

Abstract
Low‐grade inflammation, endothelial dysfunction, and platelet hyper‐reactivity to agonists are associated with an increased risk of cardiovascular events. In vitro and animal studies infer an inverse mechanistic relationship between platelet activation and the production of endothelium‐derived nitric oxide and prostacyclin. This concept is supported by evidence of an inverse relationship between endothelial function and platelet activation in high‐risk cardiac patients. The aim of this study was to investigate what relationship, if any, exists between platelet and endothelial function in healthy, middle‐aged, and elderly adults. In 51 participants (18 male, 33 post menopausal female), endothelial function was assessed by flow‐mediated dilation (FMD). Platelet function was assessed by flow cytometric determination of glycoprotein IIb/IIIa activation (measured by PAC‐1 binding), granule exocytosis (measured by surface P‐selectin expression), and monocyte‐platelet aggregates (MPAs), with and without stimulation by canonical platelet agonists adenosine diphosphate (ADP), arachidonic acid (AA), and collagen. Correlation analysis indicated there was no significant (all P => 0.05) relationship between FMD and any marker of in vivo platelet activation (MPAs R = 0.193, PAC‐1 R = −0.113, anti‐CD62P R = −0.078) or inducible platelet activation by ADP (MPA R = −0.128, anti‐CD62P R = −0.237), AA (MPA R = −0.122, PAC‐1 R = −0.045, anti‐CD62P R = −0.142), or collagen (MPA R = 0.136, PAC‐1 R = 0.174, anti‐CD62P R = −0.077). Our findings contrast with two previous studies performed in high‐risk cardiac patients, which reported inverse relationships between platelet activation and endothelial function, suggesting that some compensatory redundancy may exist in the relationship between platelet and endothelial function in preclinical populations.
Keywords: Cardiovascular disease, endothelial function, platelet function
Introduction
Thrombosis is an integral component of acute coronary syndromes such as acute myocardial infarction (MI) and ischemic stroke (Falk et al. 2013; Libby 2013). Moreover, the role of platelets in the early, dormant stages of atherosclerosis and cardiovascular disease (CVD) has recently been recognized (Lievens and von Hundelshausen 2011; Rondina et al. 2013). Low‐grade inflammation, endothelial dysfunction, and platelet hyper‐reactivity to agonists are all independently associated with an increased risk of cardiovascular events (Gurbel et al. 2007; Libby et al. 2009). An intact and healthy endothelium regulates and inhibits platelet activation (Davì and Patrono 2007) and endothelial dysfunction may play a direct role in activating platelets (Davì et al. 2002). Platelet activation results in the formation of monocyte‐platelet aggregates (MPAs) (Linden 2013), and stepwise increases in MPAs and activated platelets have been observed with cardiovascular disease progression (Linden et al. 2007). The interaction between activated platelets and monocytes can initiate the release of proinflammatory and adhesive molecules that are atherogenic (Michelson et al. 2001; McFadyen and Kaplan 2015; Yuan et al. 2015; Gerdes et al. 2016), driving a proatherogenic monocytic phenotype (Barnard et al. 2005) and facilitating the infiltration of monocytes to the subintima space (Weyrich et al. 1996; McFadyen and Kaplan 2015).
A number of in vitro experiments suggest that nitric oxide (NO) and prostacyclin (PGI2), produced by endothelial cells, directly inhibit platelet aggregation (Alheid et al. 1987, 1989; Macdonald et al. 1988). However, the functional relationship between platelet activation and endothelial function in vivo is less clear (Radomski et al. 1987; Megson et al. 2000). While inhibition of NO production in healthy young adults can increase platelet activation (Schäfer et al. 2004b) and decrease clotting time (Simon et al. 1995), this is not a universal finding (Albert et al. 1999) and these studies assessed the acute impact of pharmacological blockade. An inverse relationship between endothelium‐dependent coronary vasomotor function and MPAs measured in arterial samples has been documented in high‐risk patients undergoing cardiac angiography and angioplasty (Hamilos et al. 2011; Di Serafino et al. 2014). There is also evidence to suggest that pharmacological blockade of the platelet fibrinogen receptor (glycoprotein IIb/IIIa receptor) (Heitzer et al. 2003) and administration of the antiplatelet medications clopidogrel (Hamilos et al. 2008; Warnholtz et al. 2008; Muller et al. 2010; Patti et al. 2011) and aspirin (Husain et al. 1998; Williams et al. 2005) can acutely improve endothelial function in patients with CAD. However, these short‐term effects were abolished when administration was maintained for longer periods of time (Ostad et al. 2011). One previous study (Lanza et al. 2011) has investigated the relationship between in vivo endothelial function, assessed using brachial artery flow‐mediated dilation (FMD) and platelet function assessed via flow cytometry. This study was conducted in adolescents, with or without a positive family history of CAD, and no relationship was found between FMD and platelet activation (MPAs, PAC‐1 or CD62P binding).
Increasing age and physical inactivity are risk factors for cardiovascular disease (Sesso et al. 2000), but no previous study, to our knowledge, has investigated relationships between endothelial function and platelet activation in asymptomatic healthy older adults, or whether any such relationship extends to agonist‐induced platelet activation. Therefore, our aim was to test the relationship, if any, between platelet and endothelial function in apparently healthy, physically inactive, middle‐aged, and elderly humans. We hypothesized that FMD would be inversely associated with the presence of MPAs and activated platelets; and inversely associated with agonist‐induced platelet activation.
Materials and Methods
The study was approved by the University of Western Australia Human Research Ethics Committee, procedures were in accord with the Declaration of Helsinki and participants provided written informed consent.
Male and postmenopausal female participants were recruited from the general population in Perth, Western Australia using multiple recruitment strategies including advertisements in local newspapers, radio stations, and posters. Apparently healthy individuals aged 45 years and over were encouraged to contact the research team, resulting in initial phone screening procedures which included questionnaires to determine suitability to attend a formal screening visit. Initial exclusion criteria included serious illness such as cancer, diagnosed cognitive impairment or dementia, current or past history of ischemic heart disease, angina, stroke, persistent arrhythmias, diabetes mellitus, airway disease, epilepsy, severe mental illness, engaging in more than 1 h of physical activity per week, current or recent smokers (within 12 months), pre‐ or peri‐menopausal females and alcohol consumption >28 standard drinks/wk.
Individuals satisfying the initial criteria were invited to attend a screening session during which a number of measures were collected including: height, body mass, resting electrocardiogram (ECG), and fasting blood tests (glucose, lipid profile, full blood count, urea, and electrolytes). Participants exhibiting abnormal cardiac rhythms, blood test results suggestive of chronic kidney disease, diabetes, or total cholesterol >7 mmol/L were excluded. Included participants were then invited to perform an exercise stress test with ECG monitoring and those with evidence of exertion‐induced myocardial ischemia were excluded from further participation. In subsequent visits, participants underwent a 20 min resting blood pressure (BP) assessment and a dual‐energy x‐ray absorptiometry (DEXA) scan. For the resting BP assessment, participants arrived at the laboratory in the morning following an overnight fast and lay in a supine position in a temperature‐controlled dark room. BP was measured every 2 min by an automated device (Dinamap V100, GE Healthcare, USA). Individuals with an average systolic BP >160 mmHg or diastolic BP >100 mmHg were excluded.
For FMD and platelet function tests, participants arrived at the laboratory in the morning between 7:00 and 9:30am, following an overnight fast, having abstained from the consumption of caffeine and alcohol for 12 and 24 h, respectively, and not taken part in physical exercise for 24 h. Adherence to the protocol was confirmed by questionnaire on arrival. Prior to attending the laboratory for data collection, participants were instructed to be clear of symptoms for 7 days if they had recently suffered with acute conditions including respiratory tract infection, cold, and flu. Participants taking prescription medications were instructed to maintain their usual routine of administration. However, the use of nonprescribed medications such as anti‐inflammatory, antihistamine, antibiotic, aspirin, cold, and flu medications were ceased for at least 7 days prior to blood collection. Participants lay supine in a cool temperature‐controlled room for 15 min, after which a blood sample was collected from the dominant arm for the assessment of platelet function. Subsequent FMD tests were performed on the nondominant arm.
Blood collection
A venous blood sample was collected into a 4 mL 3.2% sodium citrate tube (Vacuette by Greiner bio‐one) and processed according to published standards (Linden 2013), as preanalytical variables during blood collection, storage, and handling are an important factor in platelet function testing. Within 10 min of collection, blood was processed to assess circulating levels of MPAs and activated platelets and their sensitivity to platelet agonists.
Monocyte‐platelet aggregates
Each MPA reaction tube included two antibodies: CD14 (monocyte identifier) conjugated to the fluorophore Brilliant Violet (BV) 421 (Clone M5E2, BioLegend, San Diego CA) and CD42b (platelet identifier) conjugated to Allophycocyanin (APC) (Clone HIP1, BioLegend) or IgG isotype control (BioLegend). Six MPA reaction tubes (Protein LoBind Eppendorf, Germany) were included: isotype control, no agonist, positive control (250 μmol/L thrombin receptor activating peptide‐6 (SFLLRN, Sigma‐Aldrich, MO)), and threshold (low) concentrations of the following three agonists: adenosine diphosphate (ADP) 1.5 μmol/L (Chrono‐Log Corp., PA), arachidonic acid (AA) 10 μg/mL (Sodium arachidonate, Bio/Data Corp., PA), and collagen 1.5 μg/mL (Chrono‐Log Corp., PA). Absence of spectral overlap was confirmed by single‐color comp bead controls (BD Biosciences). Samples were fixed and red cells lysed with 800 μL of BD FACSLyse solution (BD Biosciences) following exactly 15 min of incubation. Samples were then stored in the dark at 4°C and analyzed by flow cytometry (BD FACSCanto™ II, BD Biosciences) at a low flow rate for 10 min per tube, to avoid coincident events (Hui et al. 2015).
Platelet surface receptors
Whole blood was diluted 1:5 with HEPES saline buffer and incubated with a cocktail of the following fluorescent conjugated antibodies at saturating concentrations: PAC‐1 fluorescein (FITC), CD62P phycoerythrin (PE), CD42b PE‐Cy5, or IgG1Κ PE isotype control (all BD Pharmingen) for exactly 15 min. Six reaction tubes were used that were identical in function and agonist concentrations as those used for MPAs. However, ADP at the concentration used (1.5 μmol/L) caused maximal PAC‐1 binding in all participants, so was not included in statistical analysis. Following incubation, samples were fixed with 800 μL of stabilizing fixative (BD Biosciences) and were then stored at 4°C until analysis by flow cytometry (BD FACSCanto II) within 24 h. Samples were run at a low flow rate until 10,000 platelet positive events were counted. To account for spectral overlap between the three fluorophores, single‐stained compensation beads were used (BD Biosciences). For both MPAs and platelet surface receptor binding, samples were incubated at room temperature with the exception of tubes containing AA and collagen, which were incubated at 37°C using a dry block heater (Ratek DBH20D, Victoria, Australia).
Flow‐mediated dilation
The vascular assessments were conducted in a quiet, temperature‐controlled room in accordance to recent guidelines (Thijssen et al. 2011). To examine brachial artery FMD, the nondominant arm was extended and positioned at an angle of ~80° from the torso. A rapid inflation and deflation pneumatic cuff (D.E. Hokanson, Bellevue, WA) was positioned on the forearm, immediately distal to the olecranon process to provide a forearm ischemia stimulus. Using this approach, the brachial artery dilation represents a largely NO‐mediated, endothelium‐dependent response (Green et al. 2011). A 10‐MHz multifrequency linear array probe, attached to a high‐resolution ultrasound machine (T3200; Terason, Burlington, MA) was used to image the brachial artery in the distal 1/3rd of the upper arm. When an optimal image was obtained, the probe was held stable and the ultrasound parameters were set to optimize the longitudinal, B‐mode images of lumen–arterial wall interface. Continuous Doppler velocity assessments were also obtained using the ultrasound, and were collected using the lowest possible insonation angle (always <60°). Following a 1‐min baseline recording of brachial artery diameter and velocity (Camtasia Studio 8, TechSmith, Okemos, MI), which were used to examine baseline blood flow patterns, the forearm cuff was inflated (220 mmHg) for 5 min. Diameter and flow recordings resumed 30s prior to cuff deflation and continued for 3 min thereafter. Post‐test analysis of brachial artery diameter was performed using custom‐designed edge‐detection and wall‐tracking software, which is largely independent of investigator bias (Woodman et al. 2001). Brachial artery FMD is presented as relative (%) rise from the preceding baseline diameter. We have shown that the reproducibility of diameter measurements using this semiautomated software is significantly better than manual methods, reduces observer error significantly, and possesses an intraobserver CV of 6.7%.
Statistical methods
To test whether there was a relationship between FMD and platelet function, a Pearson product‐moment correlation test was carried out for data that were normally distributed. For data that were not normally distributed, a Spearman's Rank test was conducted. Subsequently, to determine if potential confounding factors including: age, gender, resting heart rate and blood pressure, body fat percentage, fasting blood glucose and lipids, and medication use impacted upon the results, partial correlations were conducted to account for these covariates. Participants were then divided into two groups based on gender, and correlation analysis between FMD and platelet function was conducted for males and females separately to determine if results were gender specific. To test whether any differences existed between male and female participants for FMD or any of the platelet function variables measured, independent sample T‐tests and Mann–Whitney U‐tests were conducted for data meeting and failing normality assumptions, respectively.
Results
Fifty‐one participants (18 male, 33 female) were included in the study and underwent platelet function and FMD tests. The general characteristics of participants included in the study are presented in Table 1. Two participants failed to receive a DEXA scan, so for data derived using this outcome measure n = 49. Descriptive statistics for vascular and platelet function outcome measures can be seen in Table 2. An error in the processing of platelet PAC‐1 and CD62P with no agonist occurred for 1 participant and for PAC‐1 and CD62P with AA incubation for 1 other participant. Therefore, for the direct comparison of all MPA data vs. FMD n = 51, but for PAC‐1 and CD62P NA and AA n = 50.
Table 1.
General characteristics, anthropometric, and dual‐energy x‐ray absorptiometry (DEXA) and biochemistry variables
| All participants | Male | Female | |
|---|---|---|---|
| Age (years) | 60.6 ± 7.4 | 57.1 ± 6.3 | 62.3 ± 7.6 |
| Anthropometric data | |||
| Height (cm) | 166.5 ± 8.0 | 173.8 ± 4.8 | 162.2 ± 6.0 |
| Body mass (kg) | 76.9 ± 15.8 | 89.8 ± 11.1 | 70.0 ± 13.0 |
| Body mass index (kg/m2) | 27.6 ± 4.5 | 29.8 ± 3.7 | 26.6 ± 4.6 |
| Total body fat % (DEXA) n = 49 | 39.5 ± 7.3 | 34.1 ± 1.2 | 42.4 ± 6.8 |
| Resting HR & blood pressure | |||
| Heart rate (bpm) | 62 ± 7 | ||
| Systolic BP (mmHg) | 123 ± 14 | 127 ± 12 | 121 ± 14 |
| Diastolic BP (mmHg) | 72 ± 9 | 79 ± 9 | 69 ± 6 |
| Mean arterial pressure (mmHg) | 92 ± 10 | 98 ± 9 | 89 ± 8 |
| Fasting Biochemistry (mmol/L) | |||
| Cholesterol | 5.6 ± 0.9 | 5.5 ± 1.1 | 5.6 ± 0.7 |
| Triglyceride | 1.2 ± 0.7 | 1.6 ± 0.6 | 1.1 ± 0.7 |
| LDL‐C | 3.6 ± 0.8 | 3.6 ± 0.9 | 3.6 ± 0.7 |
| HDL‐C | 1.4 ± 0.3 | 1.1 ± 0.2 | 1.5 ± 0.3 |
| Glucose | 5.1 ± 0.4 | 5.3 ± 0.1 | 5.0 ± 0.1 |
| Prescription Medication | N (dual meds) | ||
| Any medication | 12 (3) | 3 | 9 |
| Blood Pressure medication total | 6 (3) | 2 | 4 (3) |
| Ca channel block | 2 | ||
| Beta Blocker | 1 | ||
| Angiotensin II receptor antagonist | 3 | ||
| Statins | 6 (3) | 1 | 5 (3) |
| Antidepressant | 3 | 3 | |
Dual‐energy X‐ray absorptiometry DEXA, Heart rate HR, Blood pressure BP, Low‐density lipoprotein cholesterol LDL‐C, High‐density lipoprotein cholesterol HDL‐C. Values are Mean ± SD with exception of Prescription Medication presented as total N. N = 51 unless stated otherwise.
Table 2.
Descriptive statistics of FMD and platelet function tests, and results of correlation tests between FMD% and platelet function
| Mean ± SD | R | P | |
|---|---|---|---|
| FMD% | 4.6 ± 2.5 | ||
| BD (mm) | 3.6 ± 0.7 | ||
| PD (mm) | 3.8 ± 0.7 | ||
| Platelet Variable % | |||
| MPA NA | 4.1 ± 1.4 | 0.193 | 0.175 |
| MPA ADP1.5 μmol/L | 47.8 ± 16.8 | −0.128 | 0.369 |
| MPA AA10 μg/mL | 20.8 ± 25.3 | −0.122 | 0.396 |
| MPA Coll 1.5 μg/mL | 5.3 ± 5.5 | 0.136 | 0.34 |
| PAC‐1 NA (n = 50) | 4.7 ± 5.4 | −0.113 | 0.433 |
| PAC‐1 AA10 μg/mL (n = 50) | 23.3 ± 17.3 | −0.045 | 0.755 |
| PAC‐1 Coll 1.5 μg/mL | 22.3 ± 20.6 | 0.174 | 0.223 |
| CD62P NA (n = 50) | 1.8 ± 1.4 | −0.078 | 0.591 |
| CD62P ADP1.5 μmol/L | 58.7 ± 19.1 | −0.237 | 0.094 |
| CD62P AA10 μg/mL (n = 50) | 13.5 ± 11.8 | −0.142 | 0.324 |
| CD62P Coll 1.5 μg/mL | 8.8 ± 10.1 | −0.077 | 0.591 |
FMD, Flow‐mediated dilation, BD, baseline diameter, PD, peak diameter, Monocyte‐platelet aggregate MPA, No agonist NA, Adenosine diphosphate ADP, Arachidonic acid AA, Collagen Coll. N = 51 unless stated otherwise.
Relationships between FMD and platelet activation
No significant relationship was found between FMD% and circulating levels of activated platelets, whether measured by MPAs (Fig. 1), PAC‐1 (Fig. 2) or anti‐CD62P (Fig. 3) binding (all P > 0.05). No relationship was observed between FMD% and platelet reactivity to the agonists ADP (MPA & CD62P only), AA or collagen for MPAs, PAC‐1 or anti‐CD62P binding (all P > 0.05). All correlation results can be found in Table 2.
Figure 1.
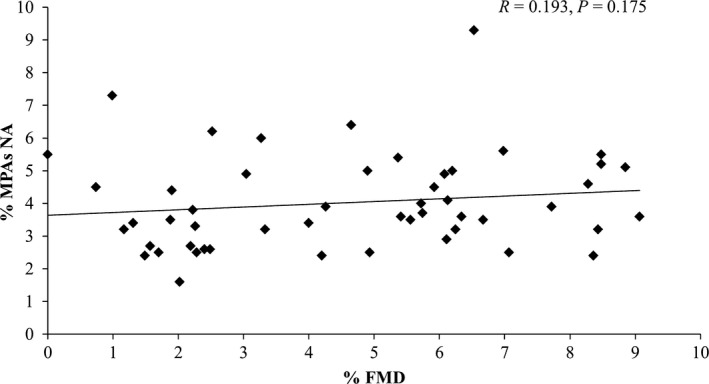
Individual responses for MPAs no agonist (NA) versus flow‐mediated dilation (FMD%) and results of correlation analysis. N = 51.
Figure 2.
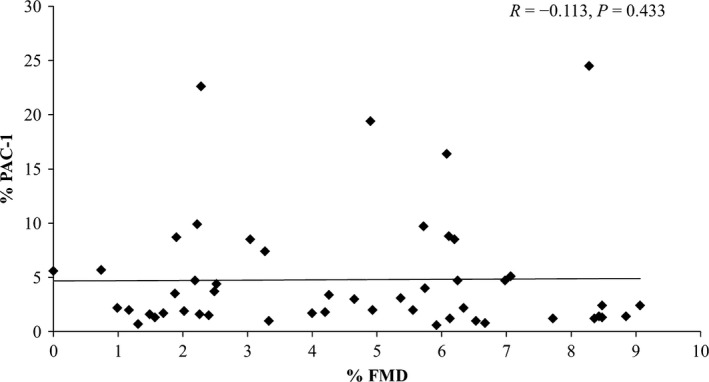
Individual responses for platelet PAC‐1 binding no agonist (NA) versus flow‐mediated dilation (FMD%) and results of correlation analysis. N = 50.
Figure 3.
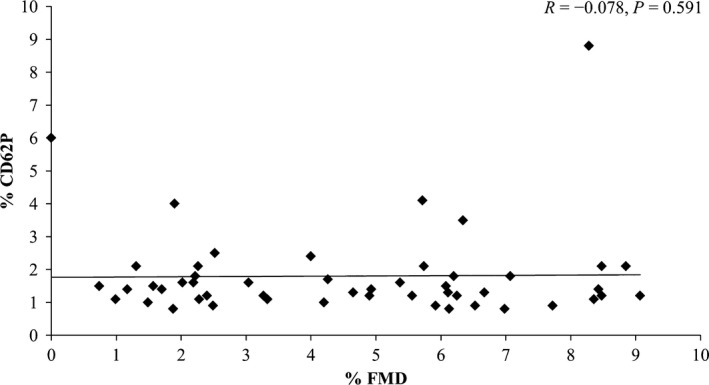
Individual responses for platelet anti‐CD62P binding no agonist (NA) versus flow‐mediated dilation (FMD%) and results of correlation analysis. N = 50.
Impact of potential confounding variables
Inspection of scatter plots suggested that no linear relationship existed between any of the potential confounding variables (age, gender, resting heart rate, blood pressure, % body fat, fasting glucose, and lipids (triglycerides, cholesterol, LDL, and HDL) and medication use) and FMD% or any of the platelet function parameters. Consequently, the results of partial correlation testing indicated that no significant relationship (all P > 0.05) existed between FMD% and any of the measures of platelet function we studied when these potentially confounding variables were included as cofactors (see Table 3).
Table 3.
Results of partial correlation tests between FMD% and platelet function accounting for the potential influence of covariates
| Platelet Variable | R | P |
|---|---|---|
| MPA NA | 0.119 | 0.484 |
| MPA ADP1.5 μmol/L | −0.071 | 0.675 |
| MPA AA10 μg/mL | 0.015 | 0.928 |
| MPA Coll 1.5 μg/mL | −0.091 | 0.592 |
| PAC‐1 NA (n = 50) | −0.075 | 0.664 |
| PAC‐1 AA10 μg/mL (n = 50) | −0.155 | 0.368 |
| PAC‐1 Coll 1.5 μg/mL | −0.005 | 0.975 |
| CD62P NA (n = 50) | 0.066 | 0.704 |
| CD62P ADP1.5 μmol/L | −0.246 | 0.143 |
| CD62P AA10 μg/mL (n = 50) | −0.091 | 0.598 |
| CD62P Coll 1.5 μg/mL | 0.034 | 0.844 |
FMD Flow‐mediated dilation, Monocyte‐platelet aggregate MPA, No agonist NA, Adenosine diphosphate ADP, Arachidonic acid AA, Collagen Coll. Covariates include: age, gender, body fat percentage, resting heart rate and blood pressure, fasting glucose and lipids and medication use. N = 51 unless stated otherwise.
Potential gender differences
Independent correlation analyses conducted for male and female participants individually revealed there were no significant relationships between FMD and any of the platelet function variables we measured for either gender (all P = >0.05). No significant differences were found between male and female participants for FMD (P = 0.767), MPA NA (P = 0.554), MPA ADP (P = 0.577), MPA AA (P = 0.419), MPA collagen (P = 0.608), PAC‐1 NA (P = 0.928), PAC‐1 AA (P = 0.524), PAC‐1 collagen (P = 0.150), CD62P NA (P = 0.146), CD62P ADP (P = 0.585), CD62P AA (P = 0.565), CD62P collagen (P = 0.086).
Discussion
Endothelial dysfunction and increased platelet activation are both associated with cardiovascular disease progression (Davì et al. 2002; Davignon J. 2004) and, based on in vitro and animal studies, it is often inferred that a direct and inverse relationship exists between the function of the endothelium and platelet activation (Alheid et al. 1987, 1989; Schäfer et al. 2004a,b). The aim of this study was to investigate the relationship between endothelial function and platelet activation in a preclinical low‐risk population of older participants with low physical activity levels. In this study, we did not find any relationship between flow‐mediated dilation, an indicator of endogenous nitric oxide and prostacyclin bioavailability (Green et al. 2011), and platelet function assessed by flow cytometry.
Our findings were unexpected, as the extant literature suggests there is an inverse relationship between platelet and endothelial function (Husain et al. 1998; Heitzer et al. 2003; Schäfer et al. 2004a,b; Hamilos et al. 2008, 2011; Warnholtz et al. 2008; Muller et al. 2010; Patti et al. 2011; Di Serafino et al. 2014). In contrast to our study, two previous investigations have reported an inverse relationship between vascular function and MPAs in high‐risk cardiac patients (Hamilos et al. 2008; Di Serafino et al. 2014). These studies utilized highly invasive vascular tests (coronary vasomotor function) and measured platelet activation in arterial blood samples. We tested endothelial function in the brachial artery following established guidelines, a common and reproducible approach (Thijssen et al. 2011; Mosawy et al. 2012). Platelet function was interrogated in venous blood samples with multiple outcomes (i.e., MPAs, PAC‐1 and anti‐CD62P binding), with and without canonical platelet agonists, using sophisticated and established flow cytometry techniques (Linden 2013) which have been associated with cardiovascular outcome. It is possible that differences in participant characteristics and/or methodological approaches between our study and these previous reports are responsible for the divergent findings. In critically ill patients with sepsis, MPAs in arterial blood were ~60% greater compared to MPAs in venous blood (Rondina et al. 2012), but to our knowledge no investigation has determined if sampling technique confers physiological or pathological relevance, or if such a difference exists in healthy individuals.
The majority of studies that have reported an inverse relationship between platelets and endothelial function have involved pharmacological modulation of these variables using acute experiments in humans (Husain et al. 1998; Heitzer et al. 2003; Schäfer et al. 2004b; Hamilos et al. 2008; Warnholtz et al. 2008; Muller et al. 2010; Patti et al. 2011). In one such study, platelet activation was increased by blockade of endothelium‐derived nitric oxide using a bolus dose of NG‐monomethyl‐L‐arginine (Schäfer et al. 2004b), whereas lower doses of blockade did not induce such changes (Albert et al. 1999). Likewise, increases in FMD have been observed following a single loading dose of clopidogrel and vasodilation responses postadministration were dose‐dependent (Warnholtz et al. 2008). Research in which a significant association was observed between platelet and endothelial function has involved participants with established coronary disease undergoing cardiac angiography and angioplasty. Such individuals exhibit large changes in platelet activation and endothelial function (Linden et al. 2007; Falk et al. 2013) and possess an inflammatory state (Libby et al. 2009) that may partly explain the inverse relationship observed. In contrast, there is some evidence to suggest upregulation of NO‐synthase‐mediated endothelial function occurs in the presence of CVD risk factors (Minor et al. 1990; Cosentino et al. 1997) and it is possible that, in subclinical and apparently healthy populations with risk factors for CVD, some compensatory redundancy exists in the interaction between the endothelium and platelets. This could account for the lack of an inverse relationship between these two variables in our study. Furthermore, there is some evidence to suggest that compensatory mechanisms that prevent spontaneous thrombosis and preserve hemostatic function occur in mice incapable of producing NO (Iafrati et al. 2005). Indeed, the percentage of circulating MPAs in our cohort (4.1 ± 1.4%) were far lower than that reported in samples taken from high‐risk cardiac (~20–38%) (Hamilos et al. 2011; Di Serafino et al. 2014) and stable CAD (~12%) patients (Linden et al. 2007), even after considering the potential differences in MPAs between arterial and venous blood (Rondina et al. 2012).
Our findings do not necessarily conflict with the established role of platelet and endothelial function in atherosclerosis, highlighted in recent reviews (Davì et al. 2002; McFadyen and Kaplan 2015; Cahill and Redmond 2016). However, because atherosclerosis evolves slowly over many decades in humans, it is possible that the relationship between endothelial and platelet function in healthy individuals is less apparent and/or detectable, even with the sensitive technical approaches used in this study. In contrast, transgenic mouse models provide a relatively accelerated and exaggerated insight into disease pathogenesis (Breslow 1996). Diabetic mice with lower NO bioavailability exhibit higher basal levels of activated platelets compared to control mice (Schäfer et al. 2004a), whereas mice with induced diabetes but preserved NO bioavailability due to overexpression of tetrahydrobiopterin possess normal levels of activated platelets. These findings suggest that in accelerated pathological models and/or in certain advanced disease states (e.g., CAD, diabetes mellitus), increased platelet activation and reduced endothelial NO may be mechanistically relevant. Although we deliberately recruited healthy middle‐aged and elderly individuals in this study, future research should include a wide range of participants, across the lifespan. A potential limitation to this study is the limited sample size, but previous studies which have reported a significant inverse relationship between FMD and platelet activation have included 30 (Di Serafino et al. 2014) and 19 (Hamilos et al. 2008) patients, so this is unlikely to be responsible for the lack of relationship in this study.
There are several limitations to our study. We constrained recruitment to >45 years and all women were postmenopausal. Our average FMD results indicate a relatively lower response than studies involving younger cohorts, in keeping with the established impact of age on vascular function. Nonetheless, we cannot generalize our findings to younger or much older age groups. Similarly, our study excluded participants with established cardiovascular diseases and, as mentioned above, correlations between platelet and endothelial function may be more apparent in those with overt disease. Finally, while some of our participants were taking medications, subgroup analysis revealed no impact of drugs on the magnitude or pattern of correlation in our study. Notably, all participants taking medications specifically known to impact upon platelet function were excluded from our study.
In summary, we did not observe any relationship between endothelial function and platelet activation in the healthy, middle‐aged, and elderly participants we recruited in this study. As our findings contrast with some previous studies conducted in high‐risk cardiac patients, our data suggest that some compensatory redundancy may exist in the relationship between platelet and endothelial function in preclinical populations.
Conflict of Interest
None.
Acknowledgments
The authors acknowledge the facilities, and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments.
Haynes A., Linden M. D., Robey E., Naylor L. H., Cox K. L., Lautenschlager N. T., Green D. J.. Relationship between monocyte‐platelet aggregation and endothelial function in middle‐aged and elderly adults, Physiol Rep, 5 (10), 2017, e13189, doi: 10.14814/phy2.13189
Funding Information
This work was supported by funding from the National Heart Foundation of Australia G12P6417 and National Health and Medical Research Council (NHMRC) APP1045204. Professor Green is a National Health and Medical Research Council Principal Research Fellow (APP1080914). Associate Professor Linden is an International Society for Advancement of Cytometry (ISAC) Marylou Ingram Scholar.
References
- Albert, J. , Wallen N. H., Nailin L., Frostell C., and Hjemdahl P.. 1999. Neither endogenous nor inhaled nitric oxide influences the function of circulating platelets in healthy volunteers. Clin. Sci. 97:345–353. [PubMed] [Google Scholar]
- Alheid, U. , Frölich J. C., and Förstermann U.. 1987. Endothelium‐derived relaxing factor from cultured human endothelial cells inhibits aggregation of human platelets. Thromb. Res. 47:561–571. [DOI] [PubMed] [Google Scholar]
- Alheid, U. , Reichwehr I., and Förstermann U.. 1989. Human endothelial cells inhibit platelet aggregation by separately stimulating platelet cyclic AMP and cyclic GMP. Eur. J. Pharmacol. 164:103–110. [DOI] [PubMed] [Google Scholar]
- Barnard, M. R. , Linden M. D., Frelinger A., Li Y., Fox M. L., Furman M. I., et al. 2005. Effects of platelet binding on whole blood flow cytometry assays of monocyte and neutrophil procoagulant activity. J. Thromb. Haemost. 3:2563–2570. [DOI] [PubMed] [Google Scholar]
- Breslow, J. L. 1996. Mouse models of atherosclerosis. Science 272:685. [DOI] [PubMed] [Google Scholar]
- Cahill, P. A. , and Redmond E. M.. 2016. Vascular endothelium–Gatekeeper of vessel health. Atherosclerosis 248:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosentino, F. , Hishikawa K., Katusic Z. S., and Lüscher T. F.. 1997. High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation 96:25–28. [DOI] [PubMed] [Google Scholar]
- Davì, G. , and Patrono C.. 2007. Platelet activation and atherothrombosis. N. Engl. J. Med. 357:2482–2494. [DOI] [PubMed] [Google Scholar]
- Davì, G. , Guagnano M. T., Ciabattoni G., Basili S., Falco A., Marinopiccoli M., et al. 2002. Platelet activation in obese women: role of inflammation and oxidant stress. JAMA 288:2008–2014. [DOI] [PubMed] [Google Scholar]
- Davignon J., Ganz P.. 2004. Role of endothelial dysfunction in atherosclerosis. Circulation 109:III–27‐III‐32. [DOI] [PubMed] [Google Scholar]
- Di Serafino, L. , Sarma J., Dierickx K., Ntarladimas I., Pyxaras S. A., Delrue L., et al. 2014. Monocyte‐Platelets Aggregates as Cellular Biomarker of Endothelium‐Dependent Coronary Vasomotor Dysfunction in Patients with Coronary Artery Disease. Cardiovasc. Transl. Res. 7:1–8. [DOI] [PubMed] [Google Scholar]
- Falk, E. , Nakano M., Bentzon J. F., Finn A. V., and Virmani R.. 2013. Update on acute coronary syndromes: the pathologists' view. Eur. Heart J. 34:719–728. [DOI] [PubMed] [Google Scholar]
- Gerdes, N. , Seijkens T., Lievens D., Kuijpers M. J., Winkels H., Projahn D., et al. 2016. Platelet CD40 Exacerbates Atherosclerosis by Transcellular Activation of Endothelial Cells and Leukocytes. Arterioscler. Thromb. Vasc. Biol. 36:482–490. [DOI] [PubMed] [Google Scholar]
- Green, D. J. , Jones H., Thijssen D., Cable N., and Atkinson G.. 2011. Flow‐mediated dilation and cardiovascular event prediction does nitric oxide matter? Hypertension 57:363–369. [DOI] [PubMed] [Google Scholar]
- Gurbel, P. A. , Becker R. C., Mann K. G., Steinhubl S. R., and Michelson A. D.. 2007. Platelet Function Monitoring in Patients With Coronary Artery Disease. J. Am. Coll. Cardiol. 50:1822–1834. [DOI] [PubMed] [Google Scholar]
- Hamilos, M. , Sarma J., Ostojic M., Cuisset T., Sarno G., Melikian N., et al. 2008. Interference of Drug‐Eluting Stents With Endothelium‐Dependent Coronary Vasomotion Evidence for Device‐Specific Responses. Circ. Cardiovasc. Interv. 1:193–200. [DOI] [PubMed] [Google Scholar]
- Hamilos, M. , Muller O., Ntalianis A., Trana C., Bartunek J., G. Sarno , et al. 2011. Relationship between peripheral arterial reactive hyperemia and residual platelet reactivity after 600 mg clopidogrel. J. Thromb. Thrombolysis 32:64–71. [DOI] [PubMed] [Google Scholar]
- Heitzer, T. , Ollmann I., Köke K., Meinertz T., and Munzel T.. 2003. Platelet glycoprotein IIb/IIIa receptor blockade improves vascular nitric oxide bioavailability in patients with coronary artery disease. Circulation 108:536–541. [DOI] [PubMed] [Google Scholar]
- Hui, H. , Fuller K., Erber W. N., and Linden M. D.. 2015. Measurement of monocyte‐platelet aggregates by imaging flow cytometry. Cytometry Part A 87:273–278. [DOI] [PubMed] [Google Scholar]
- Husain, S. , Andrews N. P., Mulcahy D., Panza J. A., and Quyyumi A. A.. 1998. Aspirin improves endothelial dysfunction in atherosclerosis. Circulation 97:716–720. [DOI] [PubMed] [Google Scholar]
- Iafrati, M. D. , Vitseva O., Tanriverdi K., Blair P., Rex S., Chakrabarti S., et al. 2005. Compensatory mechanisms influence hemostasis in setting of eNOS deficiency. Am. J. Physiol. Heart Circ. Physiol. 288:H1627–H1632. [DOI] [PubMed] [Google Scholar]
- Lanza, G. A. , Scalone G., Barone L., Infusino F., Coviello I., A. Di Monaco , et al. 2011. Platelet reactivity and endothelial function in children of patients with early acute myocardial infarction. Eur. Heart J. 32:2042–2049. [DOI] [PubMed] [Google Scholar]
- Libby, P. 2013. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 368:2004–2013. [DOI] [PubMed] [Google Scholar]
- Libby, P. , Ridker P. M., and Hansson G. K.. 2009. Leducq Transatlantic Network on A. Inflammation in atherosclerosis: from pathophysiology to practice. Journal of the American College of Cardiology 54:2129–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby, P. , Nahrendorf M., and Swirski F. K.. 2013. Monocyte heterogeneity in cardiovascular disease. Sem. Immunopathol 35:553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lievens, D. , and von Hundelshausen P.. 2011. Platelets in atherosclerosis. Thromb. Haemost. 106:827–838. [DOI] [PubMed] [Google Scholar]
- Linden, M. D . 2013. Platelet flow cytometry. Haemostasis: Methods and Protocols 241–262. [DOI] [PubMed] [Google Scholar]
- Linden, M. D. , Furman M. I., Frelinger A., Fox M. L., Barnard M. R., Li Y., et al. 2007. Indices of platelet activation and the stability of coronary artery disease. J. Thromb. Haemost. 5:761–765. [DOI] [PubMed] [Google Scholar]
- Macdonald, P. , Read M., and Dusting G.. 1988. Synergistic inhibition of platelet aggregation by endothelium derived relaxing factor and prostacyclin. Thromb. Res. 49:437–449. [DOI] [PubMed] [Google Scholar]
- McFadyen, J. D. , and Kaplan Z. S.. 2015. Platelets are not just for clots. Transfus. Med. Rev. 29:110–119. [DOI] [PubMed] [Google Scholar]
- Megson, I. L. , Sogo N., Mazzei F. A., Butler A. R., Walton J. C., and Webb D. J.. 2000. Inhibition of human platelet aggregation by a novel S‐nitrosothiol is abolished by haemoglobin and red blood cells in vitro: implications for anti‐thrombotic therapy. Br. J. Pharmacol. 131:1391–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson, A. D. , Barnard M. R., Krueger L. A., Valeri C. R., and Furman M. I.. 2001. Circulating monocyte‐platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P‐selectin studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation 104:1533–1537. [DOI] [PubMed] [Google Scholar]
- Minor, R. L. Jr , Myers P. R., Guerra R. Jr, Bates J. N., and Harrison D.. 1990. Diet‐induced atherosclerosis increases the release of nitrogen oxides from rabbit aorta. J. Clin. Invest. 86:2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosawy, S. , Jackson D. E., Woodman O. L., and Linden M. D.. 2012. Inhibition of platelet‐mediated arterial thrombosis and platelet granule exocytosis by 3′,4′‐dihydroxyflavonol and quercetin. Platelets 24:594–604. [DOI] [PubMed] [Google Scholar]
- Muller, O. , Hamilos M., Bartunek J., Ulrichts H., Mangiacapra F., Holz J.‐B., et al. 2010. Relation of endothelial function to residual platelet reactivity after clopidogrel in patients with stable angina pectoris undergoing percutaneous coronary intervention. Am. J. Cardiol. 105:333–338. [DOI] [PubMed] [Google Scholar]
- Ostad, M. A. , Nick E., Paixao‐Gatinho V., Schnorbus B., R. Schiewe , Tschentscher P., et al. 2011. Lack of evidence for pleiotropic effects of clopidogrel on endothelial function and inflammation in patients with stable coronary artery disease: results of the double‐blind, randomized CASSANDRA study. Clin. Res. Cardiol. 100:29–36. [DOI] [PubMed] [Google Scholar]
- Patti, G. , Grieco D., Dicuonzo G., Pasceri V., Nusca A., and G. Di Sciascio . 2011. High versus standard clopidogrel maintenance dose after percutaneous coronary intervention and effects on platelet inhibition, endothelial function, and inflammation: Results of the ARMYDA‐150 mg (antiplatelet therapy for reduction of myocardial damage during angioplasty) randomized study. J. Am. Coll. Cardiol. 57:771–778. [DOI] [PubMed] [Google Scholar]
- Radomski, M. , Palmer R., and Moncada S.. 1987. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet 330:1057–1058. [DOI] [PubMed] [Google Scholar]
- Rondina, M. T. , Grissom C. K., Men S., Harris E. S., Schwertz H., Zimmerman G. A., et al. 2012. Whole blood flow cytometry measurements of in vivo platelet activation in critically‐Ill patients are influenced by variability in blood sampling techniques. Thromb. Res. 129:729–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondina, M. T. , Weyrich A. S., and Zimmerman G. A.. 2013. Platelets as cellular effectors of inflammation in vascular diseases. Circ. Res. 112:1506–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer, A. , Alp N. J., Cai S., Lygate C. A., Neubauer S., M. Eigenthaler , et al. 2004a. Reduced vascular NO bioavailability in diabetes increases platelet activation in vivo. Arterioscler. Thromb. Vasc. Biol. 24:1720–1726. [DOI] [PubMed] [Google Scholar]
- Schäfer, A. , Wiesmann F., Neubauer S., Eigenthaler M., Bauersachs J., and Channon K. M.. 2004b. Rapid regulation of platelet activation in vivo by nitric oxide. Circulation 109:1819–1822. [DOI] [PubMed] [Google Scholar]
- Sesso, H. D. , Paffenbarger R. S., and Lee I.‐M.. 2000. Physical activity and coronary heart disease in men the Harvard Alumni Health Study. Circulation 102:975–980. [DOI] [PubMed] [Google Scholar]
- Simon, D. I. , Stamler J. S., Loh E., Loscalzo J., Francis S. A., and Creager M. A.. 1995. Effect of nitric oxide synthase inhibition on bleeding time in humans. J. Cardiovasc. Pharmacol. 26:339. [DOI] [PubMed] [Google Scholar]
- Thijssen, D. H. , Black M. A., Pyke K. E., Padilla J., Atkinson G., Harris R. A., et al. 2011. Assessment of flow‐mediated dilation in humans: a methodological and physiological guideline. Am. J. Physiol. Heart Circ. Physiol. 300:H2–H12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnholtz, A. , Ostad M. A., Velich N., Trautmann C., Schinzel R., Walter U., et al. 2008. A single loading dose of clopidogrel causes dose‐dependent improvement of endothelial dysfunction in patients with stable coronary artery disease: results of a double‐blind, randomized study. Atherosclerosis 196:689–695. [DOI] [PubMed] [Google Scholar]
- Weyrich, A. S. , Elstad M. R., McEver R. P., McIntyre T. M., Moore K. L., Morrissey J. H., et al. 1996. Activated platelets signal chemokine synthesis by human monocytes. J. Clin. Invest. 97:1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, P. C. , Coffey M. J., Coles B., Sanchez S., Morrow J. D., Cockcroft J. R., et al. 2005. In vivo aspirin supplementation inhibits nitric oxide consumption by human platelets. Blood 106:2737–2743. [DOI] [PubMed] [Google Scholar]
- Woodman, R. J. , Playford D. A., Watts G. F., Cheetham C., C. Reed , Taylor R. R., et al. 2001. Improved analysis of brachial artery ultrasound using a novel edge‐detection software system. J. Appl. Physiol. 91:929–937. [DOI] [PubMed] [Google Scholar]
- Yuan, M. , Fu H., Ren L., Wang H., and Guo W.. 2015. Soluble CD40 ligand promotes macrophage foam cell formation in the etiology of atherosclerosis. Cardiology 131:1–12. [DOI] [PubMed] [Google Scholar]