

Abstract
Deoxyribonuclease II (DNase II) is also known as acid deoxyribonuclease because it has optimal activity at the low pH environment of lysosomes where it is typically found in higher eukaryotes. Interestingly, DNase II has also been identified in a few genera of bacteria and is believed to have arisen via horizontal transfer. Here, we demonstrate that recombinant Burkholderia thailandensis DNase II is highly active at low pH in the absence of divalent metal ions, similar to eukaryotic DNase II. The crystal structure of B. thailandensis DNase II shows a dimeric quaternary structure which appears capable of binding double-stranded DNA. Each monomer of B. thailandensis DNase II exhibits a similar overall fold as phospholipase D (PLD), phosphatidylserine synthase (PSS) and tyrosyl-DNA phosphodiesterase (TDP), and conserved catalytic residues imply a similar mechanism. The structural and biochemical data presented here provide insights into the atomic structure and catalytic mechanism of DNase II.
INTRODUCTION
Deoxyribonuclease (DNase) enzymes perform a variety of important cellular roles by degrading DNA via hydrolysis of its phosphodiester backbone. Deoxyribonuclease I (DNase I) enzymes cleave single or double-stranded DNA and require divalent metal ions to hydrolyze DNA yielding 3΄-hydroxyl and 5΄-phosphorylated products. In contrast, deoxyribonuclease II (DNase II) hydrolyzes DNA at low pH to yield 3΄-phosphorylated and 5΄-hydroxyl products (Figure 1A) (1). Strikingly, unlike many phosphoryl transfer enzymes such as other nucleases (2) and all nucleic acid polymerases (3), DNase II does not require divalent metal ions for catalysis (4,5). Although a crystal structure of human DNase I has been reported recently (6), no crystal structure has been reported for any DNase II.
Figure 1.

Endonuclease activity of DNase II. (A) Deoxyribonuclease II (DNase II) hydrolyzes DNA in a metal ion-independent fashion at low pH to yield 3΄-phosphorylated and 5΄-hydroxyl nucleotides. (B) Cleavage of plasmid DNA by purified recombinant B. thailandensis DNase II (BthDNase II) under a variety of different pH conditions. Each cleavage buffer contained 10 mM EDTA as a chelating agent to remove divalent cations. In the absence of BthDNase II no cleavage is observed at neutral pH (lane 1) or at pH 3 (lane 9), although the lower pH results in an extra band consistent with single stranded plasmid. Smeared DNA at pH 4–5 represents BthDNase II-mediated plasmid degradation which is not observed at pH 3 and 6–9.
DNase II belongs to a unique family of nucleases that is present in most eukaryotes. Although DNase II enzymes are widely distributed in mammalian tissues (7), they are most commonly found within lysosomes or phagolysosomes (8). The acidic pH found within the lysosome organelle makes it the perfect environment for the optimum enzymatic activity of DNase II, which is also referred to as acid deoxyribonuclease for this reason. Furthermore, generation of the 3΄-phosphorylated and 5΄-hydroxyl ends has been predicted to prevent ready integration of foreign DNA into the host genome since repair enzymes typically recognize 5΄-phosphorylated and 3΄-hydroxyl substrates (9,10). Although DNase II has been shown to have preference for runs of purines, the structure of DNA appears to be more important than the actual sequence for cleavage (11,12). This was elegantly demonstrated by early experiments in which the cleavage pattern of DNase II was altered in the presence of DMSO, which significantly affects the structure of DNA (11). It has also been demonstrated that DNase II cleaves single stranded DNA and can cleave stronger on one strand (due to presence of purines) than the other (13). Due to its cleavage preference, it was previously postulated that DNase II might have a narrow binding domain that could bind one strand with minimal interference from the other strand (13).
In humans and other vertebrates, DNase II appears to have undergone a gene duplication event resulting in two closely related genes dnase IIα and dnase IIβ. DNase IIα is expressed in all tissues and mutation of DNase IIα in mice results in an embryonic lethal phenotype as this enzyme plays a critical role in phagocyte-mediated apoptotic DNA degradation (14,15). DNase IIβ is also known as DNase II like acid DNase (DLAD) (4) and is expressed in only a few tissues such as the eye lens. It has been shown to have an essential function in clearing DNA from the lens, and dnase IIβ deficient mice readily develop cataracts (16,17). Apart from its obvious role in DNA degradation, DNase II was found to be essential for activating the Toll-like Receptor 9 (TLR9) by degradation of bacterial genomic DNA and the cleavage of CpG-A DNA molecules into smaller 11–12-mers (18). These cleaved products activate TLR9 signaling and in particular, activate bone marrow-derived dendritic cells (18). In another study, mice that do not express DNase II and type I IFN receptor (double deficient) succumb to inflammatory arthritis, systemic autoimmunity, and splenomegaly due in part to defective TLR9 signaling (19). Taken together, it is clear that apart from degrading engulfed DNA, DNase II plays an important role in activating endogenous TLR9.
Our prior work demonstrated that mutation of the Caenorhabditis elegans roundworm homolog of DNase II, Nuc-1, resulted in persistent apoptotic DNA within engulfing cells as well as accumulation of DNA in the ovaries and DNA within the gut lumen (1,20,21). Mutation or down-regulation of Drosophila DNase II also resulted in the accumulation of DNA within the ovaries and lead to a highly lethal and unexpected susceptibility to bacterial infection (1,21,22). The native human DNase II enzyme has been shown to exist as a single contiguous polypeptide with intra-chain disulfide bonds and a molecular weight of 45 kDa that is reduced to ∼37 kDa after removal of N-linked glycan (23). Expression of human DNAse II in dnase II−/− mouse embryonic fibroblasts confirmed its single chain structure and the importance of the leader-peptide, intra-chain disulfide bonds, and N-glycosylation for the generation of a fully active enzyme (24).
DNase II was identified in the genus Burkholderia as well as a few other bacterial species (25). Since some Burkholderia members are known human pathogens (26), we focused on this family of putative DNase II proteins. Here, we expressed and purified recombinant B. thailandensis DNase II (BthDNase II) which exhibits activity at pH 4–5 in the absence of divalent metal ions. We solved a 1.65 Å resolution crystal structure of BthDNase II, which is the first three-dimensional crystal structure of any DNase II. This structure provides insight into the catalytic mechanism and evidence that DNase II is a member of the same protein structural family as phospholipase D (PLD), phosphatidylserine synthase (PSS), tyrosyl-DNA phosphodiesterase (TDP) and the endonuclease Nuc from Salmonella typhimurium (‘bacterial nuclease’).
MATERIALS AND METHODS
Cloning of the putative Burkholderia thailandensis DNase II gene
The putative B. thailandensis dnase II gene is comprised of an open reading frame (ORF) of 1053 bp in size, encoding for 350 amino acids polypeptide with a predicted molecular weight of 38 699.73 Da and an estimated PI of 6.65 (Protein Calculator v3.3, http://www.scripps.edu/∼cdputnam/protcalc.html). The B. thailandensis DNase II ORF (GenBank accession code NC_007650 or CP000085 REGION: 462969..464021) was amplified by using B. thailandensis genomic DNA as the template and gene-specific primers. The specific primers used to amply the gene included sequences for a six-histidine tag and thrombin cleavage site positioned at carboxyl-terminus. The forward and reverse primer sequences were 5΄-ccatggccatctcgccgcgcgac and 5΄-ctcgagctaatggtgatggtgatgatggctcgggcgcggcac-cagttcggccggttcgtgcgtcttgc-3΄, respectively. Nco I and Xho I restriction sites (in bold font) were added to the end of the amplified products, at 5΄ and 3΄, respectively, for sub-cloning purposes. The PCR reaction mixture was heat denatured 5 min at 95°C and cycled for 30 s at 95°C, 30 s at 62°C and 1.5 min at 72°C for 30 cycles, with a final extension cycle of 7 min at 72°C. PCR-amplified products were separated using agarose and the expected band was gel purified (QIAEX II, Qiagen, Valencia, CA, USA) and cloned initially into pGEM-T Easy vector (Promega, Madison, WI, USA). Cloned fragments of the expected size were verified by DNA sequencing (SequiTherm EXCEL II, Epicentre, Madison, WI, USA). One clone with the expected B. thailandensis DNase II sequence was selected for sub-cloning purposes. After digestion of pGEM-T Easy plasmid with Nco I and Xho I, the excised fragments were separated and purified as above and sub-cloned into the bacterial expression vector pET15b (Novagen, San Diego, CA, USA) using the vector's Nco I and Xho I cloning sites. The recombinant plasmid was transformed into BL21 (DE3) pLysS Escherichia coli competent cells (Novagen, San Diego, CA, USA). After addition of extra amino acids residues, the predicted molecular weight should be 40 301.48 Da for the resulting 363 amino acid product, with a calculated PI of 7.00 (as described above).
Protein induction expression and western blot analyses
Colonies containing the gene of interest were selected by their single (pGEM-T ampicillin) or double (pET15b ampicillin plus pLysS chloramphenicol) antibiotic resistance and by performing single colony PCR to amplify cloned insert (by standard techniques). Selected colonies, of BL21(DE3) pLysS and C43(DE3)pLysS, harboring pET15b and pET25b(+) plasmids, respectively, were grown at 37 °C in LB broth, containing 50 μg/ml ampicillin and 34 μg/μl chloramphenicol. Once optical density reached ∼0.35 for B21(DE3)pLysS or 0.8–1 for C43(DE3)pLysS, 1 or 0.5 mM IPTG were added to the cultures, respectively. After of induction at 37 °C for 2, 4 and 6 h, bacterial cells were harvested by centrifugation, 5000 × g at 4°C for 5 min. Cell pellets were frozen at −80 °C or processed immediately, by resuspension in PBS containing 1× complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany), followed by sonication, applying 4 pulses of 10 s on ice. Protein-containing supernatants were cleared by centrifugation at 13 000 × g for 10 min at 4 °C. Induced bacterial protein extracts (100 μg) were boiled with 1× loading sample buffer for 2–5 min and separated by SDS-PAGE essentially as previously described (27), using 12% resolving and 5% staking acrylamide gel. Separated proteins were stained with Coomassie Brilliant Blue to determine protein expression or transferred unstained for western blot analysis. Pre-stained protein markers (6–175 kDa, New England Biolabs, Ipswich, MA, USA) were used as molecular mass standards. After electrophoresis, the separated proteins were transferred from the gels onto a polyvinylidene fluoride (PVDF) microporous membrane (Pierce, Rockford, IL, USA) and blocked overnight at 4°C using 5% (w/v) bovine serum albumin (BSA) dissolved in Tris-buffered saline (TBS). The blots were then incubated for 1 h at room temperature with mouse anti-histidine monoclonal antibodies (Qiagen, Valencia, CA, USA) diluted 1:2000 in blocking solution, followed by three 15 min washes at room temperature TBS-Tween (0.1%). Bound antibody was detected by incubation for 1 h with goat anti-mouse HRP-conjugated antibodies diluted 1:3000 in blocking solution. Prior to development, an excess of secondary antibody was removed by washing the blots as previously described. The signal from antibody-reacting protein bands was subsequently visualized with chemiluminescent substrate (SuperSignal West Pico. Pierce, Rockford, IL, USA) and detected by exposure to x-ray film (BioMax. Kodak, Rochester, NY, USA).
Protein purification for enzymatic activity assays
Bacterial cell lysates containing over-expressed His-tagged BthDNase II proteins were selected from western blot results for further purification using affinity chromatography. Nickel columns (Ni-NTA, Qiagen, Valencia, CA) were loaded with 100 μl of cleared protein extract according to the manufacturer's instructions for native conditions. Bacterial crude protein extract, flow through, 50 and 250 mM imidazole elution samples were individually collected by centrifugation and used for SDS-PAGE and western blot as described above. Fractions eluted from the columns containing the recombinant protein were dialyzed overnight at 4°C against dialysis buffer (PBS containing 10% glycerol and 100 μM PMSF), using dialysis tubing with a molecular weight cut-off of 7000 Da (SnakeSkin Pierce, Rockford, IL) to remove salt excess and low molecular weight molecules.
Determining the enzymatic activity of the recombinant BthDNase II protein
Plasmid degradation assays were used to examine the nuclease activity of BthDNase II protein at varying pH conditions (from pH 3 to 9) in the absence of divalent cations. Sodium acetate (50 mM) was used for the preparation of cleavage buffers from pH 3–6 while Tris-base (10 mM) was used to prepare the pH 7–9 buffers. Each reaction mixture contained 10 mM EDTA to remove divalent cations. Additionally, 500 ng of plasmid DNA (pEGFP-N1, Clontech. Mountain View, CA) was added to each individual reaction. After addition of partially purified BthDNase II, the reaction mixtures (10 μl) were incubated at 37°C for 30 min. Cleavage reactions were terminated by addition of 2 μl of 6× loading buffer (0.25% bromophenol blue, 0.25% xylene cyanol, 30% glycerol, 50% formamide, 1 mM EDTA and 20 mM MOPS) and placed on ice. Reaction products were separated by agarose (1%) gel electrophoresis in 40 mM Tris–acetate–EDTA, pH 8.3 (1× TAE). Plasmid DNA integrity was determined by staining with ethidium bromide and visualized using a UV-light trans-illuminator (Alpha Innotech, San Leandro, CA). To examine metal ion-based inhibition of BthDNase II, enzyme in 50 mM acetate buffer pH 5.0 containing 1 mM of each divalent cation (as chlorides) was incubated for 5 min at room temperature. After this incubation, 0.5 μg of plasmid DNA was added and incubated for additional 30 min at 37°C. Digestion reactions were terminated by addition of 2 μl of loading buffer (0.25% bromophenol blue, 0.25% xylene cyanol, 30% glycerol, 50% formamide, 1 mM EDTA and 20 mM MOPS) and placed on ice. Plasmid DNA in acetate buffer with 10 mM EDTA without the addition of BthDNase II was used as negative control, whereas plasmid DNA in acetate buffer added with 10 mM EDTA and BthDNase II was used as positive control. Digestion reaction products were separated by 1% (w/v)-agarose-gel electrophoresis in Tris/acetate/EDTA buffer and stained with Ethidium bromide.
Expression and purification of BthDNase II for structure determination
The putative deoxyribonuclease II gene BTH_II0389 from B. thailandensis (UniProt Q2T8B0, target database ID: ButhA.18065.a) was cloned and expressed using general methods described previously (28). Briefly, the full-length BthDNase II sequence (residues 1–350) was PCR-amplified from B. thailendensis E264 gDNA using the primers: 5΄-CTCACCACCACCACCACCATATGGCCATCTCGCCGCGCGA and 5΄-ATCCTATCTTACTCACTTATTCGGCCGGTTCGTGCGTCTT. The gel purified PCR product was inserted into a BG1861 expression vector encoding a T7 promoter and a non-cleavable N-terminal affinity tag using ligation-independent cloning. The full sequence of the expression tag is MAHHHHHH followed by the 350-residues BthDNase II. The protein was expressed in BL21(DE3)R3 Rosetta Oxford E. coli cells as described previously in auto-induction media. BthDNase II protein for crystallography was purified using immobilized metal affinity chromatography followed by size exclusion chromatography. The expression tag was not removed prior to crystallography. The final protein sample was concentrated to 26.5 mg/ml in 20 mM HEPES (pH 7.0), 300 mM NaCl, 2 mM DTT and 5% (v/v) glycerol.
Crystallization and structure determination
Crystals of BthDNase II grew from a solution containing 0.4 μl of protein at 26.5 mg/ml and 0.4 μl of precipitant equilibrated against 80 μl of precipitant (100 mM BisTris pH 6.5, 28% (w/v) PEG MME 2000). A single crystal was cryo-protected with 15% (v/v) ethylene glycol, vitrified in liquid nitrogen, and yielded the high-resolution native data set at the ALS beamline 8.2.2 using a ADSC Quantum Q315R detector. For phasing, a crystal grown in 100 mM BisTris pH 6.0, 25% (w/v) PEG 3350 was incubated for 30 s with a reservoir solution supplemented with 750 mM NaI and 15% (v/v) ethylene glycol, vitrified, and yielded the iodide data set collected on a Rigaku FR-E+ Superbright generator and a Rigaku Saturn 944+ detector. All data sets were reduced with the XDS package (29). For the iodide data set Friedel mates were kept separate. To solve the phase problem, Phenix.hyss (30) could find eight iodide sites in the anomalous data set. Phaser_EP (31) extended the iodide substructure to 29 sites with a FOM of 0.42 for the initial phases. The phases were improved with solvent flattening, histogram matching and NCS averaging with PARROT (32) which improved the FOM to 0.75. An initial model was built automatically using ARP/wARP (33) followed by iterative cycles of manual model building in Coot (34) and refinement in Phenix (30). The structures were validated with MolProbity (35). Diffraction images are available at http://proteindiffraction.org.
Docking experiments
Docking of single-stranded and double-stranded DNA to the BthDNase II crystal structure was performed using NPDock (36) and HADDOCK (37). Ideal B-form double stranded DNA was generated in Coot, and one strand of this was used for the single-strand docking experiments. Docking of single chain DNase with single chain 9-mer DNA was first performed with HADDOCK2.2 using residues 100 and 279 of protein chain A and DNA nucleotide 5 as active residues (directly involved in the interaction). Passive residues were defined automatically around the active residues. We also attempted four additional experiments. First, 2-body docking of the protein dimer and dsDNA, each renumbered as single chains, using ambiguous restraints from protein chain A positions 100 102 279 281 (and equivalent positions in chain B) to DNA chain A nucleotide 5 (and equivalent position 14 in renumbered chain B), including center of mass restraints to prevent separation of monomers. Second, multi-body docking with C2 symmetry restraints treating each chain as separate molecules (same restraints as above). This failed due to chain A and chain B not being perfectly symmetrical (2718 atoms versus 2707). Third, we repeated this with unambiguous restraints. Finally, we repeated the experiment with a shorter DNA of only five nucleotides in length.
RESULTS
Genomics and biochemical analysis
In earlier work, we hypothesized that DNase II co-evolved with phagocytosis to degrade engulfed DNA within the acidic environment of phagolysosomes and this hypothesis was strengthened by the absence of this gene in many eukaryotic lineages that have lost phagocytic function such as in obligate autotrophs, intracellular parasites and saprophytes (8). Phylogenetic analyses have shown that dnase II genes are found in most metazoans and all vertebrates (8,25). However, one interesting finding from these phylogenetic studies was the identification of a DNase II-like gene in a single prokaryotic lineage, three members of the Gram-negative Burkholderia species B. thailandensis, B. pseudomallei and B. oklahomensis; additional microbial genes were identified after that study in other organisms (to be published). The DNase II proteins from these three organisms exhibit roughly 90% sequence identity between them and 28% sequence identity to human DNase IIα (Supplementary Figure S1). Unlike eukaryotic DNase II, Burkholderia DNase II does not contain an N-terminal secretion signal, indicating that the protein is not secreted but rather retained within the prokaryotic organism. Two independent phylogenetic studies concluded that it was highly likely that the presence of a dnase II gene in the soil-dwelling Burkholderia bacteria was most likely due to horizontal gene transfer from a eukaryote (8,25).
To demonstrate that the putative Burkholderia dnase II ORF encodes an active enzyme, we cloned the ORF from genomic DNA then expressed and purified BthDNase II to determine its enzymatic potential. The purified recombinant BthDNase II protein was found to be capable of degrading plasmid DNA at low pH and in the absence of divalent metal ions (Figure 1B). Activity at this pH in the presence of EDTA is unlikely to be the result of contaminating nucleases because the expression host E. coli does not contain a dnase II gene and other nucleases are active at neutral pH and require divalent cations. BthDNase II did not appear to be active at neutral or high pH. The results presented here provide strong evidence that BthDNase II exhibits similar enzymatic characteristics as DNase II enzymes isolated from eukaryotes (1).
Overview of BthDNase II crystal structure
The crystal structure of full-length BthDNase II was solved at 1.65 Å resolution (Table 1). Experimental phases were obtained at 2.1 Å resolution using iodide ion soaking and SAD phasing (38). BthDNase II exhibits a homodimeric quaternary structure of mixed α/β secondary structure comprised of 9 α helices and 20 β strands (Figure 2A and Supplementary Figure S1). Each protomer consists of a larger domain and a dimerization region that intertwines with the other protomer and results in a buried surface area of 3070 Å2 at the dimer interface. The two chains are nearly identical with a Cα RMSD value of 0.48 Å, although we note some asymmetry between the two chains since chain B appears slightly better ordered in general than chain A (B-factor average of 16.2 Å2 versus 19.8 Å2 in chain A). The homodimeric quaternary structure resembles a U-shaped clamp with a central cavity that is ∼16–17 Å wide.
Table 1. X-ray diffraction data and structure determination statistics.
| Native | Iodide | Cu2+ bound structure | |
|---|---|---|---|
| Data collection | |||
| Beamline | ALS 8.2.2 | Rigaku SuperBright FR-E+ | APS 21 ID-G |
| Wavelength (Å) | 0.999995 | 1.5418 | 0.97856 |
| Data reduction | |||
| Space group | P21 | P21 | P21 |
| Unit cell | a = 51.16 Å, b = 61.75 Å, c = 102.60 Å, α = γ = 90°, β = 92.125° | a = 51.21 Å, b = 61.73 Å, c = 103.49 Å, α = γ = 90°, β = 92.454° | a = 51.55 Å, b = 61.07 Å, c = 104.47 Å, α = γ = 90°, β = 90.293° |
| Solvent content (%) | 40 | 40 | |
| V m (Å3/Da) | 2.04 | 2.06 | |
| Resolution (Å) | 50–1.65 (1.69–1.65)a | 50–2.10 (2.15–2.10) | 50–1.75 (1.80–1.75) |
| I/σ | 16.7 (2.1) | 14.1 (5.6) | 21.0 (4.0) |
| Completeness (%) | 98.7 (99.5) | 99.8 (98.8) | 99.7 (96.7) |
| R merge | 0.052 (0.523) | 0.076 (0.211) | 0.051 (0.322) |
| CC 1/2 | 99.8 (65.9) | 99.6 (93.9) | 99.9 (88.7) |
| Multiplicity | 2.5 (2.5) | 3.7 (3.2) | 5.4 (4.0) |
| Reflections | 76 021 (5620)b | 73 812 (5465)b | 65 444 (4675) |
| Mosaicity | 0.3 | 0.9 | 0.6 |
| Refinement | |||
| R | 0.156 | 0.149 | |
| R free | 0.187 | 0.181 | |
| r.m.s.d. bonds (Å) | 0.006 | 0.006 | |
| r.m.s.d. angles (°) | 0.886 | 0.882 | |
| Mean B-factors (Å2) | 19.3 | 24.0 | |
| Wilson B-factors (Å2) | 12.1 | 18.2 | |
| Validation | |||
| Ramachandran Favored (%) | 96.9 | 95.8 | |
| Ramachandran allowed (%) | 100 | 99.8 | |
| Molprobity (35) score | 1.21 | 1.31 | |
| Clash score | 1.66 | 2.09 | |
| PDB ID | 5I3E | 5UNB | |
aValues in parenthesis indicate the highest resolution shell. 20 shells were used in XSCALE.
bFriedel pairs were merged for the native data set and kept separate for the anomalous data in XSCALE.
Figure 2.
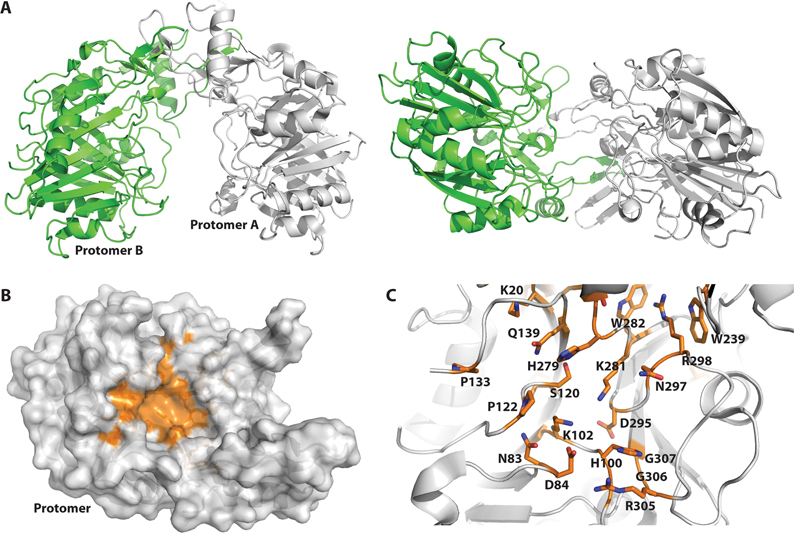
Crystal structure of B. thailandensis DNase II solved at 1.65 Å resolution. (A) Protomer A of the homodimeric BthDNase II structure is shown in gray ribbons and protomer B is shown in green ribbons either from the top at left or shown rotated 90° and viewed from the side at right. (B) Conserved residues are mapped onto the surface of the BthDNase II crystal structure and shown in orange (see supplemental Figure S1 for multiple sequence alignment). For simplicity, a single monomer of BthDNase II is shown viewed from the interior of the U-shaped clamp architecture. (C) Close-up of the active site, rotated slightly and zoomed in relative to panel B. Residues which are conserved across DNase II sequences are shown in sticks for their side chains and colored in CPK with orange carbon atoms. H100 and K102 comprise the first HxK catalytic motif whereas H279 and K281 comprise the second HxK catalytic motif.
Comparison of the BthDNase II crystal structure with other crystal structures in the Protein Data Bank via secondary structure matching (39) showed no other structures with RMSD values below 3 Å. DNase II was previously predicted to be a member of the phospholipase D (PLD) superfamily (40,41). Indeed, PLD from Streptomyces (42) is the top hit with an RMSD value of 3.09 Å versus BthDNase II. Sequence based alignment against Streptomyces PLD showed homology only over short stretches. PLD Based on secondary structure, there are a number of crystal structures with similar overall topology (RMSD ∼ 3.5 Å) from the PLD superfamily such as PSS, TDP and bacterial nuclease Nuc from which a homology model for human DNase II was previously predicted (40). Most of these do not perform well in sequence-based alignments, although human TDP contains 22% sequence identity and 40% sequence similarity with 16% gaps over 199 similar amino acids, representing <60% of the full-length proteins.
Active site analysis
Although BthDNase had not been enzymatically characterized previously, both pig DNase II (43–45) and human DNase IIα (24,46,47) have been characterized. Mutational analyses have shown that H113 and H295 of human DNase IIα are essential for activity, but H295 mutant enzymes could still interact with DNA (24). Each of these histidine residues are part of two ‘HKD’ motifs which form a pseudosymmetric active site and are hallmarks of the PLD family of enzymes (41). In BthDNase II, the equivalent residues are H100 for the first HKD motif and H279 for the second. The conserved lysine residues of the two HKD motifs are K102 and K281. As with other PLD superfamily enzymes, the BthDNase II aspartates D108 and D288 of the HKD motifs appear to be structurally important, and reside >15 Å away from the active site histidine residues. These motifs are also sometimes referred to as ‘HxK’ motifs because some PLD family members such as TDP1 contain a different amino acid than D at the equivalent position, which is not too surprising given the structural rather than functional importance of this residue (48,49). In fact, neither BthDNase II HKD motif exactly follows the standard H-x-K-(x)4-D pattern due to extra residues between the lysine and aspartate residues. Alignment of the DNase II enzymes from human, roundworms, and the three Burkholderia species (Supplementary Figure S1) highlights conserved residues which were mapped onto the surface of one molecule of the homodimeric BthDNase II crystal structure (Figure 2B). Both HxK motifs localize to the central cavity of the BthDNase II homodimer and cluster around other conserved residues (Figure 2C). One additional interesting feature conserved in DNase II enzymes appears to be the W239–W282–R298 interaction which appears to be a conserved structure feature that may be important for aligning H279 and K281 of the second HxK motif.
Many of the conserved active sites residues of DNase II enzymes overlap with those of other PLD family enzymes. Supplementary Table S1 (Supplemental information) provides equivalent catalytic residues among several different DNase and PLD family enzymes. In addition to the two HxK motifs, several other catalytic residues identified in Streptomyces PLD (Figure 3A) and human TDP1 appear to be conserved in DNase II and overlay in the BthDNase II crystal structure (Figure 3B).
Figure 3.

Comparison of B. thailandensis DNase II and phospholipase D family members. (A) Overlay of BthDNase II (gray ribbons and sticks representation) with phospholipase D from Streptomyces in complex with tungstate (magenta ribbons and sticks rendering with tungsten shown in cyan and inner sphere coordinated waters in red). (B) Overlay of BthDNase II with a ternary complex of human TDP1 (orange ribbons and sticks rendering), DNA (green carbon CPK sticks), vanadate (dark gray and red spheres) and peptide.
Inhibition by copper ions
Although other nucleases including DNase I (6) utilize divalent cations to hydrolyze DNA, DNase II does not require divalent metal ions and moreover is inhibited by zinc (50). We observed that BthDNase II was inhibited by copper ions and to a lesser extent manganese ions, but not inhibited by calcium, magnesium or zinc ions (Supplementary Figure S2). To understand this effect, we soaked a BthDNase II crystal overnight with 5 mM CuSO4 and obtained a 1.75 Å resolution crystal structure (Table 1, Figure 4A). The structure revealed the presence of a copper ion bound to H100 with one of the strongest electron density features in the structure as well as significant anomalous difference density (Figure 4A and B). This copper ion was modeled with a refined occupancy of 0.88. H100 forms an inner sphere coordinate bond to Cu2+ with a bond distance of 2.0 Å. H100 in BthDNase II is equivalent to one of the two catalytic histidine residues in human DNase II (H113) and to the catalytic histidine residue in PLD (H170) which forms a covalent intermediate with tungstate (42). H100 changes its rotamer conformation relative to the apo structure to make this interaction, and consequently, conserved residue R305 also changes its rotamer conformation (Figure 4B). Surprisingly, the region surrounding H100 was largely disordered in the second protomer and did not contain a bound copper ion off the largely disordered H100 in chain B of the structure (Figure 4A). Unlike the apo structure in which both protomers were nearly identical (Cα RMSD 0.48 Å), chain B of the Cu2+-bound structure is divergent for residues 99–101 which includes the catalytic H100 which binds Cu2+ in molecule A and for residues 119–128 (overall Cα RMSD increases to 0.68 Å, Figure 4C). A partially occupied Cu2+ ion was bound off His176 in both molecules of the BthDNase II homodimer near lattice contacts and was identified based on significant anomalous difference density (>4σ). These form outer sphere contacts, although the inner sphere water molecules are not all visualized. These interactions are more likely to be fortuitous rather than functional.
Figure 4.

Binding of Cu2+ to BthDNase II. (A) Overlay of the 1.65 Å resolution apo data set (chain A in gray, chain B in green) with the 1.75 Å resolution Cu2+ bound data set (chain A in dark gray, chain B in dark green, Cu2+ shown as copper colored spheres). Alignment was done via chain A. Anomalous maps are shown as copper colored mesh contoured at 4σ. (B) Active site of chain A showing movement of H100 upon binding Cu2+ along with movement of R305. Residues which move are labeled in gray and movement is shown with arrows. (C) Active site of chain B (via alignment of chain B) showing several loops which become disordered or change their conformation upon Cu2+ binding to H100 of chain A. Residues which move are labeled in gray and movement is shown with arrows.
DNA binding
Given that the interior of the U-shaped clamp BthDNase II structure is largely electropositive (Figure 5A), the size of the cavity correlates well with the width of double-stranded DNA (16–17 Å), and the cavity contains the catalytic HxK motifs, we hypothesize that DNase II binds double-stranded DNA through the central cavity. To determine an appropriate number of base-pairs for co-crystallization of a BthDNase II/dsDNA complex, we manually placed an ideal double-stranded DNA generated in Coot (34) within the central U-shaped cavity which guided the design of DNA constructs for co-crystallization efforts (Figure 5B). We generated a H279A mutant enzyme to prevent DNA hydrolysis and attempted co-crystallization with the six different dsDNA constructs but were unable to obtain a co-crystal structure. Future efforts may focus on H100/H279 double mutants.
Figure 5.

Hypothetical model for DNA recognition by BthDNase II. (A) Surface electrostatics of a single BthDNase II protomer viewed from the interior of the homodimeric U-shaped clamp-like structure. Catalytic residues K102, H279, and H281 are visible from this vantage point, whereas H100 is not. Additional positively charged residues which line the central cavity K20, K123 and K243 are labeled. (B) Hypothetical model for DNA recognition by BthDNase II generated via manual docking of an ideal B-form DNA into the central cavity of the BthDNase II homodimer.
To better understand how BthDNase II may interact with DNA, we performed docking experiments. When challenged with docking ideal B-form double-stranded DNA generated in Coot, both NPDock (36) and HADDOCK (37) docked the DNA to the exterior of the U-shaped homodiermic BthDNase II structure (Supplementary Figure S3). NPDock could dock the larger 11 base pair double-stranded DNA. HADDOCK failed with this length of DNA but succeeded with a 5 bp double-stranded DNA. Neither of these predictions appear likely since in both cases the nucleic acid resides far from the catalytic residues H100, K102, H279 and H281. Next, NPDock was challenged with a single protomer of BthDNase II and double stranded DNA. Here, NPDock aligns the phosphodiester backbone in close proximity to catalytic residues H100, K102, H279 and K281; however, this orientation significantly clashes with the second protomer from the homodimeric BthDNase II crystal structure (Supplementary Figure S3). NPDock produced two clusters separated by opposite polarity (5΄ to 3΄ and 3΄ to 5΄). Docking using HADDOCK was performed on a single monomer of BthDNase II with single stranded DNA derived from the same ideal double-stranded DNA used in NPDock. Docking a single stranded DNA taken from an idealized double-stranded DNA to BthDNase II while identifying H100, K102, H279 and K281 as residues involved in DNA recognition appeared to give a reasonable depiction of DNA recognition by the nuclease (Supplementary Figure S3). Again however, the docking resulted in two clusters with opposite polarity (5΄ to 3΄ and 3΄ to 5΄) but similar overall poses. These poses largely resemble that obtained by manual docking, although the distances are somewhat farther than expected from H100 for example, and may not allow binding of double-stranded DNA without clash in the current conformation.
To potentially identify local interactions, we also performed a least squares (LSQ) overlay of three central nucleotides of the ideal double-stranded DNA onto the three nucleotides in the TDP1–vanadate–DNA–peptide ternary complex (49). This may provide valuable information about local interactions in the active site, but the pitch of the double-stranded DNA appears to result in significant steric clash further away from the active site residues (Supplementary Figure S3). Combined, these overlays give some indication of how DNase II might interact with double-stranded DNA. However, nucleic acid-recognizing proteins often change their conformation in the bound state relative to the apo state; this may be a simple movement of the enzyme, or it could be a significant rearrangement as was the case for HCV polymerase (51).
DISCUSSION
The role of nucleases in a bacterial cell is not completely understood but it is believed that it assists microbial virulence and the scavenging of nucleotides from the environment. For example, it has been demonstrated that for Group A Streptococcus, the presence of extracellular bacterial DNase is a virulent factor contributing to disease progression, facilitating bacterial dissemination by evasion of the immune response (52). Furthermore, in vitro studies have demonstrated that extracellular DNases significantly impede the internalization of plasmid DNA during bacterial transformation (53–55). In addition, extracellular bacterial DNases potentially play a pivotal role as barriers to the acquisition of exogenous genetic material, which might be important in the prevention of infection or horizontal gene transfer. In the case of soil bacteria, it has been proposed that extracellular bacterial DNases can be considered as nutritional enzymes, facilitating the acquisition of DNA degradation products from their surrounding microbial environment as carbon and nitrogen sources (54). More recently, it was observed that Vibrio cholera grown at low density increase expression of DNases to obtain nucleotides from extracellular DNA to save metabolic energy necessary for de novo nucleotide synthesis (56). It is therefore possible that a soil microorganism, like B. thailandensis, uses a similar strategy to enhance its survival in a nutrient deficient environment.
Although DNase II enzymes are not similar to any known nuclease or other enzyme family of proteins, Cymerman et al. proposed that DNase II has features similar to those of the phospholipase D superfamily and a structure model was proposed using computational methods (40); however, apart from secondary structure similarities, there seemed to be little to support this view. Any alignments performed on DNase II against PLD sequences gave very low alignment scores, and much of the homology model generated by Cymerman et al. had to be aligned by hand. We therefore decided to determine the structure of a bacterial DNase II. Cloned from genomic DNA, the BthDNase II recombinant enzyme exhibits nuclease activity characteristic of other DNase II enzymes in that it is active at pH 4–5 and does not require divalent cations (Figure 1B). Secondary-structure matching of the BthDNase II crystal structure showed the highest match of Streptomyces PLD (RMSD 3.09 Å) and similar matches to other PLD family members PSS, Nuc, and TDP (RMSD ∼3.5 Å) provided structural evidence to support the hypothesis by Cymerman et al. (40). Furthermore, the known catalytic residues of each of the PLD family members align well with the known human DNase II catalytic residues in comparison with the BthDNase II active site. For example, the first HxK motif (H100 and K102) and the second HxK motif (H279 and K281) align well with the equivalent residues in other family members such as Streptomyces PLD and human TDP1 (Figure 3 and Supplementary Table S1).
In general, PLD family enzymes catalyze the hydrolysis of diverse phosphorylated substrates but all use the same general mechanism elucidated via capture of numerous states along the catalytic pathway (42). The first step of the two step mechanism involves nucleophilic attack of a catalytic histidine upon the phosphate of the substrate which generates a covalent intermediate and releases the leaving group. In the second step, the second catalytic histidine activates a water molecule which undergoes nucleophilic attack on the phosphate from the other face to release the first catalytic histidine and the product. Based on the known general reaction mechanism and structural overlays with other PLD family members, we can propose a catalytic mechanism for DNase II. First, H100 of BthDNase II undergoes nucleophilic attack on the phosphodiester backbone to generate a covalent intermediate and release the 5΄ hydroxyl-containing strand. Second, H279 activates a water molecule which attacks the phosphate from the opposite side as the H100 covalent bond to release H100 and the 3΄-phosphorylated DNA strand. Given the somewhat asymmetric nature of the apo dimer and the more significant asymmetry observed in our H100–Cu2+ bound structure, it is tempting to speculate that DNase II cleaves one strand of double-stranded DNA first before cleaving the second strand rather than both reactions centers cleaving the DNA at the same time. Comparison of our BthDNase II crystal structure with that of a vanadate intermediate of human TDP1 (49) allows for identification of other amino acids potentially involved in positioning the DNA for catalysis. For example, conserved N297 appears positioned to recognize the phosphate 5΄ to the scissile phosphate. Q302 may also be involved, although the equivalent residue in human DNase II is E318. K102 and K281 likely both coordinate the scissile phosphate which becomes the 3΄-phosphorylated DNA product. The enzyme may only weakly interact with the phosphate 3΄ of the scissile phosphate if at all. Residue 122 which is a N/Q/S in other PLD family members which catalyze cleavage of different substrates would appear in be in good position to interact with the phosphate 3΄ of the scissile phosphate; however, this residue is a conserved proline in DNase II, which may allow for release of the 5΄ hydroxyl-containing DNA product after the first step of the reaction. In general, our structure agrees well with the computational model for DNase II (40) based on the bacterial nuclease crystal structure (57) as well as a comparison to other PLD family member structures and mechanisms (42).
ACCESSION NUMBERS
Two macromolecular crystal structures: 5I3E and 5UNB.
Supplementary Material
ACKNOWLEDGEMENTS
We wish to thank the entire SSGCID team, David Smith at APS LS CAT for assistance in data collection, and David DeShazer at the United States Army Medical Research Institute of Infectious Diseases for the generous gift of B. thailandensis genomic DNA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of Allergy and Infectious Diseases, National Institute of Health, Department of Health and Human Services, under Federal Contract [HHSN272201200025C and HHSN272200700057C]; National Institute of General Medical Science (NIGMS) SCORE Grants [1SC3GM088069 and 1SC3GM103713 to R.J.A.]; The Biomolecule Analysis, Genomic Analysis and the Cytometry, Screening and Imaging Core Facilities that were used in this work were supported by the National Institute on Minority Health and Health Disparities (NIMHD) through a Research Centers for Minority Institutions grant [5G12MD007592]. Funding for open access charge: NIAID contract, although UTEP is the member institution.
Conflict of interest statement. None declared.
REFERENCES
- 1. Evans C.J., Aguilera R.J.. DNase II: genes, enzymes and function. Gene. 2003; 322:1–15. [DOI] [PubMed] [Google Scholar]
- 2. Yang W. Nucleases: diversity of structure, function and mechanism. Q. Rev. Biophys. 2011; 44:1–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steitz T.A. A mechanism for all polymerases. Nature. 1998; 391:231–232. [DOI] [PubMed] [Google Scholar]
- 4. Shiokawa D., Tanuma S.. DLAD, a novel mammalian divalent cation-independent endonuclease with homology to DNase II. Nucleic Acids Res. 1999; 27:4083–4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yamanaka M., Tsubota Y., Anai M., Ishimatsu K., Okumura M., Katsuki S., Takagi Y.. Purification and properties of acid deoxyribonucleases of human gastric mucosa and cervix uteri. J. Biol. Chem. 1974; 249:3884–3889. [PubMed] [Google Scholar]
- 6. Parsiegla G., Noguere C., Santell L., Lazarus R.A., Bourne Y.. The structure of human DNase I bound to magnesium and phosphate ions points to a catalytic mechanism common to members of the DNase I-like superfamily. Biochemistry. 2012; 51:10250–10258. [DOI] [PubMed] [Google Scholar]
- 7. Cordonnier C., Bernardi G.. A comparative study of acid deoxyribonucleases extracted from different tissues and species. Can. J. Biochem. 1968; 46:989–995. [DOI] [PubMed] [Google Scholar]
- 8. Shpak M., Kugelman J.R., Varela-Ramirez A., Aguilera R.J.. The phylogeny and evolution of deoxyribonuclease II: an enzyme essential for lysosomal DNA degradation. Mol. Phylogenet. Evol. 2008; 47:841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bergsmedh A., Ehnfors J., Kawane K., Motoyama N., Nagata S., Holmgren L.. DNase II and the Chk2 DNA damage pathway form a genetic barrier blocking replication of horizontally transferred DNA. Mol. Cancer Res. 2006; 4:187–195. [DOI] [PubMed] [Google Scholar]
- 10. Howell D.P., Krieser R.J., Eastman A., Barry M.A.. Deoxyribonuclease II is a lysosomal barrier to transfection. Mol. Ther. 2003; 8:957–963. [DOI] [PubMed] [Google Scholar]
- 11. Drew H.R., Travers A.A.. DNA structural variations in the E. coli tyrT promoter. Cell. 1984; 37:491–502. [DOI] [PubMed] [Google Scholar]
- 12. Drew H.R., Travers A.A.. Structural junctions in DNA: the influence of flanking sequence on nuclease digestion specificities. Nucleic Acids Res. 1985; 13:4445–4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drew H.R. Structural specificities of five commonly used DNA nucleases. J. Mol. Biol. 1984; 176:535–557. [DOI] [PubMed] [Google Scholar]
- 14. Krieser R.J., MacLea K.S., Longnecker D.S., Fields J.L., Fiering S., Eastman A.. Deoxyribonuclease IIalpha is required during the phagocytic phase of apoptosis and its loss causes perinatal lethality. Cell Death Differ. 2002; 9:956–962. [DOI] [PubMed] [Google Scholar]
- 15. Kawane K., Fukuyama H., Kondoh G., Takeda J., Ohsawa Y., Uchiyama Y., Nagata S.. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science. 2001; 292:1546–1549. [DOI] [PubMed] [Google Scholar]
- 16. Nishimoto S., Kawane K., Watanabe-Fukunaga R., Fukuyama H., Ohsawa Y., Uchiyama Y., Hashida N., Ohguro N., Tano Y., Morimoto T. et al. Nuclear cataract caused by a lack of DNA degradation in the mouse eye lens. Nature. 2003; 424:1071–1074. [DOI] [PubMed] [Google Scholar]
- 17. Nagai N., Takeuchi N., Kamei A., Ito Y.. Involvement of DNase II-like acid DNase in the cataract formation of the UPL rat and the Shumiya cataract rat. Biol. Pharm. Bull. 2006; 29:2367–2371. [DOI] [PubMed] [Google Scholar]
- 18. Chan M.P., Onji M., Fukui R., Kawane K., Shibata T., Saitoh S., Ohto U., Shimizu T., Barber G.N., Miyake K.. DNase II-dependent DNA digestion is required for DNA sensing by TLR9. Nat. Commun. 2015; 6:5853. [DOI] [PubMed] [Google Scholar]
- 19. Pawaria S., Moody K., Busto P., Nundel K., Choi C.H., Ghayur T., Marshak-Rothstein A.. Cutting edge: DNase II deficiency prevents activation of autoreactive B cells by double-stranded DNA endogenous ligands. J. Immunol. 2015; 194:1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lyon C.J., Evans C.J., Bill B.R., Otsuka A.J., Aguilera R.J.. The C. elegans apoptotic nuclease NUC-1 is related in sequence and activity to mammalian DNase II. Gene. 2000; 252:147–154. [DOI] [PubMed] [Google Scholar]
- 21. Mukae N., Yokoyama H., Yokokura T., Sakoyama Y., Nagata S.. Activation of the innate immunity in Drosophila by endogenous chromosomal DNA that escaped apoptotic degradation. Genes Dev. 2002; 16:2662–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seong C.S., Varela-Ramirez A., Aguilera R.J.. DNase II deficiency impairs innate immune function in Drosophila. Cell Immunol. 2006; 240:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. MacLea K.S., Krieser R.J., Eastman A.. Revised structure of the active form of human deoxyribonuclease IIalpha. Biochem. Biophys. Res. Commun. 2002; 292:415–421. [DOI] [PubMed] [Google Scholar]
- 24. MacLea K.S., Krieser R.J., Eastman A.. Structural requirements of human DNase II alpha for formation of the active enzyme: the role of the signal peptide, N-glycosylation, and disulphide bridging. Biochem. J. 2003; 371:867–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. MacLea K.S., Krieser R.J., Eastman A.. A family history of deoxyribonuclease II: surprises from Trichinella spiralis and Burkholderia pseudomallei. Gene. 2003; 305:1–12. [DOI] [PubMed] [Google Scholar]
- 26. Limmathurotsakul D., Golding N., Dance D.A., Messina J.P., Pigott D.M., Moyes C.L., Rolim D.B., Bertherat E., Day N.P., Peacock S.J. et al. Predicted global distribution of Burkholderia pseudomallei and burden of melioidosis. Nat. Microbiol. 2016; 1:15008. [DOI] [PubMed] [Google Scholar]
- 27. Miranda G.A., Chokler I., Aguilera R.J.. The murine nucleolin protein is an inducible DNA and ATP binding protein which is readily detected in nuclear extracts of lipopolysaccharide-treated splenocytes. Exp. Cell Res. 1995; 217:294–308. [DOI] [PubMed] [Google Scholar]
- 28. Choi R., Kelley A., Leibly D., Hewitt S.N., Napuli A., Van Voorhis W.. Immobilized metal-affinity chromatography protein-recovery screening is predictive of crystallographic structure success. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011; 67:998–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kabsch W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D.. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012; 68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Langer G., Cohen S.X., Lamzin V.S., Perrakis A.. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008; 3:1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 35. Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tuszynska I., Magnus M., Jonak K., Dawson W., Bujnicki J.M.. NPDock: a web server for protein-nucleic acid docking. Nucleic Acids Res. 2015; 43:W425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dominguez C., Boelens R., Bonvin A.M.. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003; 125:1731–1737. [DOI] [PubMed] [Google Scholar]
- 38. Abendroth J., Gardberg A.S., Robinson J.I., Christensen J.S., Staker B.L., Myler P.J., Stewart L.J., Edwards T.E.. SAD phasing using iodide ions in a high-throughput structural genomics environment. J. Struct. Funct. Genomics. 2011; 12:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krissinel E., Henrick K.. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2256–2268. [DOI] [PubMed] [Google Scholar]
- 40. Cymerman I.A., Meiss G., Bujnicki J.M.. DNase II is a member of the phospholipase D superfamily. Bioinformatics. 2005; 21:3959–3962. [DOI] [PubMed] [Google Scholar]
- 41. Schafer P., Cymerman I.A., Bujnicki J.M., Meiss G.. Human lysosomal DNase IIalpha contains two requisite PLD-signature (HxK) motifs: evidence for a pseudodimeric structure of the active enzyme species. Protein Sci. 2007; 16:82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leiros I., McSweeney S., Hough E.. The reaction mechanism of phospholipase D from Streptomyces sp. strain PMF. Snapshots along the reaction pathway reveal a pentacoordinate reaction intermediate and an unexpected final product. J. Mol. Biol. 2004; 339:805–820. [DOI] [PubMed] [Google Scholar]
- 43. Liao T.H. The subunit structure and active site sequence of porcine spleen deoxyribonuclease. J. Biol. Chem. 1985; 260:10708–10713. [PubMed] [Google Scholar]
- 44. Cheng Y.C., Hsueh C.C., Lu S.C., Liao T.H.. Identification of three crucial histidine residues (His115, His132 and His297) in porcine deoxyribonuclease II. Biochem. J. 2006; 398:177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang C.C., Lu S.C., Chen H.L., Liao T.H.. Porcine spleen deoxyribonuclease II. Covalent structure, cDNA sequence, molecular cloning, and gene expression. J. Biol. Chem. 1998; 273:17192–17198. [DOI] [PubMed] [Google Scholar]
- 46. Ohkouchi S., Shibata M., Sasaki M., Koike M., Safig P., Peters C., Nagata S., Uchiyama Y.. Biogenesis and proteolytic processing of lysosomal DNase II. PLoS One. 2013; 8:e59148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shiokawa D., Tanuma S.. Cloning of cDNAs encoding porcine and human DNase II. Biochem. Biophys. Res. Commun. 1998; 247:864–869. [DOI] [PubMed] [Google Scholar]
- 48. Davies D.R., Interthal H., Champoux J.J., Hol W.G.. The crystal structure of human tyrosyl-DNA phosphodiesterase, Tdp1. Structure. 2002; 10:237–248. [DOI] [PubMed] [Google Scholar]
- 49. Davies D.R., Interthal H., Champoux J.J., Hol W.G.. Crystal structure of a transition state mimic for Tdp1 assembled from vanadate, DNA, and a topoisomerase I-derived peptide. Chem. Biol. 2003; 10:139–147. [DOI] [PubMed] [Google Scholar]
- 50. Torriglia A., Chaudun E., Courtois Y., Counis M.F.. On the use of Zn2+ to discriminate endonucleases activated during apoptosis. Biochimie. 1997; 79:435–438. [DOI] [PubMed] [Google Scholar]
- 51. Appleby T.C., Perry J.K., Murakami E., Barauskas O., Feng J., Cho A., Fox D. 3rd, Wetmore D.R., McGrath M.E., Ray A.S. et al. Viral replication. Structural basis for RNA replication by the hepatitis C virus polymerase. Science. 2015; 347:771–775. [DOI] [PubMed] [Google Scholar]
- 52. Sumby P., Barbian K.D., Gardner D.J., Whitney A.R., Welty D.M., Long R.D., Bailey J.R., Parnell M.J., Hoe N.P., Adams G.G. et al. Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:1679–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dodd H.N., Pemberton J.M.. Cloning, sequencing, and characterization of the nucH gene encoding an extracellular nuclease from Aeromonas hydrophila JMP636. J. Bacteriol. 1996; 178:3926–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Focareta T., Manning P.A.. Distinguishing between the extracellular DNases of Vibrio cholerae and development of a transformation system. Mol. Microbiol. 1991; 5:2547–2555. [DOI] [PubMed] [Google Scholar]
- 55. Marcus H., Ketley J.M., Kaper J.B., Holmes R.K.. Effects of DNase production, plasmid size, and restriction barriers on transformation of Vibrio cholerae by electroporation and osmotic shock. FEMS Microbiol. Lett. 1990; 56:149–154. [DOI] [PubMed] [Google Scholar]
- 56. Blokesch M., Schoolnik G.K.. The extracellular nuclease Dns and its role in natural transformation of Vibrio cholerae. J. Bacteriol. 2008; 190:7232–7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stuckey J.A., Dixon J.E.. Crystal structure of a phospholipase D family member. Nat. Struct. Biol. 1999; 6:278–284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.