


Abstract
Accurate and efficient genotyping of simple sequence repeats (SSRs) constitutes the basis of SSRs as an effective genetic marker with various applications. However, the existing methods for SSR genotyping suffer from low sensitivity, low accuracy, low efficiency and high cost. In order to fully exploit the potential of SSRs as genetic marker, we developed a novel method for SSR genotyping, named as AmpSeq-SSR, which combines multiplexing polymerase chain reaction (PCR), targeted deep sequencing and comprehensive analysis. AmpSeq-SSR is able to genotype potentially more than a million SSRs at once using the current sequencing techniques. In the current study, we simultaneously genotyped 3105 SSRs in eight rice varieties, which were further validated experimentally. The results showed that the accuracies of AmpSeq-SSR were nearly 100 and 94% with a single base resolution for homozygous and heterozygous samples, respectively. To demonstrate the power of AmpSeq-SSR, we adopted it in two applications. The first was to construct discriminative fingerprints of the rice varieties using 3105 SSRs, which offer much greater discriminative power than the 48 SSRs commonly used for rice. The second was to map Xa21, a gene that confers persistent resistance to rice bacterial blight. We demonstrated that genome-scale fingerprints of an organism can be efficiently constructed and candidate genes, such as Xa21 in rice, can be accurately and efficiently mapped using an innovative strategy consisting of multiplexing PCR, targeted sequencing and computational analysis. While the work we present focused on rice, AmpSeq-SSR can be readily extended to animals and micro-organisms.
INTRODUCTION
Simple sequence repeats (SSRs), also known as short tandem repeats (STRs) or microsatellites, exist extensively in eukaryotic genomes. Most SSRs are non-coding and may affect gene expression, splicing, protein sequences and genome structures (1–5). The rate of SSR length mutations is estimated to be 10−7 to 10−3 per locus per generation in eukaryotes (6), which is much higher than the rate of ∼10−9 for base mutations (7,8) and accounts for the high diversity of SSRs. Despite the high variability of SSR sequences, their flanking regions are often conserved within the same species, occasionally among closely related species (9,10) and even across species (11,12). SSRs have several advantages over other genetic variations, including co-dominance, high reproducibility and requiring a small amount of template DNA for experiment (13–16). Importantly, the combination of the diversity of SSR sequences and the conservation of their flanking regions makes SSRs ideal genetic markers. Indeed, SSRs have been successfully adopted in various applications such as DNA fingerprinting, gene mapping, forensic analysis, marker assisted breeding and assessment of seed purity (16–20).
The most notable source of SSR diversity is the innate slippage of DNA polymerases during SSR replication (21–27). Such a slippage is also inherent in in vitro SSR polymerase chain reaction (PCR) amplification, which results in erroneous SSR alleles and makes accurate SSR genotyping difficult. Moreover, gel electrophoresis, the currently most popular approach to detecting SSR PCR products, is inaccurate due to its low resolution and is inefficient as it can only handle a small number of SSRs at a time (20). For example, only 28, 25 and 48 SSR loci are analyzed by gel electrophoresis and used to construct DNA fingerprints of jute (Corchorus spp.) (28), winter mushroom (Flammulina velutipes) (29) and pigeonpea (30), respectively. Such limited SSRs are not sufficient or robust to construct high-quality SSR fingerprints to discriminate close kinships. Whole genome resequencing can profile a large number of SSR loci at once (25,31,32). Nevertheless, since SSR sequences only constitute a small percentage of a whole genome, e.g. ∼3% of the human genome (33), whole genome resequencing dilutes the sequencing reads on SSRs with a huge number of other genomic reads, making it difficult to reach a required coverage of more than 10- to 100-folds (25) for accurate SSR genotyping with an acceptable cost. In addition, whole genome resequencing introduces additional problems to SSR genotyping, e.g. preferential amplification of specific SSR loci (34) and difficulties in data analysis with repeats in SSRs (35,36).
We developed a novel sequencing based SSR genotyping method by combining multiplex amplification of target SSRs, high-throughput sequencing of the amplicons (Ampli-Seq) and comprehensive computational and statistical analyses. For convenience, we call our new method AmpSeq-SSR. AmpSeq-SSR overcomes nearly all difficulties in the existing methods. In particular, AmpSeq-SSR is able to efficiently genotype a large number of SSR loci at once with ultra-deep coverages and has an accuracy close to 100% and a single-base resolution. We applied AmpSeq-SSR to genotyping a total of 3105 SSRs in eight rice varieties and validated all of them. We further demonstrated the utility and power of the new approach with two additional applications. The first is construction of rice fingerprints that contain 449 differential SSRs on average, which can accurately distinguish a variety under test. The second application is mapping of Xa21, a gene that confers a broad and persistent resistance against rice bacterial blight (BB), a devastating rice disease causing a substantial annual rice yield reduction worldwide. While the development of AmpSeq-SSR and our current work focus on rice, the new method is general and applicable to animal species and eukaryotic micro-organisms.
MATERIALS AND METHODS
Rice plants used
The eight homozygous rice varieties (A–H in Supplementary Table S1) are commercially released varieties in China and are representatives of indica and japonica rice plants. The three pairs of nearly isogenic lines (NILs, I-N in Supplementary Table S1) share similar genetic backgrounds except the Xa21 gene and its linkage regions.
Target SSRs and design of multiplex primers
Forty-eight SSRs that are listed in the National Agricultural Standard of China (NY/T 1433–3014) and 3057 randomly selected SSRs from the Japanica reference genome (irgsp1.0) were chosen as target SSRs (Supplementary Table S2, the online file http://www.cse.wustl.edu/∼zhang/SSR_ST2.pdf has the full list of primers). The service at https://ampliseq.com/ was used to design multiplex primers for 3105 target SSRs, which have amplicon lengths <250 bp on reference genome. The designed primers were synthesized by Thermo Company, USA.
Construction and high-throughput sequencing of Ampli-Seq libraries
We describe here the major steps for the construction and high-throughput sequencing of Ampli-Seq libraries. A step-by-step protocol is given in Supplementary Method. Whole plants at the first leaf stage were harvested for extraction of genomic DNA, following the protocol of E-Z 96® Mag-Bind® Plant DNA Kit (Cat. No. M1027, Omega bio-tek, USA). Varieties A-N in Supplementary Table S1 were used to construct Applied Biosystems (ABI) S5 Ampli-Seq libraries according to the user guide of Ion AmpliSeq™ Library Kit 2.0 (Cat No. 4475345, Thermo, USA). The resulting libraries were sequenced on S5 system using the single-end sequencing with a length of 300 bp. Varieties A–H in Supplementary Table S1 were also used to construct Illumina MiSeq Ampli-Seq libraries according to the user guide with modification. That is, additional PCR with 14 cycles were introduced for DNA amplification. The resulting libraries were sequenced on MiSeq system using the paired-end sequencing with a length of 2 × 300 bp. The library construction and sequencing for S5 and MiSeq systems were respectively performed in our lab and BestNovo Co., Ltd, China within a 30-day interval.
SSR genotyping by gel electrophoresis and Sanger sequencing
A 25 μl PCR reaction system was used, which includes 0.05 μM primer R and 0.05 μM primer F, 10 ng template DNA and 12.5 μl AmpliTaq Gold® 360 Master Mix (4398876, Applied Biosystems™, USA). The PCR reaction procedure is 95°C/5 min; 40 cycles of 95°C/30 s and 60°C/1 min; 72°C/25 min. The amplicons were sent for Sanger sequencing (TsingKe Company, China) or were separated by 2% agarose gel electrophoresis, 6% polyacrylamide gel electrophoresis (PAGE) or capillary electrophoresis (CE). The CE was performed on ABI 3500 Genetic Analyzer (Applied Biosystems, USA) and the fragments were analyzed using GeneMapper softerware v4.1. The major and minor alleles of an SSR of CE were defined as the lengths of the largest and the second largest bands on electropherogram. The stutter ratio of an SSR was estimated as the ratio of the areas between the second allele and the major alleles of the SSR.
Processing of sequencing reads and SSR genotype calling
Here, we described the rationale and criteria for processing sequencing reads from homozygous varieties for SSR profiling. The related scripts were provided in Supplementary materials. The methods for heterozygous varieties were provided in the last section of ‘Materials and Methods’ section.
Reads were mapped to the reference genome by Bowtie2 (37) to determine which target SSRs they belonged to. However, many reads were left unmapped because of the variations in SSR lengths. To resolve this problem, sub-sequences of 40 bp were taken from the unmapped reads by a scheme of a sliding window of 40 bp. The sub-sequences were then mapped to the reference genome by BLASTN to determine potential SSRs they belonged to. To avoid the interference of SSR length variations on mapping, the SSR sequences on the reference genome were replaced with ‘N’. After a read was located, the two boundaries of the SSR in the read were determined by aligning the read to the flanking sequences of the corresponding SSR on the reference genome (ref-SSR). Then, the tandem repeats of the ref-SSR motif between the two boundaries were determined as the SSR allele of the read.
The reads were tallied for each kind of allele at an SSR locus. The alleles with the largest and the second largest numbers of reads were designated as the major and minor alleles of the SSR locus, respectively. The major allele was also taken as the genotype because only a homozygous allele was expected for an SSR locus in the homozygous plants used in the current study. The ratio between the numbers of reads of the minor and major alleles was taken as the stutter ratio of the SSR locus.
To determine the amplicon length of a target SSR, the 20 bp upstream and downstream sequences of the amplicon on the reference genome were extracted and mapped to each sequencing read of the target SSR. The distance between the upstream and downstream sequences of each sequencing read was calculated and classified. The average distance in the class with the most reads plus 40 bp and the primer lengths was taken as the amplicon length of the target SSR, which corresponds to the amplicon length on electropherogram.
Estimation of AmpSeq-SSR accuracy and reproducibility
A valid SSR has a coverage of no less than a specified fold c and a stutter ratio no greater than a specified value s. For convenience, the specific genotyping condition of the valid SSRs is denoted as (c, s). A valid SSR is consistent if it has an identical genotype from the MiSeq and S5 systems, otherwise it is inconsistent. An inconsistent SSR is deemed to be incorrectly genotyped by the MiSeq or S5 system. The probability that a consistent SSR is the result of the same erroneous genotype from the two systems is negligible and therefore is ignored. Then, the genotyping errors of AmpSeq-SSR can be represented by half of the inconsistent SSRs. Therefore, the accuracy A(c,s) and the reproducibility R(c,s) of AmpSeq-SSR under a specific genotyping condition (c,s) are
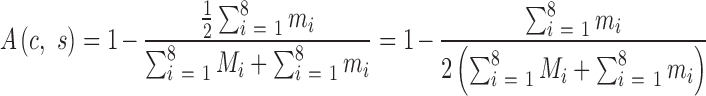 |
(1) |
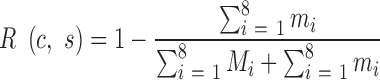 |
(2) |
where Mi and mi are the numbers of consistent and inconsistent SSRs in the ith variety, respectively.
For a particular sequencing platform, the exact accuracy A(c,s) and reproducibility R(c,s) may slightly differ from the results from Equations (1) and (2) because different platforms have different error rates and sequence biases.
Improvement to the accuracy of AmpSeq-SSR by removing non-random error-prone SSRs
When an SSR is valid in two varieties by the two sequencing systems, we call the SSR comparable for the two varieties. If a comparable SSR is inconsistent for both of the two varieties, it is considered to be an error-prone SSR. Let 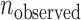 ,
, 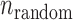 and
and 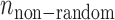 be the numbers of observed, random and non-random error-prone SSRs, respectively. Then,
be the numbers of observed, random and non-random error-prone SSRs, respectively. Then, 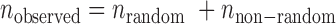 .
.
We introduce a null hypothesis H0 that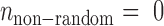 , i.e. all the observed error-prone SSRs occur randomly and the event that an SSR is inconsistent in one variety is independent of the event that the SSR is inconsistent in another variety. Let Nij be the number of comparable SSRs for the ith and jth varieties. Then, the total number of comparable SSRs in all
, i.e. all the observed error-prone SSRs occur randomly and the event that an SSR is inconsistent in one variety is independent of the event that the SSR is inconsistent in another variety. Let Nij be the number of comparable SSRs for the ith and jth varieties. Then, the total number of comparable SSRs in all  pairs of K varieties is
pairs of K varieties is 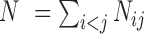 (K = 8 in the current study). The probability of a random error-prone SSR can be estimated as
(K = 8 in the current study). The probability of a random error-prone SSR can be estimated as 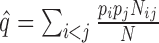 , where
, where 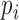 and
and 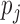 are the ratios between the inconsistent and comparable SSRs in the ith and jth varieties, respectively. The probability of having k random error-prone SSRs is binomially distributed, i.e.
are the ratios between the inconsistent and comparable SSRs in the ith and jth varieties, respectively. The probability of having k random error-prone SSRs is binomially distributed, i.e. 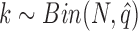 . Then, the probability of having no less than n random error-prone SSRs in N comparable SSRs is
. Then, the probability of having no less than n random error-prone SSRs in N comparable SSRs is
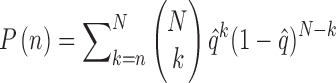 |
(3) |
Let  be the significance level for accepting H0 (
be the significance level for accepting H0 (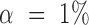 in our study). When
in our study). When 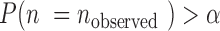 , we accept H0. Otherwise, we reject it, meaning that non-random error-prone SSRs exist. When all non-random error-prone SSRs were identified and removed from the target SSRs,
, we accept H0. Otherwise, we reject it, meaning that non-random error-prone SSRs exist. When all non-random error-prone SSRs were identified and removed from the target SSRs, 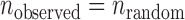 . Then, the inconsistent SSRs in the K varieties can be estimated as
. Then, the inconsistent SSRs in the K varieties can be estimated as 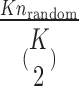 , where the value of
, where the value of 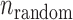 is determined by the equations of
is determined by the equations of 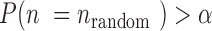 and
and 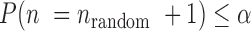 . Following Equation (1) the accuracy of AmpSeq-SSR can be improved to:
. Following Equation (1) the accuracy of AmpSeq-SSR can be improved to:
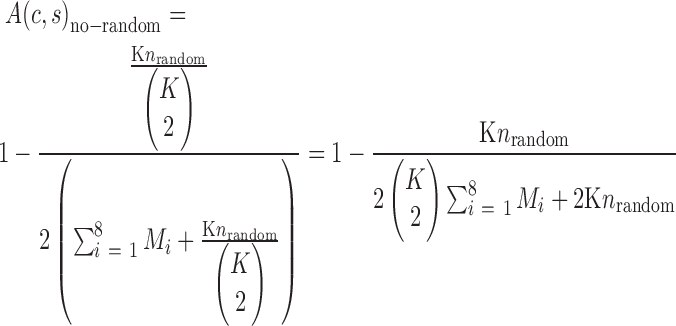 |
(4) |
where Mi is the number of consistent SSRs in the ith variety. As an example, we used seven of the eight varieties to identify and remove the error-prone SSRs from the target SSRs, and calculated the improved accuracy of AmpSeq-SSR in the eighth variety.
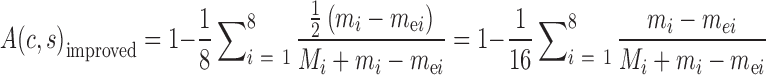 |
(5) |
where mei is the number of the error-prone SSRs for the ith variety and one of the other seven varieties.
Estimation of the accuracy of Sanger sequencing
The MiSeq pair-end reads with identical forward and reverse sequences have negligible sequencing errors and have a quality comparable to Sanger sequencing. Therefore, the accuracy of SSR genotyping by Sanger sequencing can be estimated as the ratio between the reads for the correct (or major) allele of a valid SSR and all the reads of the SSRs.
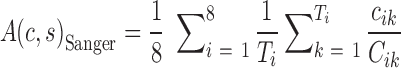 |
(6) |
where Ti is the number of valid SSRs in the ith variety, cik is the number of the reads which account for the major allele in the kth valid SSR of the ith variety and represents the times of correct SSR genotyping by Sanger sequencing, Cik is the total number of the reads of this SSR and represents all the times of SSR genotyping by Sanger sequencing.
Extension to heterozygous varieties and estimation of AmpSeq-SSR accuracy
To extend AmpSeq-SSR to heterozygous varieties and estimate its performance, we created a pseudo-heterozygous variety ij and generated a set of in silico sequencing data by randomly sampling and mixing 0.8 M reads from each of the ith and jth varieties. The genotypes from the ith and jth varieties were combined as the reference genotypes of the pseudo-heterozygous variety ij. To ensure the accuracy on the reference genotypes, we only considered those SSR loci with stutter ratios being lower than 0.5 and major alleles being covered by at least 50 reads in the ith and jth varieties.
An error correction model was introduced to determine the homozygosis or heterozygosis of an SSR locus using the sequencing data from homozygous SSR loci following the principles outlined in (25). The model may include, for example, all the loci in rice varieties of the current study or the loci on the human X chromosome. In the current study, the sequencing data of variety G were used to build an error correction model to determine the heterozygosis of the SSR loci in pseudo-heterozygous varieties. The reads of all the homozygous SSR loci with the same genotypes were pooled to generate a set of in silico sequencing data of pseudo-SSR loci. For a pseudo-SSR locus of a specific genotype, the percentage of the reads for each observed allele among all the reads at this pseudo-SSR locus were used to estimate the probability that an allele would be detected at an SSR locus of this genotype. These probabilities were then used to calculate the probabilities of the six possible homozygous and heterozygous pseudo-genotypes from the three alleles with the most reads at an SSR locus of the pseudo-heterozygous variety ij. The homozygosis or heterozygosis of an SSR locus of the pseudo-heterozygous variety ij was determined by its pseudo-genotype with the highest probability.
The scripts in Supplementary materials were used to call for the alleles of an SSR locus in pseudo-heterozygous variety ij. The allele or the two alleles with the most reads were taken as the test genotypes for the homozygous or heterozygous SSR loci in pseudo-heterozygous variety ij, respectively. If a test genotype was the same as the reference genotype, the test genotype was taken as correct for the pseudo-heterozygous variety. The accuracy of AmpSeq-SSR on the pseudo-heterozygous variety ij was then estimated as:
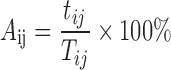 |
(7) |
where 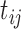 and
and 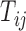 were the numbers of the correct genotypes and reference genotypes in pseudo-heterozygous variety ij, respectively.
were the numbers of the correct genotypes and reference genotypes in pseudo-heterozygous variety ij, respectively.
RESULTS
The AmpSeq-SSR method for SSR genotyping
We developed a novel method for large-scale SSR genotyping which is able to overcome the shortcomings of the existing methods (Table 1). The new method, named as AmpSeq-SSR, has a higher resolution, can be more efficiently deployed, and is able to provide more accurate results that can also be compared from different experiments and/or methods. AmpSeq-SSR combines multiplexing PCR, targeted deep sequencing and comprehensive analysis (Figure 1A). The current existing multiplex amplification techniques, such as that for whole exome sequencing, can amplify over 20 000 target loci in a single PCR reaction (38). Such a power of multiplex amplification was utilized in AmpSeq-SSR and 3105 SSRs were profiled in the current study. Since the genetic identities of SSR loci are mainly determined by their length variations, the SSR PCR products should be deeply sequenced on platforms (e.g. ABI S5 and Illumina MiSeq) that can produce long reads (e.g. 300 bp in the current study) to accommodate long SSR alleles. Sequencing reads were then mapped to target SSR loci to call SSR alleles. For homozygous samples, e.g. the rice varieties studied in the current study, only one allele was expected for a SSR locus and the allele with the most reads was taken as the SSR genotype (see ‘Materials and Methods’ section). For heterozygous samples, an additional step as described by (25) or (39) could be used to determine the SSR locus to be homologous or heterologous. Finally, the allele or the two alleles with the most reads were taken as the genotype of a homologous or heterologous SSR locus, respectively. Following the convention, an SSR genotype was represented as the amplicon length (see ‘Materials and Methods’ section), or the motif plus its repeat times, e.g. (AT)10.
Table 1. Comparisons of AmpSeq-SSR with the existing methods on a hypothetical scenario of genotyping 3000 SSR loci.
| Stages | Key points of the applicability | AmpSeq-SSR1 | Multiplex PCR-capillary electrophoresis2 | Singleplex PCR-capillary electrophoresis1 | Singleplex PCR-PAGE electrophoresis1 | Singleplex PCR-Agarose Elcectropheresis1 | Whole genome sequencing3 | Sanger sequencing1 |
|---|---|---|---|---|---|---|---|---|
| SSR loci amplification | Template DNA needed (μg) | 0.01 | 3000/5 × 0.01 = 6 | 3000 × 0.01 = 30 | 3000 × 0.01 = 30 | 3000 × 0.01 = 30 | 0.2–1 | 3000 × 0.01 = 30 |
| PCR reaction times | 1 | 3000/5 = 600 | 3000 | 3000 | 3000 | 1 | 3000 | |
| Number of PCR cycles | 16 | 35 | 40 | 40 | 40 | 5–20 | 40 | |
| Slippage chance4 | Low | high | high | high | high | Low | high | |
| Manual workload5 | Light | Heavy | Very heavy | Very heavy | Very heavy | Light | Very heavy | |
| Time consuming5 | Short | Very long | Very long | Very long | Very long | Short | Very long | |
| Cost ($) | 40 | 3000/5 × 1 = 600 | 3000 × 1 = 3000 | 3000 × 1 = 3000 | 3000 × 1 = 3000 | ∼100 | 3000 × 1 = 3000 | |
| SSR loci detection and genotyping | Automation level | High | High | High | Low | Low | High | High |
| Detection times | 1 | 3000/5 = 600 | 3000 | 3000 | 3000 | 1 | 3000 | |
| Signal for detection | Digital | Analog | Analog | Analog | Analog | Digital | Digital | |
| Base mutation | Can detect | Cannot detect | Cannot detect | Cannot detect | Cannot detect | Can detect | Can detect | |
| Different SSR genotypes with identical amplicon lengths | Can discriminate | Cannot discriminate | Cannot discriminate | Cannot discriminate | Cannot discriminate | Can discriminate | Can discriminate | |
| Reference6 | No need | Need | Need | Need | Need | No need | No need | |
| Genotyping resolution (bp) | 1 | 1 | 1 | Several | >10 | 1 | 1 | |
| Genotyping accuracy7 | Very high | High | High | Low | Very low | High | High | |
| Manual workload8 | Light | Heavy | Heavy | Very heavy | Very heavy | light | Very heavy | |
| Time consuming8 | Short | Very long | Very long | Very long | Very long | Short | Very long | |
| Cost ($) | 5 | 3000/5 × 5 = 3000 | 3000 × 5 = 15000 | 3000 × 0.1 = 300 | 3000 × 0.1 = 300 | >2000 | 3000 × 3 = 9000 | |
| Comparability of genotypes from different experiments | High | Low | Low | Low | Very low | High | High | |
1Referred to the current study for detailed parameters;
2Referred to the ‘Materials and Methods’ section in (39);
3Referred to the ‘Materials and Methods’ section in (25) and assumed that the average sequencing depth is 60-folds;
4The greater number of the PCR cycles, the greater chance to incur DNA polymerase slippage;
5The more PCR reaction times, the heavier workload and more time needed to perform PCR amplification;
6Molecular ladder or reference sample;
7For sequencing, the more highly the SSR loci are covered, the higher accuracy for SSR genotyping;
8The more manual operation and the more detection times, the heavier workload and more time needed to detect and genotype SSR loci.
Figure 1.

AmpSeq-SSR: its procedure, characteristics and comparison with capillary electrophoresis (CE). (A) A sketch of AmpSeq-SSR procedure applied to genotyping 3105 SSRs within 24 h. (B) Validation of an example SSR genotype in variety F using Sanger sequencing. (C, D and E) Profiles of AMPL1118228 (C) and AMPL1122663 (D) in variety H by CE and the comparisons of their genotypes with the genotypes from AmpSeq-SSR (E). (F) Amplicons with 40–70% GC contents have relatively uniform coverages. (G) The length distributions of all of the target SSRs on the reference genome. (H) The length distributions of the motifs of the target SSRs on the reference genome.
Genotypes identified by AmpSeq-SSR are highly accurate
The SSR amplicons were designed to be <250 bp and could be sequenced through by MiSeq 2 × 300 bp paired-end sequencing mode from both forward and backward directions. On average, 1.38 million (M) (58.72%) of the 2.35 M paired-end reads for each of varieties A–H had identical forward and backward sequences and were expected to have negligible sequencing errors (Table 2). As a result, 2427.75 (78.19%) of the 3105 target SSRs were genotyped from the 1.38 M highly accurate reads with a coverage of 594.69-folds and stutter ratio of 0.15 on average for each detected SSR (Table 2).
Table 2. The coverages and stutter ratios of the MiSeq-detected SSRs in rice varieties A–H.
| Varieties SSRs | A | B | C | D | E | F | G | H | Average | Standard deviation |
|---|---|---|---|---|---|---|---|---|---|---|
| Total reads (M) | 2.12 | 2.31 | 2.74 | 2.42 | 2.53 | 2.23 | 3.11 | 2.33 | 2.35 | 0.47 |
| Consistent reads (M) 1 | 1.56 | 1.73 | 1.98 | 1.78 | 1.87 | 1.55 | 2.34 | 1.69 | 1.81 | 0.26 |
| Reads mapped to SSR amplicons (M) | 1.17 | 1.40 | 1.61 | 1.35 | 1.30 | 1.06 | 1.73 | 1.40 | 1.38 | 0.22 |
| Detected SSRs | 2480 | 2290 | 2476 | 2334 | 2297 | 2480 | 2597 | 2468 | 2427.75 | 108.83 |
| Detected SSRs (%)2 | 79.87 | 73.75 | 79.74 | 75.17 | 73.98 | 79.87 | 83.64 | 79.48 | 78.19 | 3.51 |
| Coverage per detected SSR | 508.90 | 646.83 | 669.96 | 629.30 | 597.77 | 426.90 | 687.18 | 590.68 | 594.69 | 87.57 |
| Stutter ratio per detected SSR (%) | 16.07 | 12.83 | 15.96 | 14.31 | 14.95 | 16.18 | 17.27 | 14.42 | 15.25 | 1.40 |
| SSRs with coverage ≥10× and stutter ratio ≤0.5 | 1874 | 1756 | 1920 | 1755 | 1753 | 1874 | 1989 | 1922 | 1855.38 | 90.71 |
| SSRs with coverage ≥10× and stutter ratio ≤0.5 (%)3 | 75.56 | 76.68 | 77.54 | 75.19 | 76.32 | 75.56 | 76.59 | 77.88 | 76.42 | 0.96 |
1Consistent reads are the MiSeq reads with identical forward and backward sequences.
2Detected SSRs (%) = Detected SSRs/3105.
3SSRs with coverage ≥10× and stutter ratio ≤0.5 (%) = SSRs with coverage ≥10× and stutter ratio ≤0.5/Detected SSRs.
To assess the accuracy and reproducibility of AmpSeq-SSR, all of the 3105 target SSRs in varieties A–H were genotyped on the MiSeq and S5 sequencing systems by different labs (see ‘Materials and Methods’ section). An inconsistent SSR, which has distinct genotypes from the two sequencing systems (see ‘Materials and Methods’ section), was taken as a mistaken genotyping and used to calculate the accuracy of AmpSeq-SSR according to Equation (1). Even under a relaxed error-controlling criterion of the coverage no <10-folds and the stutter ratio no >0.5, AmpSeq-SSR had an accuracy of A (10, 0.5) = 99.73% when 10 584 of 10 641 SSRs had identical genotypes in the two sequencing systems (Table 3). With a lower stutter ratio of no >0.1 and a higher coverage of no <50-folds, AmpSeq-SSR had 100.00% accuracy when all of the 4940 SSRs had identical genotypes in the two sequencing systems (Table 3).
Table 3. The accuracy of AmpSeq-SSR is very high and positively correlated with SSR coverage and negatively correlated with SSR stutter ratio.
| Stutter ratio | ≤0.1 | ≤0.3 | ≤0.4 | ≤0.5 | ≤1.0 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Coverage | Total1 | Consistent2 | Accuracy3 | Total1 | Consistent2 | Accuracy3 | Total1 | Consistent2 | Accuracy3 | Total1 | Consistent2 | Accuracy3 | Total1 | Consistent2 | Accuracy3 |
| ≥1× | 8027 | 8233 | 98.75% | 11 699 | 12 035 | 98.60% | 12 471 | 12 924 | 98.25% | 13 033 | 13 687 | 97.61% | 13 991 | 15 564 | 94.95% |
| ≥10× | 6839 | 6841 | 99.99% | 9789 | 9808 | 99.90% | 10 270 | 10 306 | 99.83% | 10 584 | 10 641 | 99.73% | 11 064 | 11 303 | 98.94% |
| ≥20× | 6281 | 6282 | 99.99% | 8719 | 8733 | 99.92% | 9074 | 9099 | 99.86% | 9298 | 9333 | 99.81% | 9644 | 9799 | 99.21% |
| ≥30× | 5799 | 5800 | 99.99% | 7841 | 7851 | 99.94% | 8124 | 8142 | 99.89% | 8290 | 8317 | 99.84% | 8563 | 8680 | 99.33% |
| ≥40× | 5330 | 5331 | 99.99% | 7082 | 7090 | 99.94% | 7305 | 7320 | 99.90% | 7449 | 7473 | 99.84% | 7672 | 7767 | 99.39% |
| ≥50× | 4940 | 4940 | 100.00% | 6445 | 6449 | 99.97% | 6636 | 6646 | 99.92% | 6751 | 6768 | 99.87% | 6863 | 6895 | 99.77% |
| ≥100× | 3382 | 3382 | 100.00% | 4141 | 4143 | 99.98% | 4228 | 4230 | 99.98% | 4284 | 4288 | 99.95% | 4382 | 4410 | 99.68% |
| ≥200× | 1654 | 1654 | 100.00% | 1896 | 1896 | 100.00% | 1935 | 1935 | 100.00% | 1957 | 1958 | 99.97% | 1994 | 2002 | 99.80% |
| ≥500× | 180 | 180 | 100.00% | 200 | 200 | 100.00% | 205 | 205 | 100.00% | 208 | 208 | 100.00% | 208 | 208 | 100.00% |
1Total: the number of SSRs in varieties A–H whose coverages and stutter ratios satisfy the specified values for both MiSeq and S5 sequencing platforms.
2Consistent: the number of SSRs in varieties A–H whose coverages and stutter ratios satisfy the specified values and whose genotypes are identical between MiSeq and S5 sequencing platforms.
3Refer to Equation (1) for calculation of accuracy.
A total of 21 pseudo-heterozygous varieties were generated (see ‘Materials and Methods’ section) from every pair of the 7 varieties A–H in Supplementary Table S1. By Equation (7), AmpSeq-SSR had an average accuracy of 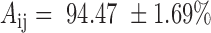 (Supplementary Table S3) on pseudo-heterozygous varieties, which is comparable to the accuracy of SNP genotyping arrays on heterozygous varieties (40). The lower accuracy of AmpSeq-SSR on heterozygous varieties can be attributed to DNA polymerase slippage, especially when the real and slippage alleles had the same or similar lengths (25,39). It is noteworthy that AmpSeq-SSR should be more accurate on heterozygous varieties than other methods, such as whole genome sequencing (WSG) that is difficult to achieve a high sequencing coverage in order to statistically exclude errors from DNA polymerase slippage.
(Supplementary Table S3) on pseudo-heterozygous varieties, which is comparable to the accuracy of SNP genotyping arrays on heterozygous varieties (40). The lower accuracy of AmpSeq-SSR on heterozygous varieties can be attributed to DNA polymerase slippage, especially when the real and slippage alleles had the same or similar lengths (25,39). It is noteworthy that AmpSeq-SSR should be more accurate on heterozygous varieties than other methods, such as whole genome sequencing (WSG) that is difficult to achieve a high sequencing coverage in order to statistically exclude errors from DNA polymerase slippage.
We randomly selected three SSR loci in two varieties for validation of AmpSeq-SSR using Sanger sequencing, the gold standard for genotyping. All of the six genotypes of the chosen SSR loci were proven to be correct (see Figure 1B for an example). We also compared AmpSeq-SSR with CE, the currently most popular and accurate electrophoresis technique, on two randomly chosen SSRs (Figure 1C and D). The lengths of the two SSRs detected by CE were respectively 5.53 and 4.47 bp shorter than those by AmpSeq-SSR (Figure 1E), suggesting that the SSR genotypes from different batches of CE might be inconsistent and incomparable. The stutter ratios for CE were respectively 28.83- and 2.11-folds greater than that for AmpSeq-SSR (Figure 1E), suggesting that the interference from CE on SSR genotyping was more serious than that from AmpSeq-SSR.
Improvement to the accuracy of AmpSeq-SSR
The accuracy and reproducibility of AmpSeq-SSR can be improved on SSRs with adequate sequencing coverages and lower stutter ratios (see ‘Materials and Methods’ section, Table 3). The stutter ratios of SSRs in varieties B-H were significantly decreased by 50.88% on average after removing the target SSRs with stutter ratios >0.2 in variety A (Supplementary Table S4). The SSRs with moderate GC contents of 40–70% had relatively uniform coverages of 0.2- to 5.0-folds (Figure 1F). Lower coverages on regions of low GC content have also been observed in WSG of sugar beet (41) and transcriptome sequencing (42). Therefore, the phenomenon of low coverages on low GC content regions may be due to PCR during library construction and sequencing since it is not limited to special amplicon types, sequencing platforms or specific species.
We called an SSR error-prone when it was inconsistent in two varieties. An error-prone SSR can occur randomly or non-randomly. Random error-prone SSRs are evidently unavoidable. A non-random error-prone SSR in one variety tends to be error-prone in another variety. Therefore, non-random error-prone SSRs, if exist, can be identified from the genotyping data of the tested varieties using a statistical analysis (see ‘Materials and Methods’ section). Following Equation (3), the probability of no non-random error-prone SSRs for the eight rice varieties in our current study is 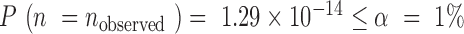 (Supplementary Table S5). Therefore, non-random error-prone SSRs exist and the accuracy of AmpSeq-SSR can be improved by removing them from the target SSRs (see ‘Materials and Methods’ section). Following Equation (4), the accuracy of AmpSeq-SSR can be improved to
(Supplementary Table S5). Therefore, non-random error-prone SSRs exist and the accuracy of AmpSeq-SSR can be improved by removing them from the target SSRs (see ‘Materials and Methods’ section). Following Equation (4), the accuracy of AmpSeq-SSR can be improved to 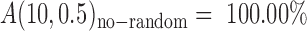 when all non-random error-prone SSRs are removed, which can be realized when a sufficient number of varieties were analyzed. As an example, we used seven of the eight varieties to identify as many as 70.18% error-prone SSRs (Supplementary Table S6). Following Equation (5), the accuracy of AmpSeq-SSR in the eighth variety was actually improved to
when all non-random error-prone SSRs are removed, which can be realized when a sufficient number of varieties were analyzed. As an example, we used seven of the eight varieties to identify as many as 70.18% error-prone SSRs (Supplementary Table S6). Following Equation (5), the accuracy of AmpSeq-SSR in the eighth variety was actually improved to 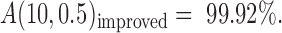 (For the calculation parameters, referred to Supplementary Table S6).
(For the calculation parameters, referred to Supplementary Table S6).
Most of the target SSRs were randomly selected from the reference genome (see ‘Materials and Methods’ section) and their distributions did not skew toward short SSRs (Figure 1G) or long motifs (Figure 1H), which can be more accurately genotyped as suggested in (25) and our analysis (data not shown). Therefore, the high accuracy of AmpSeq-SSR did not come from the over-representativeness of short SSRs or long motifs.
Highly discriminative fingerprints derived via AmpSeq-SSR
The discriminative power of fingerprints on rice varieties is proportional to the number of SSRs included in the fingerprints. In this study, we employed as many as 3105 SSRs for fingerprinting rice varieties A–H, which are much more than the 48 SSRs widely used for rice identification currently adopted in the National Agricultural Standard of China (Standard No. NY/T 1433–3014) (Figure 2A). The power of the fingerprints is also positively correlated with the discernable differences between the SSR genotypes of two rice varieties. The resolution of SSR genotypes derived from AmpSeq-SSR is single base and therefore any subtle difference between SSR genotypes can be clearly observed. As a result, 449.71 SSRs on average were identified by AmpSeq-SSR to possess differential genotypes between two varieties (Figure 2B and Supplementary Table S7). To our knowledge, 449.71 differential SSRs between two fingerprints are the largest ever reported, suggesting the great power of AmpSeq-SSR to unambiguously distinguish any rice variety under test.
Figure 2.

Highly discriminative fingerprints discovered by AmpSeq-SSR. (A) AmpSeq-SSR employed as many as 3105 target SSRs for rice fingerprint construction, which is ∼65 times of the 48 SSRs listed in rice National Standard. (B) AmpSeq-SSR could detect on average 449.71 differential SSR pairs between any two varieties. Among them, 48.28, 44.65 and 7.07% had amplicon length differences over 5, 1–5 and 0 bp, respectively, and therefore, are respectively distinguishable, hardly distinguished and indistinguishable on electropherograms. (C) An example of SSR with differential SSR genotypes but the same amplicon length. The SSR sequences are shown by letters in yellow background and the nucleotide substitution variations are shown by black arrows. The numbers of sequencing reads are on the left of the sequencing reads. (D) The SSRs in (C) show no difference on agarose gel electropherograms even though they have distinct SSR genotypes. (E) Examples of SSR amplicons on agarose gel electropherograms. On each electropherogram, from left to right are rice varieties A–H marked by their SSR genotypes.
Although electrophoresis can discern the differences of SSR amplicon lengths, it cannot distinguish base changes or base differences. Among the 449.71 differential SSRs, 33.68 (7.07%) had different bases but the same amplicon lengths between two varieties so that they were deemed to be mistaken as identical on electropherograms. For example, amplicon AMPL1141969 has the same amplicon length but distinct SSR genotypes in varieties F and G, which could be clearly identified by AmpSeq-SSR (Figure 2C) but not by electrophoresis (Figure 2D). Furthermore, when the differences of amplicon lengths were below the resolution, they might also be indiscernible on electropherograms (e.g. Figure 2E). Among the 449.71 differential SSRs, 221.32 (51.72%) had amplicon length differences no >5 bp (Figure 2B and Supplementary Table S7), which was the resolution of CE (Figure 1E), the most accurate electrophoresis technique.
Mapping of Xa21 gene by AmpSeq-SSR
Rice BB is a devastating disease that causes a significant annual rice yield reduction worldwide and Xa21 is one of the most effective genes to control BB (43,44). Through back cross of over six generations, we introduced Xa21 from the donor variety IRBB21 into three receptor parental varieties, IRBB24, 9311 and D62B, and developed their respective NILs, R24, R11 and R62 (Supplementary Table S1). Each parental variety and its NIL share similar genomic background except for the target gene Xa21 and its linked region (hereinafter referred to as ‘target region’). Used as genetic markers, the SSRs applicable to Xa21 mapping should be in the target regions of the NILs, have distinct genotypes between a receptor parent and its NIL but the identical genotype among the three NILs, R24, R11 and R62.
The 3105 target SSRs that were used for fingerprinting were adopted to map Xa21. Among them, 9, 14 and 55 SSRs have different genotypes between IRBB24 and R24, 9311 and R11, and D62B and R62, respectively (Figure 3A and Supplementary Table S8). One of these SSRs, in amplification AMPL1562757 (hereinafter referred to as ‘SSR21’), has the same genotype of (CGC)7 in the three NILs (Figure 3A and B). Therefore, using AmpSeq-SSR Xa21 can be easily mapped to the region near SSR21, which is about 0.6 Mbp from the actual locus of Xa21 (45). This gene mapping process was done within 24 h (Figure 1A), which is significantly faster than any conventional method. However, PAGE electrophoresis failed to detect the key differences of SSR21 genotypes between the receptor parents and their NILs (Figure 3C). A close inspection of the Ampli-Seq reads revealed that the length differences of the SSR21 genotypes were compensated by non-SSR sequences in amplicons (Figure 3D).
Figure 3.

Mapping gene Xa21 by AmpSeq-SSR. (A) SSR21 is the only SSR with differential genotypes between every pair of the three nearly isogenic lines (NILs). (B) Identical genotypes of (CGC)7 among the receptor parental varieties of the NILs. (C) No genotype difference between NILs can be detected by PAGE electrophoretogram. The molecular weight standards are marked on the left of the electrophoretogram. (D) The sequences of SSR21 amplicons in the three pairs of NILs. Letters in yellow background are the sequences of SSR21. The nucleotide substitution variations are marked by black arrow. Numbers of sequencing reads are listed on the left of the sequences.
DISCUSSION
As a classic molecular marker, SSRs have been employed in broad applications, such as fingerprinting, gene mapping, forensic analysis and variety identification (16–20). Compared to the existing methods, AmpSeq-SSR is high applicable to those applications thanks to its high accuracy, high efficiency and low cost.
AmpSeq-SSR is more accurate than the existing methods for SSR genotyping
Accuracy is always the primary consideration for any genotyping method. The current study demonstrated that AmpSeq-SSR had a much higher accuracy than the existing electrophoresis-based methods for SSR genotyping (Figures 1E, 2B–E, 3C and D; Supplementary Table S7). Except gel electrophoresis, Sanger sequencing is occasionally used for SSR genotyping. By double sequencing of the same sequences, AmpSeq-SSR could detect and avoid most of the sequencing errors to a level comparable with Sanger sequencing, i.e. ∼10−6 per base. As a by-product, the accuracy of SSR genotyping by Sanger sequencing could be estimated with AmpSeq-SSR reads by Equation (6) as A(10, 0.5)Sanger = 83.89%, which is much lower than the AmpSeq-SSR accuracy of A(10, 0.5) = 99.73%. The whole genome resequencing can also be used for SSR genotyping (25,31,32). However, for a high accuracy that AmpSeq-SSR has, the target SSRs need to be covered for ∼500-folds (Table 2), which requires coverage of the whole genome for thousands of folds, resulting in an unacceptable cost.
By overcoming most of the shortcomings in the existing methods for SSR genotyping, the accuracy of AmpSeq-SSR reached A(10, 0.5) = 99.73% (Table 3), which can be further improved to 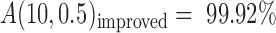 (for the parameters used, refer to Supplementary Table S6) or even potentially
(for the parameters used, refer to Supplementary Table S6) or even potentially 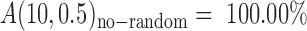 . The high accuracy makes AmpSeq-SSR capable to accurately identify a few or even one distinct gene from the plant varieties developed by transgene, backcrossing and mutation. However, a few distinct genes can be elusive within a background of hundreds of uncertain SSRs on electrophoresis profiles (Figure 2B and Supplementary Table S7). Moreover, AmpSeq-SSR has potential advantages for legal applications such as forensic analysis or granting rights under plant variety protection (PVP), for which any mistake may have serious consequences.
. The high accuracy makes AmpSeq-SSR capable to accurately identify a few or even one distinct gene from the plant varieties developed by transgene, backcrossing and mutation. However, a few distinct genes can be elusive within a background of hundreds of uncertain SSRs on electrophoresis profiles (Figure 2B and Supplementary Table S7). Moreover, AmpSeq-SSR has potential advantages for legal applications such as forensic analysis or granting rights under plant variety protection (PVP), for which any mistake may have serious consequences.
Factors that made AmpSeq-SSR accurate
The slippage of DNA polymerases during PCR amplification is the major source of error for SSR genotyping (21–27). The more PCR cycles, the higher chance for slippage errors to accumulate. Because visual inspection requires sufficiently bright bands, gel electrophoresis for SSR genotyping needs more PCR cycles than AmpSeq-SSR, i.e. 40 versus 16 PCR cycles in this study, resulting in more serious slippage (i.e. higher stutter ratios) for gel electrophoresis (Figure 1E). Note that a sequencing library can be constructed using a PCR-free protocol (25), so that polymerase slippage during library construction can be avoided for AmpSeq-SSR.
Duplex sequencing can improve the accuracy of SSR genotyping by reducing sequencing errors. In addition to inherent and unavoidable sequencing error, the PCR cycle number (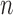 ) and sequencing coverage (
) and sequencing coverage (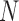 ) are primary factors contributing to the genotyping error probability (
) are primary factors contributing to the genotyping error probability (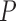 ). An SSR locus cannot be genotyped correctly when slippage reads take up more than 50% of the total coverage of the locus. Therefore, the error probability can be estimated as:
). An SSR locus cannot be genotyped correctly when slippage reads take up more than 50% of the total coverage of the locus. Therefore, the error probability can be estimated as:
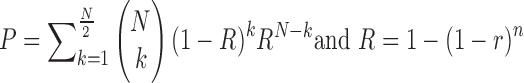 |
where 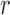 is the slippage probability of a read during a single PCR cycle, and
is the slippage probability of a read during a single PCR cycle, and 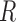 is the percentage of slippage reads in the final PCR product.
is the percentage of slippage reads in the final PCR product.
The PCR cycle number (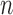 ) smaller than the electrophoresis based methods and the sequencing coverage (
) smaller than the electrophoresis based methods and the sequencing coverage (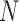 ) greater than WSG lead to a potentially higher accuracy of AmpSeq-SSR for SSR genotyping. The SSR often slips for a motif (2–4 bases) (25), making it difficult to distinguish the allele from slippage on electropherogram. Without slippage, accurately comparing SSR genotypes on electropherogram might still be nontrivial. For example, the SSRs in amplicon AMPL1141969 of variety F had negligible stutter ratio of no >0.01 (Supplementary Table S2) but migrated differently between two neighboring wells of the same gel (Figure 2D).
) greater than WSG lead to a potentially higher accuracy of AmpSeq-SSR for SSR genotyping. The SSR often slips for a motif (2–4 bases) (25), making it difficult to distinguish the allele from slippage on electropherogram. Without slippage, accurately comparing SSR genotypes on electropherogram might still be nontrivial. For example, the SSRs in amplicon AMPL1141969 of variety F had negligible stutter ratio of no >0.01 (Supplementary Table S2) but migrated differently between two neighboring wells of the same gel (Figure 2D).
High efficiency of AmpSeq-SSR extends its applications
We simultaneously amplified 3105 SSRs in eight rice varieties in a single PCR reaction and sequenced them in a single sequencing run. At present, such a combination of multiplex amplification and high-throughput sequencing can examine even more SSRs. Consider the MiSeq sequencing platform, for example, which can produce M = 25 million reads in a single run. When covered by m = 10 reads, an SSR can be genotyped with almost 100% accuracy (Table 3). Therefore, AmpSeq-SSR using the MiSeq platform can accurately genotype M/m = 2.5 million SSRs at once. In contrast, gel electrophoresis based SSR genotyping individually amplifies and separately examines each SSR, and thus is labor intensive and inefficient. Furthermore, thousands of SSR PCR reactions require a significant amount of template DNAs, e.g. an excessive ∼31 μg for 3105 SSRs.
The ability to genotype a large number of SSRs is the key to the success of AmpSeq-SSR in many applications. The 3105 target SSRs used by AmpSeq-SSR for fingerprinting are critical for the great discriminative power of the resulting fingerprints. Another good example is the mapping of Xa21 gene. Because of the successive backcrossing and selection, the target region of Xa21 gene became rather small, i.e. ∼2 Mbp, as estimated in our previous study (46). This small region reduces the chance to have usable SSRs for gene mapping. On average, 449.71 (29.61%) SSRs of the 1518.61 comparable SSRs on the ∼400 Mbp rice genome had differential genotypes between two fingerprints (Supplementary Table S8). Therefore, 7.59 (2 × 1518.61/400) SSRs are expected to exist in the target region. The probability of having at least one differential SSR genotype between the three pairs of NILs within the target region is 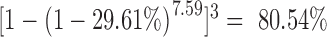 , suggesting a great chance for AmpSeq-SSR to successfully map a target gene.
, suggesting a great chance for AmpSeq-SSR to successfully map a target gene.
High reproducibility of AmpSeq-SSR extends its applications
Reproducibility is important for a new technique. Based on Equation (2), AmpSeq-SSR had a reproducibility close to 100% (Supplementary Table S9), ensuring it to produce highly consistent and comparable results under various conditions. Therefore, AmpSeq-SSR can be used to collaboratively construct fingerprint database without sharing the original biological resources (e.g. specific rice varieties), which are often taken as trade secrets or national strategic resources. More importantly, a fingerprint from AmpSeq-SSR can be freely and accurately compared with all records in a database to determine its distinctness from all the existing varieties, which is the legal precondition for a PVP grant. However, the fingerprints from electrophoresis can be accurately compared only when they are constructed in parallel, making it nearly infeasible to compare one variety against thousands of existing varieties.
AmpSeq-SSR is economical
The cost of AmpSeq-SSR is low, i.e. ∼$0.015 per SSR in this study, which is more affordable than other techniques (47). The high consensus of SSR genotypes between MiSeq and S5 (Table 3) indicated that the cost could be further decreased by the more economical single-end sequencing strategy.
The advantages of AmpSeq-SSR over the existing methods
AmpSeq-SSR has comparative advantages over the existing methods for large-scale SSR genotyping, including accuracy, resolution, throughput, efficacy and comparability of results from different experiments and/or methods (Table 1). Besides, comparing with WGS, AmpSeq-SSR is able to avoid the interference of homologous sequences on target SSRs by designing multiplex primers that do not anneal to homologous SSRs, which would be difficult if not impossible to realize for WGS. To overcome this shortcoming, WGS reads have to be discarded when the regions flanking SSRs are not perfectly matched to the reference, e.g. that appeared in (25), resulting in a genotyping failure for SSRs with variations in their flanking regions. In order to correct genotyping errors, a WGS-based strategy needs to resort to an ultra-high-throughput sequencing to deeply sequence target SSRs (Table 3). On the other hand, ultra-high-throughput sequencing typically produces short reads, producing a skewed distribution of reads toward short SSRs. AmpSeq-SSR has evident advantages and shows no obvious shortcomings over the other methods (Table 1), suggesting that AmpSeq-SSR is the most preferable method for large-scale SSR genotyping.
ACCESSION NUMBERS
All sequencing data of the eight rice varieties produced and analyzed in the current study have been deposited into NCBI Sequence Read Archive (SRA) under accession number of SRP089769.
AVAILABILITY
The package of scripts that implement the AmpSeq-SSR method is freely available at a public repository https://github.com/SystemsBiologyOfJianghanUniversity/AmpSeq-SSR.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: H.P. conceived the project, drafted the manuscript and supervised the study. W.Z. supervised the study and wrote the paper. L.Li and Z.F. analyzed the data. J.Z. performed the sequencing experiments. H.C. and Z.H. prepared the materials. L.G., L.C. and H.M. performed the validation experiments. L.Lu and S.R. participated in the statistical analysis. All of the authors discussed the results and commented on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The National Key Research and Development Program of China [2016YFF0202300]; Projects on EDVs by Ministry of Agriculture; Projects of Wuhan yellow crane talents; Youth and Technology Morning Program in Wuhan [2014072704011257]; Guidance project by Hubei Ministry of Education [B2015229]; United States National Institutes of Health [R01GM100364]. Funding for open access charge: JSPS Grants-in-aid for Scientific Research (KAKENHI) [16H04743].
Conflict of interest statement. None declared.
REFERENCES
- 1. Li Y.-C., Korol A.B., Fahima T., Nevo E.. Microsatellites within genes: structure, function, and evolution. Mol. Biol. Evol. 2004; 21:991–1007. [DOI] [PubMed] [Google Scholar]
- 2. Iglesias A.R., Kindlund E., Tammi M., Wadelius C.. Some microsatellites may act as novel polymorphic cis-regulatory elements through transcription factor binding. Gene. 2004; 341:149–165. [DOI] [PubMed] [Google Scholar]
- 3. Martin P., Makepeace K., Hill S.A., Hood D.W., Moxon E.R.. Microsatellite instability regulates transcription factor binding and gene expression. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:3800–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Krishnan J., Mishra R.K.. Code in the Non-Coding. Proc. Indian Natl. Sci. Acad. 2015; 81:609–628. [Google Scholar]
- 5. Gymrek M., Willems T., Guilmatre A., Zeng H., Markus B., Georgiev S., Daly M.J., Price A.L., Pritchard J.K., Sharp A.J. et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet. 2016; 48:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buschiazzo E., Gemmell N.J.. The rise, fall and renaissance of microsatellites in eukaryotic genomes. Bioessays. 2006; 28:1040–1050. [DOI] [PubMed] [Google Scholar]
- 7. Yang S., Wang L., Huang J., Zhang X., Yuan Y., Chen J.-Q., Hurst L.D., Tian D.. Parent-progeny sequencing indicates higher mutation rates in heterozygotes. Nature. 2015; 523:463–467. [DOI] [PubMed] [Google Scholar]
- 8. Ossowski S., Schneeberger K., Lucas-Lledo J.I., Warthmann N., Clark R.M., Shaw R.G., Weigel D., Lynch M.. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science. 2010; 327:92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moore S., Sargeant L., King T., Mattick J., Georges M., Hetzel D.. The conservation of dinucleotide microsatellites among mammalian genomes allows the use of heterologous PCR primer pairs in closely related species. Genomics. 1991; 10:654–660. [DOI] [PubMed] [Google Scholar]
- 10. Moodley Y., Baumgarten I., Harley E.. Horse microsatellites and their amenability to comparative equid genetics. Anim. Genet. 2006; 37:258–261. [DOI] [PubMed] [Google Scholar]
- 11. Dawson D.A., Horsburgh G.J., KÜPPER C., Stewart I.R., Ball A.D., Durrant K.L., Hansson B., Bacon I., Bird S., Klein A.. New methods to identify conserved microsatellite loci and develop primer sets of high cross-species utility–as demonstrated for birds. Mol. Ecol. Resour. 2010; 10:475–494. [DOI] [PubMed] [Google Scholar]
- 12. Moodley Y., Masello J.F., Cole T.L., Calderon L., Munimanda G.K., Thali M.R., Alderman R., Cuthbert R.J., Marin M., Massaro M. et al. Evolutionary factors affecting the cross-species utility of newly developed microsatellite markers in seabirds. Mol. Ecol. Resour. 2015; 15:1046–1058. [DOI] [PubMed] [Google Scholar]
- 13. Selkoe K.A., Toonen R.J.. Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol. Lett. 2006; 9:615–629. [DOI] [PubMed] [Google Scholar]
- 14. Guichoux E., Lagache L., Wagner S., Chaumeil P., Leger P., Lepais O., Lepoittevin C., Malausa T., Revardel E., Salin F. et al. Current trends in microsatellite genotyping. Mol. Ecol. Resour. 2011; 11:591–611. [DOI] [PubMed] [Google Scholar]
- 15. Schlötterer C. Evolutionary dynamics of microsatellite DNA. Chromosoma. 2000; 109:365–371. [DOI] [PubMed] [Google Scholar]
- 16. Kaur S., Panesar P.S., Bera M.B., Kaur V.. Simple sequence repeat markers in genetic divergence and marker-assisted selection of rice cultivars: a review. Crit. Rev. Food Sci. Nutr. 2015; 55:41–49. [DOI] [PubMed] [Google Scholar]
- 17. Jarne P., Lagoda P.J.. Microsatellites, from molecules to populations and back. Trends Ecol. Evol. 1996; 11:424–429. [DOI] [PubMed] [Google Scholar]
- 18. Kim K.S., Sappington T.W.. Microsatellite data analysis for population genetics. Methods Mol. Biol. 2013; 1006:271–295. [DOI] [PubMed] [Google Scholar]
- 19. Chambers G.K., Curtis C., Millar C.D., Huynen L., Lambert D.M.. DNA fingerprinting in zoology: past, present, future. Nvestig. Genet. 2014; 5:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Borsting C., Morling N.. Next generation sequencing and its applications in forensic genetics. Forensic Sci. Int. Genet. 2015; 18:78–89. [DOI] [PubMed] [Google Scholar]
- 21. Ellegren H. Microsatellites: simple sequences with complex evolution. Nat. Rev. 2004; 5:435–445. [DOI] [PubMed] [Google Scholar]
- 22. Webster M.T., Hagberg J.. Is there evidence for convergent evolution around human microsatellites?. Mol. Biol. Evol. 2007; 24:1097–1100. [DOI] [PubMed] [Google Scholar]
- 23. Brandström M., Bagshaw A.T., Gemmell N.J., Ellegren H.. The relationship between microsatellite polymorphism and recombination hot spots in the human genome. Mol. Biol. Evol. 2008; 25:2579–2587. [DOI] [PubMed] [Google Scholar]
- 24. Kelkar Y.D., Tyekucheva S., Chiaromonte F., Makova K.D.. The genome-wide determinants of human and chimpanzee microsatellite evolution. Genome Res. 2008; 18:30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fungtammasan A., Ananda G., Hile S.E., Su M.S., Sun C., Harris R., Medvedev P., Eckert K., Makova K.D.. Accurate typing of short tandem repeats from genome-wide sequencing data and its applications. Genome Res. 2015; 25:736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abdulovic A.L., Hile S.E., Kunkel T.A., Eckert K.A.. The in vitro fidelity of yeast DNA polymerase δ and polymerase ε holoenzymes during dinucleotide microsatellite DNA synthesis. DNA Repair. 2011; 10:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baptiste B.A., Eckert K.A.. DNA polymerase kappa microsatellite synthesis: two distinct mechanisms of slippage-mediated errors. Environ. Mol. Mutagen. 2012; 53:787–796. [DOI] [PubMed] [Google Scholar]
- 28. Zhang L., Cai R., Yuan M., Tao A., Xu J., Lin L., Fang P., Qi J.. Genetic diversity and DNA fingerprinting in jute (Corchorus spp.) based on SSR markers. Crop J. 2015; 3:416–422. [Google Scholar]
- 29. Liu X.B., Feng B., Li J., Yan C., Yang Z.L.. Genetic diversity and breeding history of Winter Mushroom (Flammulina velutipes) in China uncovered by genomic SSR markers. Gene. 2016; 591:227–235. [DOI] [PubMed] [Google Scholar]
- 30. Njung’e V., Deshpande S., Siambi M., Jones R., Silim S., De Villiers S.. SSR genetic diversity assessment of popular pigeonpea varieties in Malawi reveals unique fingerprints. Electron. J. Biotechnol. 2016; 21:65–71. [Google Scholar]
- 31. Kim K.-S., Noh C.H., Moon S.-J., Han S.-H., Bang I.-C.. Development of novel tetra-and trinucleotide microsatellite markers for giant grouper Epinephelus lanceolatus using 454 pyrosequencing. Mol. Biol. Rep. 2016; 43:541–548. [DOI] [PubMed] [Google Scholar]
- 32. Bozzi J.A., Liepelt S., Ohneiser S., Gallo L.A., Marchelli P., Leyer I., Ziegenhagen B., Mengel C.. Characterization of 23 polymorphic SSR markers in Salix humboldtiana (Salicaceae) using next-generation sequencing and cross-amplification from related species. Appl. Plant Sci. 2015; 3:1400120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W. et al. Initial sequencing and analysis of the human genome. Nature. 2001; 409:860–921. [DOI] [PubMed] [Google Scholar]
- 34. Star B., Hansen M.H., Skage M., Bradbury I.R., Godiksen J.A., Kjesbu O.S., Jentoft S.. Preferential amplification of repetitive DNA during whole genome sequencing library creation from historic samples. STAR. 2016; 2:36–45. [Google Scholar]
- 35. Treangen T.J., Salzberg S.L.. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 2012; 13:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gymrek M., Golan D., Rosset S., Erlich Y.. lobSTR: a short tandem repeat profiler for personal genomes. Genome Res. 2012; 22:1154–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pater J.A., Benteau T., Griffin A., Penney C., Stanton S.G., Predham S., Kielley B., Squires J., Zhou J., Li Q.. A common variant in CLDN14 causes precipitous, prelingual sensorineural hearing loss in multiple families due to founder effect. Hum. Genet. 2017; 136:107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berg K.D., Glaser C.L., Thompson R.E., Hamilton S.R., Griffin C.A., Eshleman J.R.. Detection of microsatellite instability by fluorescence multiplex polymerase chain reaction. J. Mol. Diagn. 2000; 2:20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Unterseer S., Bauer E., Haberer G., Seidel M., Knaak C., Ouzunova M., Meitinger T., Strom T.M., Fries R., Pausch H.. A powerful tool for genome analysis in maize: development and evaluation of the high density 600 k SNP genotyping array. BMC Genomics. 2014; 15:823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Minoche A.E., Dohm J.C., Himmelbauer H.. Evaluation of genomic high-throughput sequencing data generated on illumina HiSeq and genome analyzer systems. Genome Biol. 2011; 12:R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li S., Labaj P.P., Zumbo P., Sykacek P., Shi W., Shi L., Phan J., Wu P.Y., Wang M., Wang C. et al. Detecting and correcting systematic variation in large-scale RNA sequencing data. Nat. Biotechnol. 2014; 32:888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mew T. Current status and future prospects of research on bacterial blight of rice. Annu. Rev. Phytopathol. 1987; 25:359–382. [Google Scholar]
- 44. NiÑo D.O., Ronald P.C., Bogdanove A.J.. Xanthomonas oryzae pathovars: model pathogens of a model crop. Mol. Plant Pathol. 2006; 7:303–324. [DOI] [PubMed] [Google Scholar]
- 45. Song W.Y., Wang G.L., Chen L.L., Kim H.S., Pi L.Y., Holsten T., Gardner J., Wang B., Zhai W.X., Zhu L.H. et al. A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science. 1995; 270:1804–1806. [DOI] [PubMed] [Google Scholar]
- 46. Peng H., Chen Z., Fang Z., Zhou J., Xia Z., Gao L., Chen L., Li L., Li T., Zhai W.. Rice Xa21 primed genes and pathways that are critical for combating bacterial blight infection. Sci. Rep. 2015; 5:12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jennings T., Knaus B., Mullins T., Haig S., Cronn R.. Multiplexed microsatellite recovery using massively parallel sequencing. Mol. Ecol. Res. 2011; 11:1060–1067. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.