) and insulation score (34), as all the five features are associated with marks of chromatin modifications and gene transcription.
We first compared SSD and A/B compartment score in terms of their genome-wide correlations with 10 ChIP-seq experiments of histone modification marks in GM12878. SSD shows higher correlations with most ChIP-seq signals than A/B compartment score does (Figure 4A). We also compared SSD with the data of FAIRE-seq (20) and DNase-seq (19), two sequencing-based method to measure chromatin accessibility. SSD is significantly and more positively correlated with both FAIRE-seq (Spearman correlation 0.51, P-value < 2.2e-16) and DNase-seq (Spearman correlation 0.37, P-value < 2.2e-16) than A/B compartment score is. We also compared SSD and A/B compartment score in terms of their genome-wide correlations with transcriptional activity. Chromatin regions with differential SSD between two cell lines show more significantly differential expression than those located in changed A/B compartments do (Supplementary Figure S6). These results suggest that SSD better measures chromatin accessibility and correlates with transcriptional changes than Hi-C derived A/B compartments.
Figure 4.

Comparing SSD with compartment profile and hub. (A) GM12878 SSD's and compartment score's Spearman correlations with ChIP-seq signals of histone modifications, FAIRE-seq and DNase-seq at the whole genome scale. (B) GM12878 SSD's, insulation score's and directionality index's Spearman correlations with ChIP-seq signals of histone modifications and DNA-binding proteins at the whole genome scale. (C) Comparing histone mark signatures between hubs and SSD hubs. X-axis represents the relative distance from hubs/median center (−5 to 5 Mb) and Y-axis represents averaged ChIP-seq signals.
Next we compared SSD with two other measures that represent chromatin organization and transcriptional activities, directionality index (10) and insulation score (34). We found that the correlation between SSD and directionality index was not significant (Spearman correlation = 0.002, P-value = 0.61). However, SSD correlated well with insulation score (Spearman correlation = 0.32, P-value < 2.2e-16), suggesting that insulation score and directionality index were orthogonal data representing chromatin domains. Then we correlated the three measures with 18 epigenetic marks and proteins occupancy on chromatin. We found that SSD and insulation score correlated with these marks and factors significantly better than directionality index did (Figure 4B). When compared with insulation score, SSD correlated better in 15/18 comparisons and SSD's mean correlation was 4.1-fold higher (0.316 versus 0.077, t-test P-value = 9.788e-6). These results suggest that SSD better measures chromatin accessibility than Hi-C derived insulation score and directionality index.
A recently identified feature of genome organization from Hi-C data are chromatin hubs (17), defined as chromatin regions with the highest interaction frequencies with other regions. Like chromatin regions with high SSD, chromatin hubs have specifically associated signatures of histone marks. Following chromatin hubs’ definition, we defined SSD hubs and SSD median using the top 10 and 45% quantiles of SSD as thresholds, and calculated these regions’ histone mark signatures (‘Material and Methods’ section). There are generally larger differences of histone mark signals between chromatin hubs and median defined by SSD than those defined by Hi-C interaction frequencies (Figure 4C), suggesting that a chromatin accessibility measure derived from both 3D chromatin structure data and dynamic equilibration process may better predict epigenetic features than using physical chromatin structures alone.
Besides comparing SSD with compartment score in human cell lines, we also compared them in Drosophila embryos. The Drosophila's Hi-C data were obtained from Sexton et al. (22). Similar to GM12878's SSD, Drosophila's SSD also followed a long tailed distribution (Supplementary Figure S7A). We correlated SSD and compartment score with a public Drosophila's DNase-Seq dataset from ENCODE (24). The Spearman correlation between SSD and DNase-Seq was 0.111 (P-value = 0.01), while the correlation between compartment score and DNase-Seq was 0.006 (P-value = 0.82) (Supplementary Figure S7B). These results suggest that SSD better captures chromatin accessibility information in Drosophila than compartment score does. However, although SSD's correlation is significant, the absolute value is relative low compared with human data (Figure 4A). This indicates that either there is difference between Drosophila's genome structural organization and human's, or the SSD model's parameters need to be recalibrated for Drosophila Hi-C data. Further study is needed to resolve the issue.
SSD reveals two stages of spatial genome organization during stem cell differentiation
Next we asked how SSD may help study genome reorganization during biological processes. The development and differentiation processes are accompanied with changes of both 3D genome structures and gene expression (37), but how genome structure and transcription affects each other remains unresolved (38,39). To investigate genome structure reorganization during differentiation, we analyzed a neuron differentiation dataset containing Hi-C data for three differentiation stages: mouse embryonic stem cells (ESC), neuronal progenitor cells (NPC) and neurons (21). We defined each stage's marker genes as those located in its high SSD regions and other two stages’ low SSD regions, so that the marker genes are only highly accessible in one differentiation stage. GO enrichment analysis for each stage's marker genes reveals that in ESC, the genes are enriched in general and house-keeping GO terms such as biological regulation and cell cycle. After cells differentiate into NPC, marker genes are not only enriched in terms associated with neuron differentiation as expected, such as cell proliferation in forebrain, but also enriched in chromatin modification and chromatin organization terms. These genes include DNMT1 and SMARCC1, which are known critical regulators of chromatin structure's remodeling (40,41). In contrast, marker genes in neurons are mainly enriched in terms related to neuronal functions, such as nervous system development (Figure 5A).
Figure 5.

Dynamic change of genome organization and gene expression during stem cell differentiation. (A) GO biological process enrichment for NPC's and Neuron's marker genes defined by SSD. (B) GO biological process enrichment for NPC's and Neuron's marker genes defined by expression level. In A and B, only selected terms are shown, and see Supplementary Table S1 for the full list. The red line indicates the position of –log10(0.05). (C) Model for the functional roles that genome 3D structure plays during stem cell differentiation. As stem cells differentiate, chromatin organization genes are first made accessible and transcribed at the progenitor stage, which help to open up the chromatin of lineage-specific genes when progenitor cells differentiate into downstream cell types.
For comparison, we also analyzed changes of gene expression during mouse neuron differentiation using another dataset (23). We compared gene expression levels between stem cells, neuron progenitor cells and neurons, and defined each stage's marker genes as those only highly expressed in one stage (top 10% expression level). Through GO enrichment analysis of marker genes, we found that NPC's marker genes are also enriched in chromosome organization terms, and neuron's marker genes are enriched in terms related to both nervous system development and chromosome organization (Figure 5B). Combining the changes of genome organization and gene expression, we propose a two-stage genome organization model during stem cell differentiation (Figure 5C). Genome structures first change from stem cells to neuron progenitor cells to make chromatin organization genes’ loci more accessible to be transcribed. Then the chromatin organization genes are highly expressed in both progenitor cells and neurons to further remodel genome structures to make accessible and transcribe gene loci associated with neuronal functions.
DISCUSSION
The 3D genome structure influences chromatin accessibility and transcription via nuclear structures such as heterochromatin, lamina associated domains and nucleolus (42). Protein molecules move within the nucleus randomly in search of their interacting partners and DNA-binding sites (43), for example, telomerase uses three-dimensional diffusion to search for telomeres and forms either dynamic or static interactions with them (44). We propose that genome structure influences protein distributions on the whole genome through dynamic chromatin accessibility. Here we have developed a Markov process model of stochastic collisions between molecules and chromatin and computed the equilibrium distribution of molecules locating on a chromosome. The model extracts dynamic chromatin accessibility information from 3D genome structure, allowing us to compare and integrate it with other genomic datasets such as histone modification or gene expression data and to better understand the roles genome structure plays in epigenetic and transcriptional processes.
Our results suggest a model where chromatin regions with different SSD have different accessibilities to transcription factors and histone modification enzymes, leading to different transcriptional activities between regions (Figure 3F). Our results are consistent with a previous study on glucocorticoid receptor binding patterns (14), which concludes that the distribution of exposed chromatin dictates the genomic distribution of DNA-binding proteins. A unique feature of our model is that the chromatin interactions in the 3D genome serve as the traffic network of randomly moving protein molecules, and chromatin accessibility is the dynamic equilibrium resulting from this network through a Markov process. This dynamic feature may explain the better agreement between SSD and transcription and epigenetic marks than Hi-C derived static chromatin features such as A/B compartments and chromatin hubs. We also observe that the correlations between histone repression marks and SSD are weak and variable between cell types (Figure 2B and Supplementary Figure S4). We propose the possible reason may be that gene repression marks are regulated more actively by means other than random movements than gene activation marks, and therefore a random collision model cannot capture their distributions well. Our results highlight the importance of genome structure in DNA–protein interaction. It suggests that all the molecules in a nucleus share a basal interacting distribution on the chromatin, and could explain the reason why some molecules with opposite functions distribute similarly. For example, histone acetyltransferase and histone deacetylase are both positively correlated with histone acetylation levels (45).
SSD transforms Hi-C matrices to one-dimensional vectors, making it easier to compare multiple Hi-C profiles and integrate with other genomic datasets. We have analyzed stem cell differentiation by jointly analyzing the change of genome structure and gene expression in three differentiation states, stem cell, progenitor cell and neuron. The increasing of chromatin organization genes’ SSD and expression levels during differentiation leads to a two-stage model of genome structure's functional role (Figure 5C). Our results confirm that cells in different differentiation states have characteristic chromatin structure features (37), suggesting a potential application of our method to identify chromatin structure features associated with cell states. It may provide more sensitive markers to measure cell reprogramming or differentiation states and classify cancer subtypes (46).
To interpret Hi-C data, several models such as TAD (10), hub (17) and hotspot (47) have been proposed and improved our understanding of genome structure and its functional roles. The main difference between SSD and these models is its dynamic property. TAD and hotspot focus on functional interactions among static chromosome regions such as promoters and enhancers or transcription factors and targets. In addition to these physical interactions among chromosome regions, SSD suggests a complementary mechanism of genome structure's functional role, leading to the hypothesis that 3D structure could influence gene expression by affecting the dynamic distribution of epigenetic modifications and transcription factors. We observe high correlations between SSD and histone modifications and validate their robustness in different cell types and Hi-C resolutions. Similar with the hubs (17), we find that chromatin regions with high SSD have enriched histone modification patterns. Our results provide a possible explanation of hubs’ histone modification patterns, which could result from random collisions between histone modification enzymes and hubs.
We could only infer correlative rather than causal relationship between chromatin structure and protein binding. The inference of causal relationship from observed correlation data is still a challenging and active research field in statistics (48), and the causal relationship between chromatin accessibility and protein binding is also an unsolved question (42). Chromatin structure is a complex network influenced by DNA sequence features, transcription factors, nucleosome remodelers and histone modifiers (42). The pioneer transcription factor model proposes that the initial binding of transcription factors on chromatin could remodel chromatin structure and influence the binding of more proteins (49). Therefore, there may be a complex and feedback relationship rather than a directional relationship between protein binding and chromatin structure. Our model captures the dynamic equilibrium of this complex process and could predict the equilibrium distribution of proteins for given chromatin structures. Moreover, in Figure 5C we propose a model to explain the change of chromatin structure during stem cell differentiation. Several studies also found that genes like PARP1 could affect gene expression by influencing the condensation of chromosome during cellular development (50). Although we find that SSD is highly correlated with gene expression, it is possible that the change of gene transcription leads to the reorganization of chromosome structure during cell differentiation. Further experiments and analysis are needed to unravel the complex and dynamic relationship between chromatin structure and gene expression during biological processes.
Markov models have been widely applied in biological science, such as chromatin-state characterization (51), TAD identification (10) and detection of long-range chromosomal interactions from Hi-C data (52). These existing methods mainly use hidden Markov models, which infer hidden states from observed states, such as the inference of TAD boundaries from Hi-C data or enhancers from ChIP-seq data. Similar to these methods, we take Markov property as an assumption of our model. But instead of using a hidden Markov model, we directly apply Markov process to model molecule–chromatin interactions and compute SSD.
Our method has several aspects that can be improved in the future. First, we compute SSD for each intra-chromosome Hi-C contact matrix instead of the whole genome contact matrix, and the probability that a molecule moves from one chromosome to another cannot be computed. The present version of our model allows the whole genome contact matrix as input but the computation time will increase significantly, especially for high resolution Hi-C data. We will develop more efficient algorithms in the future version. Second, we use a single parameter θ to control the probability that a molecule detaches and leaves a chromosome (‘Material and Methods’ section). However, for different chromatin regions the detaching probability may differ. A possible solution is to learn parameters from biological experiments which can measure or monitor the movement of biological molecules, such as single-molecule imaging (53). Finally, here we only show that SSD could be derived from Hi-C, but other methods which detect genome structure and generate distance matrix such as ChIA-PET or high-resolution imaging of chromatins could also be served as the input of our model.
AVAILABILITY
The R code to compute SSD from Hi-C can be accessed at https://github.com/ChengLiLab/markov3d.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Yujie Sun, Jian Ge, Yanhui Xu and members of Cheng Li lab for helpful discussion. We thank Guo-Cheng Yuan, Xiaowo Wang and reviewers for critical advices on the manuscript.
Author contributions: Y.W. and C.L. designed the study and wrote the manuscript. Y.W. analyzed the data and prepared the figures. Y.Z. participated in figure preparation. C.F. participated in data analysis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Peking-Tsinghua Center for Life Sciences, School of Life Sciences and Center for Statistical Science of Peking University; National Natural Science Foundation of China Key Research [71532001]; Chinese National Key Projects of Research and Development [2016YFA0100103]. Funding for open access charge: Peking-Tsinghua Center for Life Sciences.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lanctôt C., Cheutin T., Cremer M., Cavalli G., Cremer T.. Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat. Rev. Genet. 2007; 8:104–115. [DOI] [PubMed] [Google Scholar]
- 2. Gibcus J.H., Dekker J.. The hierarchy of the 3D genome. Mol. Cell. 2013; 49:773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cohen B.A., Mitra R.D., Hughes J.D., Church G.M.. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat. Genet. 2000; 26:183–186. [DOI] [PubMed] [Google Scholar]
- 4. Jin F., Li Y., Dixon J.R., Selvaraj S., Ye Z., Lee A.Y., Yen C.-A., Schmitt A.D., Espinoza C.A., Ren B.. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013; 503:290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dostie J., Richmond T.A., Arnaout R.A., Selzer R.R., Lee W.L., Honan T.A., Rubio E.D., Krumm A., Lamb J., Nusbaum C. et al. . Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006; 16:1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fullwood M.J., Liu M.H., Pan Y.F., Liu J., Xu H., Mohamed Y.B., Orlov Y.L., Velkov S., Ho A., Mei P.H. et al. . An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009; 462:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. et al. . Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rao S.S.P., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. et al. . A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Laat W., Dekker J.. 3C-based technologies to study the shape of the genome. Methods. 2012; 58:189–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zuin J., Dixon J.R., van der Reijden M.I.J.A., Ye Z., Kolovos P., Brouwer R.W.W., van de Corput M.P.C., van de Werken H.J.G., Knoch T.A., van IJcken W.F.J. et al. . Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsompana M., Buck M.J.. Chromatin accessibility: a window into the genome. Epigenetics Chromatin. 2014; 7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsiung C.C.-S., Morrissey C.S., Udugama M., Frank C.L., Keller C.A., Baek S., Giardine B., Crawford G.E., Sung M.-H., Hardison R.C. et al. . Genome accessibility is widely preserved and locally modulated during mitosis. Genome Res. 2015; 25:213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. John S., Sabo P.J., Thurman R.E., Sung M.-H., Biddie S.C., Johnson T.A., Hager G.L., Stamatoyannopoulos J.A.. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 2011; 43:264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shlyueva D., Stampfel G., Stark A.. Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 2014; 15:272–286. [DOI] [PubMed] [Google Scholar]
- 16. Boettiger A.N., Bintu B., Moffitt J.R., Wang S., Beliveau B.J., Fudenberg G., Imakaev M., Mirny L.A., Wu C.-T., Zhuang X.. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature. 2016; 529:418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang J., Marco E., Pinello L., Yuan G.-C.. Predicting chromatin organization using histone marks. Genome Biol. 2015; 16:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fortin J.-P., Hansen K.D.. Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome Biol. 2015; 16:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Song L., Crawford G.E.. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring. Harb. Protoc. 2010; 2010, doi:10.1101/pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giresi P.G., Kim J., McDaniell R.M., Iyer V.R., Lieb J.D.. FAIRE (formaldehyde-assisted isolation of regulatory elements) isolates active regulatory elements from human chromatin. Genome Res. 2007; 17:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fraser J., Ferrai C., Chiariello A.M., Schueler M., Rito T., Laudanno G., Barbieri M., Moore B.L., Kraemer D.C.A., Aitken S. et al. . Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol. Syst. Biol. 2015; 11:852–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sexton T., Yaffe E., Kenigsberg E., Bantignies F., Leblanc B., Hoichman M., Parrinello H., Tanay A., Cavalli G.. Three-dimensional folding and functional organization principles of the drosophila genome. Cell. 2012; 148:458–472. [DOI] [PubMed] [Google Scholar]
- 23. Linares A.J., Lin C.-H., Damianov A., Adams K.L., Novitch B.G., Black D.L.. The splicing regulator PTBP1 controls the activity of the transcription factor Pbx1 during neuronal differentiation. Elife. 2015; 4:e09268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sloan C.A., Chan E.T., Davidson J.M., Malladi V.S., Strattan J.S., Hitz B.C., Gabdank I., Narayanan A.K., Ho M., Lee B.T. et al. . ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016; 44:D726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Servant N., Varoquaux N., Lajoie B.R., Viara E., Chen C.-J., Vert J.-P., Heard E., Dekker J., Barillot E.. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015; 16:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Servant N., Lajoie B.R., Nora E.P., Giorgetti L., Chen C.-J., Heard E., Dekker J., Barillot E.. HiTC: exploration of high-throughput ‘C’ experiments. Bioinformatics. 2012; 28:2843–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lesne A., Riposo J., Roger P., Cournac A., Mozziconacci J.. 3D genome reconstruction from chromosomal contacts. Nat. Methods. 2014; 11:1141–1143. [DOI] [PubMed] [Google Scholar]
- 28. Codling E.A., Plank M.J., Benhamou S.. Random walk models in biology. J. R. Soc. Interface. 2008; 5:813–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cairns J., Freire-Pritchett P., Wingett S.W., Várnai C., Dimond A., Plagnol V., Zerbino D., Schoenfelder S., Javierre B.-M., Osborne C. et al. . CHiCAGO: robust detection of DNA looping interactions in capture Hi-C data. Genome Biol. 2016; 17:390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barbieri M., Chotalia M., Fraser J., Lavitas L.M., Dostie J., Pombo A., Nicodemi M.. Complexity of chromatin folding is captured by the strings and binders switch model. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:16173–16178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li B., Dewey C.N.. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011; 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leng N., Dawson J.A., Thomson J.A., Ruotti V., Rissman A.I., Smits B.M.G., Haag J.D., Gould M.N., Stewart R.M., Kendziorski C.. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013; 29:1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maere S., Heymans K., Kuiper M.. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005; 21:3448–3449. [DOI] [PubMed] [Google Scholar]
- 34. Crane E., Bian Q., McCord R.P., Lajoie B.R., Wheeler B.S., Ralston E.J., Uzawa S., Dekker J., Meyer B.J.. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 2015; 523:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Knight S.C., Xie L., Deng W., Guglielmi B., Witkowsky L.B., Bosanac L., Zhang E.T., El Beheiry M., Masson J.-B., Dahan M. et al. . Dynamics of CRISPR-Cas9 genome interrogation in living cells. Science. 2015; 350:823–826. [DOI] [PubMed] [Google Scholar]
- 36. Imakaev M., Fudenberg G., McCord R.P., Naumova N., Goloborodko A., Lajoie B.R., Dekker J., Mirny L.A.. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods. 2012; 9:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dixon J.R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J.E., Lee A.Y., Ye Z., Kim A., Rajagopal N., Xie W. et al. . Chromatin architecture reorganization during stem cell differentiation. Nature. 2015; 518:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Misteli T. Self-organization in the genome. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:6885–6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cavalli G., Misteli T.. Functional implications of genome topology. Nat. Struct. Mol. Biol. 2013; 20:290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martinowich K., Hattori D., Wu H., Fouse S., He F., Hu Y., Fan G., Sun Y.E.. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003; 302:890–893. [DOI] [PubMed] [Google Scholar]
- 41. Schaniel C., Ang Y.-S., Ratnakumar K., Cormier C., James T., Bernstein E., Lemischka I.R., Paddison P.J.. Smarcc1/Baf155 couples self-renewal gene repression with changes in chromatin structure in mouse embryonic stem cells. Stem Cells. 2009; 27:2979–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bell O., Tiwari V.K., Thomä N.H., Schübeler D.. Determinants and dynamics of genome accessibility. Nat. Rev. Genet. 2011; 12:554–564. [DOI] [PubMed] [Google Scholar]
- 43. Pederson T. Protein mobility within the nucleus–what are the right moves. Cell. 2001; 104:635–638. [DOI] [PubMed] [Google Scholar]
- 44. Schmidt J.C., Zaug A.J., Cech T.R.. Live cell imaging reveals the dynamics of telomerase recruitment to telomeres. Cell. 2016; 166:1188–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang Z., Zang C., Cui K., Schones D.E., Barski A., Peng W., Zhao K.. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009; 138:1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rousseau M., Ferraiuolo M.A., Crutchley J.L., Wang X.Q., Miura H., Blanchette M., Dostie J.. Classifying leukemia types with chromatin conformation data. Genome Biol. 2014; 15:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Capurso D., Bengtsson H., Segal M.R.. Discovering hotspots in functional genomic data superposed on 3D chromatin configuration reconstructions. Nucleic Acids Res. 2016; 44:2028–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pearl J. Causal inference in statistics: an overview. Stat. Surv. 2009; 3:96–146. [Google Scholar]
- 49. Zaret K.S., Mango S.E.. Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr. Opin. Genet. Dev. 2016; 37:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ji Y., Tulin A.V.. The roles of PARP1 in gene control and cell differentiation. Curr. Opin. Genet. Dev. 2010; 20:512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ernst J., Kellis M.. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012; 9:215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xu Z., Zhang G., Jin F., Chen M., Furey T.S., Sullivan P.F., Qin Z., Hu M., Li Y.. A hidden Markov random field-based Bayesian method for the detection of long-range chromosomal interactions in Hi-C data. Bioinformatics. 2016; 32:650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Harada Y., Funatsu T., Murakami K., Nonoyama Y., Ishihama A., Yanagida T.. Single-molecule imaging of RNA polymerase-DNA interactions in real time. Biophys. J. 1999; 76:709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.