


Abstract
Non-homologous end joining (NHEJ) repairs DNA double strand breaks in non-cycling eukaryotic cells. NHEJ relies on polynucleotide kinase/phosphatase (PNKP), which generates 5΄-phosphate/3΄-hydroxyl DNA termini that are critical for ligation by the NHEJ DNA ligase, LigIV. PNKP and LigIV require the NHEJ scaffolding protein, XRCC4. The PNKP FHA domain binds to the CK2-phosphorylated XRCC4 C-terminal tail, while LigIV uses its tandem BRCT repeats to bind the XRCC4 coiled-coil. Yet, the assembled PNKP-XRCC4–LigIV complex remains uncharacterized. Here, we report purification and characterization of a recombinant PNKP–XRCC4–LigIV complex. We show that the stable binding of PNKP in this complex requires XRCC4 phosphorylation and that only one PNKP protomer binds per XRCC4 dimer. Small angle X-ray scattering (SAXS) reveals a flexible multi-state complex that suggests that both the PNKP FHA and catalytic domains contact the XRCC4 coiled-coil and LigIV BRCT repeats. Hydrogen-deuterium exchange indicates protection of a surface on the PNKP phosphatase domain that may contact XRCC4–LigIV. A mutation on this surface (E326K) causes the hereditary neuro-developmental disorder, MCSZ. This mutation impairs PNKP recruitment to damaged DNA in human cells and provides a possible disease mechanism. Together, this work unveils multipoint contacts between PNKP and XRCC4–LigIV that regulate PNKP recruitment and activity within NHEJ.
INTRODUCTION
DNA double strand breaks (DSBs) are among the most deleterious DNA lesions. In mammalian cells, the major pathway for the repair of DSBs in both cycling and non-cycling cells is non-homologous end joining (NHEJ) (1,2). NHEJ is active in both G1 and G2 stages of the cell cycle. In contrast, the alternate DSB repair pathway homologous recombination repair (HRR) is active only in S and G2 cells when a replicated and undamaged sister chromatid is available as a template for repair. Thus, non-cycling cells and cells in the G1 phase of the cell cycle rely on NHEJ, sometimes called classical-NHEJ (c-NHEJ) or an alternative-NHEJ pathway (Alt-NHEJ). Classical NHEJ uses a network of proteins that recognize the break, tether the damaged ends, interrogate the chemical structure of the ends for further processing, and finally ligate the DNA. The broken DNA ends are first recognized and bound by the Ku70–Ku80 heterodimer, which recruits additional proteins including the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), XRCC4-like factor (XLF) and the XRCC4-DNA ligase IV complex. XRCC4 plays a central scaffolding role in NHEJ and interacts not only with DNA LigIV but also with XLF to form filamentous structures that bind and organize the DNA near the DSB (3,4). Moreover, XRCC4 interacts with several DNA end-processing enzymes including polynucleotide kinase/phosphatase (PNKP), aprataxin (APTX) and APTX, and/or PNKP-related protein (APLF), discussed below. Additional factors such as the Werner's syndrome helicase (5), DNA polymerase X family members (6) and paralog of XRCC4 and XLF (PAXX) (7,8) are likely also involved in NHEJ but their functions are less clear.
LigIV, like all DNA ligases, requires 5΄-phosphate and 3΄-hydroxyl strand termini for ligation. Yet, many forms of damage, such as those resulting from ionizing radiation and reactive oxygen species, yield non-ligatable ends such as 3΄-phosphate, 3΄-phosphoglycolate and, more rarely, 5΄-hydroxyl termini (9–12). Furthermore, abortive ligation can result in the release of a ligation intermediate containing a 5΄-adenylate group (13). To complete ligation, these non-ligatable or ‘dirty’ ends must be processed by DNA processing factors that are specifically recruited to XRCC4. XRCC4 is phosphorylated by the acidophilic kinase CK2 at a conserved set of Ser/Thr residues immediately C-terminal to the N-terminal structured region. This phosphorylated region specifically binds three related proteins that play important roles in NHEJ and DNA end processing: PNKP, APTX, and APLF (14–19). PNKP is a bi-functional 3΄ DNA phosphatase/5΄ DNA kinase and processes 5΄-hydroxyl and 3΄-phosphate termini to the ligatable 5΄-phosphate/3΄-hydroxyl ends (20). APTX specifically recognizes and hydrolyzes 5΄-adenylated DNA ends for subsequent ligation (21). APLF interacts directly with Ku80 (5,22), binds poly(ADP)-ribosylated proteins near the lesion (23,24), and has also been reported to have nuclease activity that may play a role in limited processing of the DNA ends (17,25). The phosphorylation-dependent recruitment of each factor to XRCC4 relies on a conserved forkhead-associated (FHA) domain in each protein that binds and recognizes the multiply phosphorylated XRCC4 C-terminus (14,26,27). Interestingly, a conserved CK2 phosphorylation sequence is also found in the single strand break scaffold protein, XRCC1, and this region has been shown to be responsible for the recruitment of PNKP, APTX and APLF to the sites of single strand break repair (15–19,28–30).
PNKP is critical for strand break repair in both nuclear and mitochondrial DNA (20,31,32). Knockdown of PNKP in human cells leads to dramatically elevated sensitivities to a range of genotoxic agents and marked elevation in the rates of spontaneous mutation (33), and inhibitors of PNKP also sensitize human cells to DNA damaging agents (34–36). This suggests that inhibition of PNKP could potentiate tumor cell killing by DNA targeting therapies. Structural investigations show PNKP consists of a catalytic domain with separate phosphatase and kinase sub-domains joined by a flexible linker to a regulatory FHA domain (27). The phosphatase and kinase active sites are positioned on the same side of the catalytic domain and can potentially interact with a single substrate (27). Structural studies of PNKP–substrate complexes together with binding and activity assays indicate PNKP can efficiently bind and process DNA double strand break ends, as well as nicked and gapped DNAs, expected in diverse strand break repair pathways (27,37–40). The phosphatase active site cleft is deep and narrow and can only bind single-stranded DNA termini. PNKP-substrate binding may drive limited DNA base pair destabilization to allow the unwinding and active site engagement of 3΄ termini within double-stranded DNAs (39). These collective observations suggest that complexes between PNKP and its partners in DNA repair pathways may have multiple functional consequences including specificity and efficiency for end processing.
End processing factors such as PNKP present a dilemma for DNA repair since the unprotected DNA ends produced by the reaction are potentially as dangerous to the cell as the unprocessed substrates. Such DNA repair factors may in fact remain bound to the product until the product can be passed to the next enzyme in the repair pathway (41). Indeed, PNKP does show appreciable affinity for its products and exhibits only very low rates of product turnover (42). The ordered transfer of DNA end repair intermediates between different DNA repair enzymes may be mediated by the XRCC1 and XRCC4 scaffolding proteins and therefore play a more complex role in repair than the simple recruitment of repair proteins. Interestingly, both XRCC1 and XRCC4 stimulate the kinase and phosphatase activities of PNKP (28,43,44). While there is some evidence that XRCC1 may enhance the affinity of PNKP for its DNA substrates (45), other studies have instead suggested that the major effect of XRCC1 and XRCC4 may be on substrate turnover (46–48). The observed effects of XRCC1/4 on PNKP catalytic activity are in accord with a more complex interaction of the scaffolding protein with PNKP. Indeed, in vitro both XRCC1 and XRCC4 weakly interact with PNKP in the absence of phosphorylation, likely involving the PNKP catalytic domain (46–48). This interaction could explain the finding that XRCC1 and XRCC4 can both enhance PNKP catalytic activities in the absence of CK2 phosphorylation (43,44).
To resolve interactions important to function and regulation of the PNKP-XRCC4–LigIV repair complex beyond the PNKP FHA–XRCC4 phosphopeptide interaction, we developed a system to purify a stoichiometrically defined complex of PNKP bound to CK2-phosphorylated XRCC4 together with the tandem BRCT domains of LigIV. Assessment of the structure and dynamics of the purified complex using small angle X-ray scattering (SAXS) suggests flexible tethering of PNKP to XRCC4 via the FHA-phosphopeptide interaction. However, this analysis also indicates that the complex adopts compact conformations that imply dynamic interactions between the PNKP catalytic domain and the N-terminal structured region of XRCC4 and/or the LigIV BRCT domain. Hydrogen-deuterium mass spectrometry (HX-MS) reveals that this additional interaction likely involves a surface of the PNKP phosphatase domain whose mutation (E326K) is associated with a severe form of the neuro-developmental disorder termed MCSZ, which is characterized by microcephaly, infant onset seizures and developmental delay (49). We show that the E326K mutation in human cells reduces PNKP recruitment to sites of UV laser-induced DNA damage and its interaction with XRCC4. We propose that dynamic interactions between PNKP and XRCC4–LigIV modulate PNKP catalytic function in the context of XRCC4, and may be critical to stabilize PNKP within functioning DNA repair foci.
MATERIALS AND METHODS
In vitro phosphorylation of XRCC4 by CK2α
XRCC4 was phosphorylated in vitro as part of the XRCC4/LigIV heterodimer with purified GST-CK2α. The phosphorylation reaction was carried out in 50 mM Tris pH 7.5, 150 mM NaCl, 10 mM MgCl2 and 1 mM DTT 1× activity buffer for 20 min at 30°C with ATP and GST-CK2α at 30× and 0.05× final XRCC4 concentration, respectively. Maximal phosphorylation of XRCC4 by GST-CK2α was suggested by a complete band shift of XRCC4 in Phos-Tag SDS-PAGE assays (Wako Pure Chemical Industries, Ltd) and saturated 32P-incorporation into XRCC4 despite an increase in GST-CK2α concentration or increased time of CK2α phosphorylation reaction (Supplementary Figure S1A and C). Addition of fresh CK2 following a 20-min reaction at 1× CK2 concentration did not increase phosphorylation (not shown). Exclusive phosphorylation of the XRCC4 protein and of S232 and T233 residues in its C-terminal CK2α consensus sequence was verified by SDS-PAGE 32P-incorporation assays in XRCC4wt, XRCC4S232A, XRCC4T233A, XRCC4S232A/T233A. 32P-signal was completely absent in the XRCC4S232A/T233A double mutant dimerized with LigIV and present in both XRCC4S232A and XRCC4T233A single mutants dimerized with LigIV (Supplementary Figure S1B). Phosphorylation of both S232 and T233 in the XRCC4/LigIV heterodimer was also confirmed by LC-MS/MS as part of HX-MS experiments (not shown).
Multi-angle laser light scattering (MALLS)
Central peak fractions from the final pXRCC4/LigIV/PNKP purification step were used for molecular weight determination by inline size-exclusion chromatography-multi-angle laser light scattering (SEC-MALLS). 100-μl samples were loaded at ∼5 mg/ml onto the SEC-MALLS and Buffer U (150 mM KCl, 20 mM Tris pH 8.0, 1 mM EDTA and 1 mM DTT) was used for elution and for differential refractometry. Peak fractions were collected, visualized by SDS-PAGE, and data was analyzed with ASTRA V software (Wyatt Technology). BSA in Buffer U was used to normalize the MALLS detector. Experiments were performed at 25°C on an FPLC-managed Superose 6 10/300 size exclusion chromatography column (GE Healthcare), coupled to a Wyatt DAWN EOS light scattering detector, Optilab® rEX Refractive Index Detector, QELS (Quasi Elastic Light Scattering), UV detector (Wyatt Technology) and fraction collector.
Electrophoretic mobility shift assay (EMSA)
Different concentrations of PNKP (0, 0.04, 0.08, 0.2, 0.4, 0.8, 2 μM) were titrated against three different DNA substrates at 20 nM concentration in the presence or absence of 2 μM of either phosphorylated or non-phosphorylated XRCC4–LigIV complex. The DNA substrates (Integrated DNA Technology) each were synthesized to contain a single 5΄-fluorescein (6-FAM) label and a single 3΄-phosphate. The DNAs used were a single stranded 20 nucleotide DNA (FAM-20p: 5΄-(6-FAM)-TAGCACCTACCGATTGTATGp-3΄), a double stranded blunt-ended 20 bp DNA (FAM-20p with complementary strand 20c: 5΄-pCATACAATCGGTAGGTGCTA-3΄), and a double stranded substrate carrying a 16-nucleotide 5΄ overhang (FAM-20p with complementary strand 36c: 5΄-pCGACACAAAAACGTATCATACAATCGGTAGGTGCTA-3΄).
In each reaction 1 μl of (phosphorylated/non-phosphorylated) XRCC4–LigIV (20 μM), 1 μl of PNKP in storage buffer (150 mM KCl, 10 mM Tris pH 7.5, 1 mM DTT), 1 μl of poly(dI-dC) (1 mg/ml), 5 μl of EMSA binding buffer (storage buffer + 1 mM MgSO4, 10% glycerol) and 1 μl of H2O were mixed and pre-incubated together for 5 min. At the final step, the FAM-labeled DNA substrate was added to the reaction mixture. After 10 minutes of additional incubation time, 2 μl of 4× EMSA loading dye (40% glycerol, 240 mM Tris–HCl pH 7.5 and 4 mg/ml bromophenol blue) was added to the reaction mixture and loaded on a pre-run 6% native polyacrylamide gel (19:1 acrylamide/bisacrylamide). Electrophoresis was carried out at 100 V for 55 min at 4°C. Free and bound DNA substrates were visualized by Typhoon™ phosphoimager.
Hydrogen–deuterium exchange mass spectrometry (HX-MS)
Peptide-level HX-MS data were measured for full-length free hPNKP and hPNKP in the ternary complex with pXRCC4/LigIV. Briefly, the two protein states were deuterated at 20°C in Buffer U, using 50% D2O and the continuous labeling method (50,51) Samples were quenched at various time-points using cold glycine–HCl (100 mM, pH 2.5) to arrest exchange, and then digested using purified nepenthes digestive fluid (10°C for 2 min). The resulting digests were processed using a research-grade HX-MS2 platform, consisting of a temperature-controlled autosampler and chilled chamber for reversed phase chromatography (HDX PAL, Leap Technologies), an LC system (nanoLC Ultra, Sciex) and a QqTOF mass spectrometer (TripleTOF 5600, Sciex). Using non-deuterated controls, hPNKP peptides were fragmented by recursive data-dependent MS/MS acquisition, and identified using Mascot 2.3 (Matrix Science). The resulting feature lists (peptide sequence, retention time and m/z) were used to extract mass shifts (in the form of average deuterium incorporation, D) from peptides arising from the actual deuteration experiments. Data extraction was conducted with Mass Spec Studio (52), and summarized in a Woods plot. It was determined that hPNKP was labeled extensively after 5 min of deuteration, therefore the comparative analysis of bound and free forms was conducted at this time-point. Binding-induced alterations of deuteration (Delta value, expressed as percent of maximum deuteration) were deemed significant if they passed a two-tailed t test based on quadruplicate analyses of free and bound states (P < 0.05), and if Delta values exceed a threshold mass shift value (±2 SD of the shift noise). Peptide isotopomer envelopes were also required to pass a distribution analysis to guard against spectral overlap. PNKP peptides with significant Delta values upon binding pXRCC4/LigIV were mapped onto the homologous residues of the mouse PNKP crystal structure.
Please see Supplementary Materials for additional detailed Materials and Methods.
RESULTS
Assembly and characterization of PNKP–pXRCC4–LigIV complexes
Previous work suggested that stable association of PNKP with XRCC4 relied on phosphorylation of XRCC4 Ser232/Thr233 by CK2 (14). To prepare complexes of full-length PNKP with phosphorylated XRCC4, we phosphorylated a C-terminal truncation mutant of XRCC4 corresponding to residues 1–238 (Supplementary Figure S1). This fragment contains the structured N-terminal domain of XRCC4, as well as the minimal portion of the C-terminal tail of XRCC4 that is phosphorylated by CK2 and is required for binding PNKP (Figure 1A). XRCC41-238 was co-purified with a fragment of LigIV corresponding to the tandem BRCT domains (residues 654–911). The XRCC4–LigIV complex purified as a single peak on gel filtration chromatography (Figure 1B), unlike the XRCC4 fragment alone which eluted as multiple peaks. This is consistent with previous results that showed that XRCC4 can adopt a number of oligomeric forms but that interaction with the LigIV BRCT domain stabilizes the XRCC4 dimer (53). The XRCC4–LigIV complex was then phosphorylated using purified recombinant CK2 and purified by gel filtration chromatography. One mole equivalent of PNKP was added and the mixture was separated by gel filtration chromatography. The results revealed that addition of PNKP resulted in the formation of a single peak containing PNKP, XRCC4 and LigIV. In contrast, mixtures containing PNKP and the non-phosphorylated XRCC4–LigIV complex did not associate within a single peak, indicating that phosphorylation of XRCC4 is required to form a complex with PNKP that is stable in gel filtration chromatography (Figure 1A–C). To define the stoichiometry of the PNKP–pXRCC4–LigIV complex, we subjected this complex to gel filtration chromatography with in-line multi-angle laser light scattering (MALLS), which can reveal the molecular mass of complexes independent of shape. The results of this experiment gave a mass of 143 ± 4 kDa consistent with the formation of 1:2:1 PNKP:pXRCC4:LigIV complex (theoretical mass: 141 kDa; Figure 1D).
Figure 1.

Formation of a phosphorylation-dependent 1:2:1 PNKP–pXRCC4–LigIV complex. (A) Overview of the PNKP, XRCC4 and LigIV protein constructs used in this study. (B) Analysis of PNKP interactions with XRCC4–LigIV by Superdex 200 analytical gel filtration chromatography. ‘A’ indicates the elution position of the PNKP–pXRCC4–LigIV complex, ‘B’ indicates the elution position of the XRCC4–LigIV complex, and ‘C’ indicates elution position of free PNKP. (C) Analysis of the peaks in (B) by SDS-PAGE. Molecular weight markers are shown in the left lane for each gel. (D) Analysis of PNKP–pXRCC4–LigIV by MALLS. PNKP–pXRCC4–LigIV, premixed at a 1:2:1 molar ratio was run on a Superose6 gel filtration column with in-line multi-angle laser light scattering (MALLS). The refractive index readout is shown in blue, and the light scattering is shown in red with the eluted peaks labelled as in (B). (E) Titration of PNKP against pXRCC4–LigIV assessed by Superdex 200 analytical gel filtration chromatography as in (B). The numbers refer to the ratio of PNKP:pXRCC4–LigIV for each trace. The magenta trace is a pXRCC4–LigIV control and the red trace is the PNKP control.
To further probe the stoichiometry of binding, we next titrated increasing concentrations of PNKP against pXRCC4–LigIV and monitored complex formation by gel filtration chromatography (Figure 1E). At one mole PNKP per mole pXRCC4–LigIV (i.e. 1 PNKP:2 pXRCC4:1 LigIV), we observed the expected peak corresponding to the PNKP–pXRCC4–LigIV complex (green trace). As the amount of PNKP was increased to two (orange), four (cyan) or eight (blue) moles of PNKP per mole pXRCC4–LigIV, the height and position of the PNKP–pXRCC4–LigIV peak remained unchanged, while the free PNKP peak increased in height. This result suggested that even under these conditions of excess PNKP, additional PNKP molecules are not loaded onto the 1:2:1 PNKP–pXRCC4–LigIV complex. This experiment taken together with the MALLS experiment demonstrates that a single PNKP protomer binds the heterotrimeric pXRCC4–LigIV complex with high affinity. These findings suggest that only one of the two phosphorylated tails of the XRCC4 dimer is utilized for high affinity interactions with PNKP.
Phosphorylated XRCC4–LigIV binds PNKP–DNA complexes
To assess whether interactions between PNKP and DNA substrates are affected by XRCC4–LigIV, we used an electrophoretic mobility shift assay (EMSA, Figure 2). These experiments employed a murine PNKP harbouring a mutation in the key catalytic Asp170 in the phosphatase active site (mPNKPD170A). We previously showed that this mutant binds with high affinity to 3΄-phosphorylated DNA substrates (39). We titrated this mutant protein against DNAs containing a single 3΄-phosphate terminus in three different contexts: a single stranded DNA, a blunt, double-stranded end, and a 5΄ overhanging double-stranded end. In control experiments in the absence of added XRCC4–LigIV, the mPNKPD170A bound to the DNA to result in lower mobility species in EMSA. The single-stranded DNA yielded a single shifted species consistent with binding of the PNKP to the 3΄-phosphate DNA end. The double stranded DNAs yielded two shifted species, one with a high apparent affinity, likely due to binding of the 3΄-phosphate, and a second, lower affinity and lower mobility species, likely corresponding to non-specific binding of PNKP to other regions of the DNA. Addition of non-phosphorylated XRCC4–LigIV to these binding reactions did not result in the appearance of new shifted species or in a change in the apparent affinity of PNKP for the DNA substrate, suggesting that XRCC4–LigIV does not interact with PNKP or the DNA under these conditions. However, addition of phosphorylated XRCC4–LigIV to either of the titrations of the double-stranded substrates resulted in the shifting of the PNKP–DNA complexes to a lower mobility species migrating very close to the well. This low mobility species was not observed with the single-stranded DNA, however addition of the pXRCC4–LigIV did result in a loss in intensity of the PNKP–DNA species. It is possible that in this case, pXRCC4–LigIV can bind the PNKP–ssDNA complex, but that the resulting complex does not enter the gel due to the reduced negative charge of the complex compared to those formed with the dsDNA substrates. These results imply that XRCC4–LigIV binds to PNKP in a phosphorylation-dependent manner, and that these interactions do not significantly modulate the affinity of PNKP for its DNA substrates.
Figure 2.

CK2-phosphorylated XRCC4–LigIV can bind PNKP–DNA substrate complexes. EMSA experiments were carried out using the FAM-labeled DNA constructs illustrated to the left in the absence of XRCC4–LigIV (left panels), with 2 μM XRCC4–LigIV (center panels), or with 2 μM pXRCC4–LigIV (right panels). The PNKP concentrations increased from left to right in each gel. The PNKP concentrations are: 0, 40, 80, 200, 400, 800, 2000 nM.
Solution scattering shows dynamic interactions of PNKP catalytic domain with XRCC4–LigIV
We used small angle X-ray scattering (SAXS) to gain insight into the structure and dynamics of the PNKP–pXRCC4–LigIV complex in solution (54). Previously we had used similar methods to probe the interactions of PNKP with DNA substrates of the kinase domain (38), interactions of XRCC4 with XLF and the LigIV BRCT domain (3,55), as well as the full length complex XRCC4–LigIV (56).
SAXS measurements were carried out on a PNKP–pXRCC4–LigIV complex that was freshly purified by gel filtration chromatography immediately prior to data collection. Guinier plots of pXRCC4–LigIV with and without bound PNKP showed unbiased residuals indicating a lack of aggregation in the samples (Supplementary Figure S2). Molecular mass of the complex was determined from the SAXS data using the QR parameter as previously described (57). Scattering data from the XRCC4–LigIV complex (Figure 3A) gave a molecular mass of 83 kD using the reciprocal space approach, and 77 kD using a real space approach, similar to the theoretical mass of 84.5 kD. Scattering data for the PNKP–pXRCC4–LigIV complex gave a molecular mass of 160 kD using the reciprocal space approach, and 140 kD using the real space approach. These data are consistent with the 1:2:1 PNKP:pXRCC4:LigIV stoichiometry (theoretical mass: 141.6 kD) and our MALLS analysis (Figure 1D). Kratky plots for both complexes failed to reach a baseline plateau at higher-q indicating the presence of conformational flexibility rather than a rigid, globular domain (Supplementary Figure S3). Conformational flexibility was further corroborated with the presence of an asymptopic plateau for higher-q data after the Guinier region in a q3.I(q) versus q3 plot, indicating partially-folded particles (58) (Supplementary Figure S4).
Figure 3.

SAXS ensemble analyses indicate PNKP–pXRCC4–LigIV adopts compact and elongated conformations. (A) XRCC41–238/LigIV654–911 experimental X-ray scattering (black) is displayed with the calculated scattering of the XRCC41–203/LigIV654–911 X-ray crystal structure (pdb code 3II6, red), and the scattering calculated from a five model ensemble in which XRCC4 residues 202–238 were allowed to be flexible (green). Residuals are shown below. (B) PNKP–pXRCC4–LigIV experimental X-ray scattering is shown (black) with calculated scattering of a five-model ensemble with χ2 1.7 (red). Four of the theoretical scattering curves for the individual models that constitute the ensemble are shown with curves corresponding to elongated structures in blue, and the single curve corresponding to a compact structure in green. (C) Frequency of occurrence of models extracted in multiple runs of the genetic algorithm plotted against RG. A bi-modal distribution is evident relative to that of the model library (black) indicating a mixture of compact (green) and extended conformations (blue). (D) Models from the ensemble with their RG and the frequency of occurrence. χ2 for fit of individual models to data, from left to right: 13.9; 9.6; 9.5; 15.2; 22.5. Each model is coloured with XRCC4 (blue), Ligase IV (green), polypeptide linkers (orange), PNKP FHA domain (wheat), PNKP catalytic domain (white).
We used the molecular dynamics based conformational sampling as implemented in BILBOMD (59) along with the ensemble optimization methods in GAJOE (60) to model the population of conformations adopted by the PNKP–pXRCC4–LigIV complex. BILBOMD was used to generate a library of conformers representing the conformational space for the pXRCC4–LigIV-PNKP ternary complex. Existing crystal structures for mouse PNKP, pXRCC4–LigIV and mouse PNKP–FHA bound to cognate human XRCC4 phosphopeptide linked by flexible polypeptide tethers were used to generate model libraries sampling complexes containing either one or two PNKP protomers bound per pXRCC4–LigIV (Materials and Methods). GAJOE uses a genetic algorithm to search a library of possible conformers to find ensembles of conformations whose combined theoretical scattering intensities have a best fit to the experimental SAXS data. Despite the asymmetry in the pXRCC4–LigIV crystal structure, ensembles from pools containing single PNKP complexes on chain A or chain B showed similar fits of χ2 1.6 and 1.8 respectively. In contrast, the ensemble derived from the pool containing complex models with two PNKP molecules produced a poorer fit to the data with a χ2 of 8.7. Combining both single-PNKP model sets produced an ensemble fit similar to the individual sets at χ2 of 1.7. Adding the doubly-bound PNKP models to this combined pool produced no improvement in fit of the ensemble to the data, although conformations where both PNKP molecules are bound to pXRCC4–LigIV contributed to the final ensemble at around 20%. The theoretical scattering and fit to experimental data for both the pXRCC4–LigIV and PNKP–pXRCC4–LigIV ensembles are shown in Figure 3A, B. Representative models from the best pXRCC4/LigIV/PNKP ensemble are shown in Figure 3D. The radius of gyration (RG) frequency distributions averaged from multiple runs of the genetic algorithm show a clear bimodal distribution relative to the pool (Figure 3C), suggesting a mixture of compact and elongated conformations in solution. Inspection of the models from the elongated group revealed structures in which the PNKP FHA and catalytic domain extend away from XRCC4–LigIV (Figure 3D).
In contrast, the compact group contains models where PNKP catalytic and FHA domains are folded back and in contact with the XRCC4 structured domain and/or the LigIV BRCTs. Comparison of the RG distribution of the selected structures with that of the model set reveals that while the extended selected group is near the center of the RG distribution of the entire model set, the compact selected group is at the small end of the RG distribution range. Together, this analysis suggests that while the PNKP–XRCC4–LigIV complex is flexible and samples dynamic extended conformations, it also samples compact conformations more frequently than would be expected based on MD simulation. Analysis of the compact states suggest that they may be stabilized by interactions between PNKP and the structured N-terminal domain of XRCC4 and/or the BRCT domain of LigIV in addition to the interaction of the phosphorylated C-terminal tail of XRCC4 and the PNKP FHA domain.
HX-MS suggests interactions between PNKP catalytic domain and XRCC4–LigIV
While the SAXS study suggested an interaction between PNKP and XRCC4–LigIV beyond the FHA–phosphopeptide interaction, the modest resolution of SAXS precluded an unambiguous identification of specific contact surfaces. We used HX-MS to detect changes in the deuteration profile of PNKP and XRCC4–LigIV upon complex formation. We were specifically interested in peptide regions with decreased hydrogen–deuterium exchange behavior that might represent interaction surfaces. When attempting controlled proteolysis of the pXRCC–LigIV–PNKP complex for HX-MS we were unable to generate usable fragments from either XRCC4 or LigIV and analysis was limited to PNKP. We detected several discrete fragments from PNKP that were protected against exchange when in the ternary complex. As expected from the mPNKP FHA-cognate XRCC4 phosphopeptide crystal structure (27), multiple peptides from the PNKP FHA domain, and particularly the loops that form the XRCC4 phosphopeptide binding surface, were the most strongly protected in the complex (Figure 4). The only other significant region of protection was a set of overlapping peptides in the phosphatase domain between residues 313 and 327 (Delta values between ∼–3 and –2). These peptides correspond to the α6–β14–α7 region near the C-terminus of the phosphatase domain, which is exposed on a surface of the domain distant from the catalytic site (Figure 4C). Interestingly, each of these peptides contains Glu326. Glu326 mutation to Lys (E326K) has previously been associated with MCSZ (49). We propose that the compact conformation of the PNKP–pXRCC4–LigIV complex implicates an interaction of the Glu326 surface with XRCC4–LigIV and that mutation of Glu326 may disrupt this interaction and explain the molecular basis for MCSZ.
Figure 4.

HX-MS reveals regions of PNKP FHA and phosphatase domain are protected by pXRCC4–LigIV. (A) Left panel, Woods plot of HX-MS PNKP peptide protection data in which peptides that are significantly protected in complex with pXRCC4–LigIV (Delta value < –1.4) are colored blue while those that show significantly enhanced exchange in the complex (Delta value > 1.4) are colored red. Right panel, overview of the PNKP structure with peptides showing significant protection colored blue. The phosphatase and kinase active sites are colored yellow (PDB ID: 1YJ5). (B) View of protected peptides within the FHA domain. The CK2-phosphorylated peptide from XRCC4 is coloured green (PDB ID: 1YJM). (C) View of the protected peptide within the PNKP phosphatase domain.
E326K mutation does not disrupt interactions with XRCC4–LigIV
We used gel filtration chromatography to test if the E326K mutation impacts the ability of PNKP to form complexes with XRCC4–LigIV (Figure 5). Recombinant PNKP E326K was expressed and purified in similar yield to the WT, suggesting that this mutation does not result in a gross misfolding of the protein, consistent with previous findings (61). Addition of one mole equivalent of PNKP to one mole equivalent of phosphorylated XRCC4–LigIV yielded a complex that ran as a single peak on gel filtration chromatography with the same elution volume as the WT control. Mixtures of the E326K mutant with non-phosphorylated XRCC4–LigIV did not form a single peak. These results suggest that like the WT protein, PNKP E326K is capable of forming a stable phosphorylation-dependent 1:2:1 complex with pXRCC4–LigIV.
Figure 5.
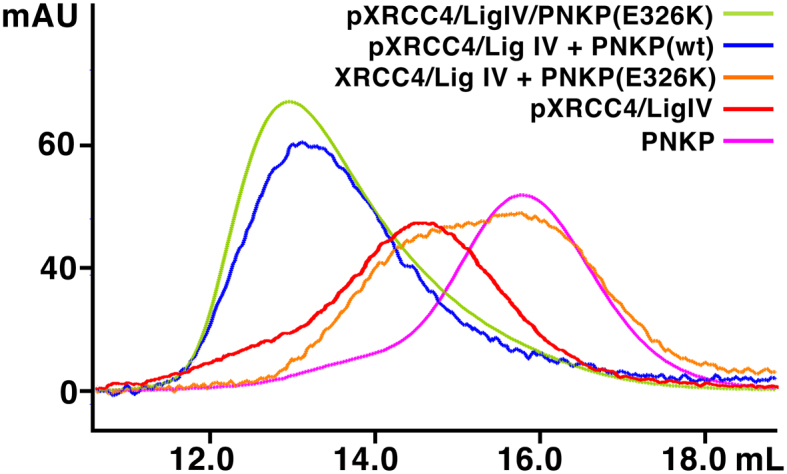
PNKP E326K is competent to bind pXRCC4–LigIV. One mole equivalent PNKP (wt or E326K) was mixed with one mole equivalent of XRCC4–LigIV complex (+/– phosphorylation) and separated by Superdex 200 analytical gel filtration chromatography as in Figure 1B.
E326K mutation modulates the effect of pXRCC4–LigIV on PNKP kinase activity
Previous work had suggested that interactions between XRCC4 and PNKP regulate PNKP kinase activity (48). These results suggested that phosphorylated XRCC4 inhibits PNKP kinase activity, while the non-phosphorylated XRCC4 enhanced product turnover. The XRCC4–LigIV complex, however displayed a different behavior in that both the phosphorylated and non-phosphorylated forms appeared to enhance PNKP kinase product turnover. To test the effects of our XRCC4–LigIV complex on PNKP kinase activity, and to assess the effects of the PNKP E326K mutation, we measured ATP to ADP conversion using a coupled pyruvate kinase/lactate dehydrogenase ATPase assay (see Supplementary Materials).
Unlike the previous work, this assay allowed the quantitation of the kinetic parameters of the kinase reaction. The results show that the KM is not significantly affected by complex formation with either non-phosphorylated or phosphorylated XRCC4–LigIV suggesting that substrate binding by wild type PNKP is not impacted by XRCC4–LigIV (Table 1). Substrate turnover as assessed by kcat was enhanced by XRCC4–LigIV, most significantly for pXRCC4–LigIV. Overall, these results resemble previous qualitative studies, however, the magnitudes of the effects are more subtle than anticipated by prior work. We compared these measurements on WT PNKP to measurements obtained for PNKP E326K. The E326K mutant showed a slightly reduced kcat compared to WT, and a similar KM, suggesting similar substrate binding affinity but reduced product turnover. The effect of XRCC4–LigIV complex on the kcat of the mutant was less significant than the WT, suggesting that the mutation may reduce the ability of XRCC4–LigIV to effect product turnover. In contrast, pXRCC4–LigIV significantly increased the KM of PNKP E326K, suggesting that the interaction of pXRCC4–LigIV reduces the binding of substrate by the mutant. Taken together, pXRCC4/LigIV enhances the catalytic efficiency (kcat/KM) of WT PNKP, while pXRCC4/LigIV actually impairs the catalytic efficiency of the E326K mutant. While these results are subtle, taken together they suggest that the E326K mutation interferes with both substrate binding and product turnover in the context of pXRCC4–LigIV.
Table 1. Impact of XRCC4–LigIV on PNKP kinase activity.
| K M (μM) | k cat (s−1) | k cat/KM (μM−1•s−1) | |
|---|---|---|---|
| PNKP | 34 ± 5 | 0.45 ± 0.03 | 0.013 ± 0.003 |
| PNKP-XRCC4–LigIV | 41 ± 6 | 0.47 ± 0.03 | 0.012 ± 0.002 |
| PNKP–pXRCC4–LigIV | 33 ± 5 | 0.60 ± 0.04 | 0.018 ± 0.004 |
| PNKPE326K | 26 ± 3 | 0.33 ± 0.014 | 0.013 ± 0.0019 |
| PNKPE326K–XRCC4–LigIV | 25 ± 4 | 0.38 ± 0.02 | 0.015 ± 0.003 |
| PNKPE326K–pXRCC4–LigIV | 44 ± 6 | 0.42 ± 0.03 | 0.0096 ± 0.0019 |
| XRCC4–LigIV | 0.4 ± 3 | 0.029 ± 0.009 | 0.07 ± 0.4 |
| pXRCC4–LigIV | 0.2 ± 0.2 | 0.38 ± 0.011 | 2.1 ± 2.0 |
K m and kcat values are derived from measurements of ATP to ADP conversion rates for the indicated protein complexes, as measured with the coupled pyruvate kinase/lactate dehydrogenase ATPase assay. The indicated errors represent the standard deviations from nine experiments.
PNKP Glu326 and FHA domain act in PNKP recruitment to sites of damage
To determine the effects of the E326K mutation on PNKP function in vivo, U2OS cells were transiently transfected with EGFP–PNKP and the kinetics of recruitment to sites of DNA damage in vivo examined using UV laser microirradiation. Briefly, GFP–PNKP transfected cells were grown on coverslips then pre-sensitized with BrdU. DNA damage was introduced using a 355 nm CryLas FTSS355-50 UV-Laser PALM MicroBeam system as described previously (62). Localization of GFP–PNKP to sites of DNA damage was determined at 30-s intervals for a 20-min time period in 30 cells for each time point. Relative fluorescence intensity was determined by subtracting the background fluorescence from the fluorescence intensity at the GFP–PNKP track for each cell as described in Supplementary Materials (62). Wild-type PNKP was rapidly recruited to sites of damage, reaching maximum intensity within 3 minutes and remaining at ∼70% maximum intensity over a 20-min period (Figure 6). We confirmed the importance of the FHA-group in recruitment to sites of DNA damage using PNKP in which R35 in the FHA domain had been ablated (EGFP–PNKP–R35A) (14). In agreement with a previous study in MEFs (63), mutation of the FHA domain virtually abolished PNKP recruitment to DNA damage sites. We next transfected cells with EGFP–PNKP–E326K. Significantly, recruitment of GFP–PNKP–E326K to laser tracks was <50% of that of GFP–PNKP-wt, indicating that Glu326 plays an important role in recruitment of PNKP to sites of DNA damage in vivo (Figure 6).
Figure 6.

E326K mutation abrogates recruitment of PNKP to sites of DNA damage. (A) U2OS cells were transiently transfected with EGFP–PNKP–wt (circles), EGFP–PNKP–R35A (triangles) or EGFP–PNKP–E326K (squares). Laser tracks were introduced after BrdU sensitization and the relative fluorescence determined at 30-s intervals. The means of 30 cells with standard error of the mean at each time point is shown. All data points for EGFP–PNKP–R35A and EGFP–PNKP–E326K were statistically significant (P < 0.05) with respect to EGFP–PNKP–wt. (B) Representative images of laser tracks in cells expressing EGFP–PNKP–wt, EGFP–PNKP–R35A and EGFP–PNKP–E326K. The images show GFP-tagged proteins expressed in the nuclei of individual cells. See Supplementary Figure S5 for an expanded view.
To determine whether GFP–PNKP–E326K was able to interact with XRCC4, U2OS cells were transiently transfected with GFP–PNKP–wt, GFP–PNKP–R35A or GFP–PNKP–E326K and cell extracts were made from either unirradiated cells or after irradiation at 10 Gy (Figure 7). GFP–PNKP was immunoprecipitated and immunoprecipitates were probed for GFP (PNKP) and XRCC4. 50 μg of total cell extract was run on the same gels (inputs, Figure 7). The amount of PNKP immunoprecipitated from cells expressing PNKP–E326K was slightly reduced (compared to that in cells expressing wild type-PNKP) in both the presence and absence of DNA damage (compare lanes 1 and 2 to lanes 5 and 6). Similarly, less XRCC4 immunoprecipitated with GFP–PNKP–E326K in unirradiated cell (compared to GFP-PNKP wt) but after DNA damage, the amount of XRCC4 interacting with GFP–PNKP–E326K decreased significantly (Figure 7, lane 6). Together, these data suggest that PNKP-E326K is defective in its ability to be recruited to sites of DNA damage, possibly though decreased interaction with XRCC4 after DNA damage.
Figure 7.
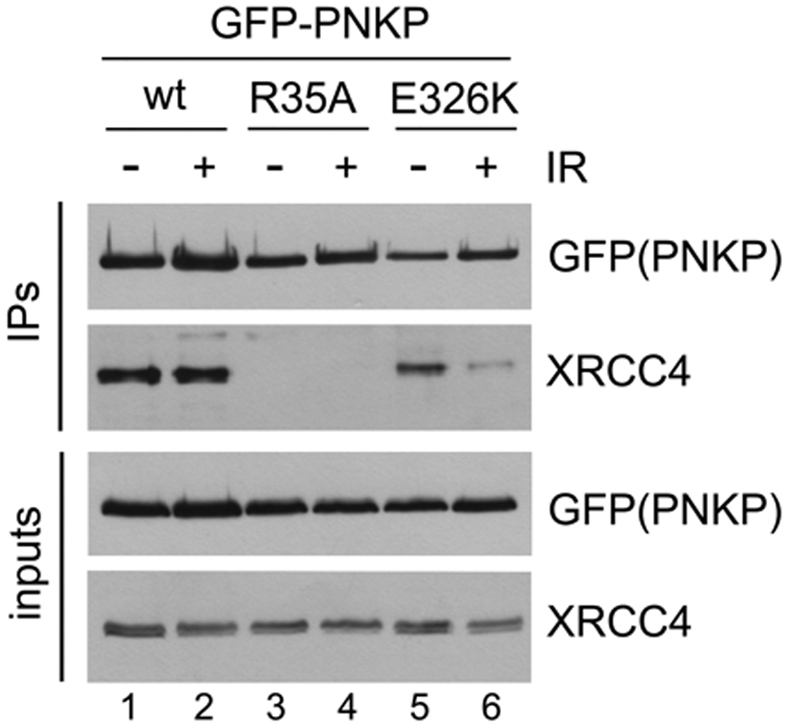
E326K mutation reduces the interaction of PNKP with XRCC4. U2OS cells were transfected with EGFP–PNKP–wt, R35A or E326K and after 24 h, either unirradiated (–) or irradiated at 10 Gy and harvested after 1 h. PNKP was immunoprecipitated using anti-GFP antibody and immunoprecipitates were probed with antibodies to GFP (GFP-PNKP) or XRCC4 as shown. The doublets in blots of XRCC4 are likely due to phosphorylation as shown previously (48) and data not shown. The experiment shown is representative of three separate experiments.
DISCUSSION
PNKP is among the most well-studied of the DNA end processing factors, but defining its interactions with the core NHEJ complex has been challenging. Yet, these protein partner interactions are fundamental to insights into efficient and specific DNA end recognition and processing for ligation. Such end processing is under selective pressure to avoid releasing mutagenic intermediates that could cause genome instability. Here, we find that PNKP forms a flexible, phosphorylation-dependent complex with XRCC4 and the BRCT repeat region of DNA ligase IV. Integration of the data indicates that there are complex interactions between PNKP and XRCC4–LigIV in addition to that between the PNKP FHA domain and the phosphorylated C-terminal tail of XRCC4. Modeling the available conformational states based on our SAXS data indicates that the complex adopts discrete compact conformations in which the PNKP catalytic and FHA domains pack against the coiled-coil of XRCC4 and the LigIV BRCT repeats. These solution conformations suggest that while the complex is anchored by the FHA–phosphopeptide interaction, added weak modular interactions among other surfaces may impact the population of compact conformations. Further evidence for these interactions comes from HX-MS experiments, which reveal significant protection of a peptide on the surface of the PNKP phosphatase domain when in complex with XRCC4–LigIV. Thus, combined SAXS and HX-MS results suggest weak additional interactions that involve, at least in part, interactions of the protected PNKP peptide with XRCC4–LigIV.
These results support and extend previous work analyzing interactions of PNKP with isolated XRCC4 (48). Using a fluorescently-tagged PNKP mutant, we showed binding of PNKP to both phosphorylated and non-phosphorylated XRCC4, although binding to pXRCC4 was tighter than to non-phosphorylated XRCC4 (48). However, using either gel filtration chromatography or EMSA, we do not see evidence for an interaction in the absence of phosphorylation, suggesting that for the XRCC4–LigIV complex, binding of PNKP to the non-phosphorylated complex is very weak. Our previous work also suggested that XRCC4 could stimulate PNKP catalytic activity by enhancing substrate turnover. Using our quantitative assay of PNKP kinase activity, we find that the enhancement of PNKP kcat is modest. Our analysis of the effects of XRCC4–LigIV on PNKP substrate interactions indicates that while PNKP–DNA complexes are capable of binding pXRCC4–LigIV, DNA binding affinity is not significantly impacted by this interaction. We therefore believe that the role of pXRCC4–LigIV in the regulation of PNKP may involve other mechanisms besides the simple enhancement of product turnover.
In the context of NHEJ, XRCC4 is thought to be a central scaffold that coordinates handoff of DNA repair intermediates between various DNA repair enzymes. The CK2-phosphorylated tail of XRCC4 governs the interactions with PNKP, APTX and APLF through their structurally-related FHA domains. XRCC4 also interacts with XLF to form DNA-binding filaments that have been suggested to mediate synapsis of the damaged DNA ends (3,56,64–67). APLF may also play a scaffolding role, utilizing its poly(ADP)-ribose binding PDZ domain (68,69) and its Ku80 binding motif (22,70) to potentially link chromatin and other key components of NHEJ to XRCC4 (71). Further complicating this picture is the recent finding that interactions with Ku may also be facilitated by K63-linked poly-ubiquitylation of XRCC4 (72). The idea of a coordinated handoff between these enzymes is an attractive one that can explain how the potentially toxic intermediates can be protected until repair is finalized (41).
The 1:2 PNKP:XRCC4 stoichiometry within the PNKP–pXRCC4–LigIV complex suggests that PNKP only binds one of the phosphorylated XRCC4 C-terminal tails and opens the possibility for simultaneous binding of either APLF or APTX to the other XRCC4 C-terminal tail. Weak interactions between the PNKP catalytic domain and XRCC4–LigIV, like those in the proposed PNKP Glu326 region, also seem suitable to position PNKP to gain access to DNA within the XRCC4-XLF filament. These secondary interactions could regulate and facilitate the ordered sampling of the DNA termini by the tethered catalytic domains of PNKP, APTX and LigIV.
Furthermore, PNKP not only participates in NHEJ, but is also important for single-strand break repair (SSBR) in the base excision repair (BER) pathway. It is especially prominent in repair involving the NEIL class of DNA glycosylases (70). Analogous to XRCC4 in NHEJ, BER and SSBR in general is coordinated by XRCC1. Like XRCC4, XRCC1 is also phosphorylated by CK2 at multiple sites that are targets for PNKP binding (28–30). While XRCC1 bears no structural similarity to XRCC4, it does contain BRCT domains which may play a role in poly(ADP)-ribose binding at sites of damage (73) as well as other domains that may be important in stabilizing PNKP binding (45,46). It is then possible that the Glu326 region also plays a role in recruitment of PNKP to single strand break lesions and that the E326K mutation disrupts directed PNKP activity in BER and SSBR.
Clinically, our results may help to explain the severe phenotype associated with the E326K mutation in MCSZ (49). The severity of the mutant phenotype has been puzzling since the mutation itself does not dramatically destabilize the folding of PNKP and does not significantly reduce PNKP catalytic efficiency in assays with purified recombinant protein (Table 1 and (61)). Moreover, we find that the purified E326K mutant is competent to form the 1:2:1 PNKP:pXRCC4:LigIV complex (Figure 5). In contrast to this in vitro data, cells isolated from E326K MCSZ patients show extremely low levels of PNKP (49,61). Lower levels of PNKP-E326K were also observed after transient transfection (Figure 7), but PNKP-E326K still interacted with XRCC4 in unirradiated cells although the amount decreased after DNA damage. Similarly, less GFP-PNKP E326K was recruited to sites of DNA damage in laser micro-irradiation experiments (Figure 6). We therefore suggest that at least part of the defect associated with this mutation may be to destabilize the interaction of PNKP with its partner proteins specifically under conditions of DNA damage. The dissociation of the PNKP E326K variant from DNA repair complexes might result in its degradation and loss in MCSZ patient cells.
Supplementary Material
ACKNOWLEDGEMENTS
We thank our many colleagues for creative ideas and advice including those in the Structural Biology of DNA Repair program and Dr Anne Vaahtokari and the Charbonneau Microscopy Facility at the University of Calgary for assistance with laser micro-irradiation experiments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
PO1CA092584 (to J.A.T., J.N.M.G., S.P.L.M.); CIHR [114975 to J.N.M.G., M.W.]; CIHR [MOP13639 to S.P.L.M.]; CIHR [MOP15385 to M.W.]; NSERC Discovery [298351-2010 to D.C.S.]. J.A.T. acknowledges added support of a Robert A. Welch Chemistry Chair, plus startup funds from the Cancer Prevention and Research Institute of Texas, and the University of Texas STARs program. D.C.S. acknowledges the additional support of Alberta Ingenuity—Health Solutions and the Canada Foundation for Innovation. S.A.X.S. efforts at the SIBYLS beamline of the Advanced Light Source (Lawrence Berkeley National Laboratory) are supported in part by United States Department of Energy program IDAT. Funding for open access charge: PO1CA092584.
Conflict of interest statement. None declared.
REFERENCES
- 1. Radhakrishnan S.K., Jette N., Lees-Miller S.P.. Non-homologous end joining: emerging themes and unanswered questions. DNA Repair (Amst.). 2014; 17:2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang C., Lees-Miller S.P.. Detection and repair of ionizing radiation-induced DNA double strand breaks: new developments in nonhomologous end joining. Int. J. Radiat. Oncol. Biol. Phys. 2013; 86:440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hammel M., Yu Y., Fang S., Lees-Miller S.P., Tainer J.A.. XLF regulates filament architecture of the XRCC4.ligase IV complex. Structure. 2010; 18:1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brouwer I., Sitters G., Candelli A., Heerema S.J., Heller I., de Melo A.J., Zhang H., Normanno D., Modesti M., Peterman E.J. et al. . Sliding sleeves of XRCC4-XLF bridge DNA and connect fragments of broken DNA. Nature. 2016; 535:566–569. [DOI] [PubMed] [Google Scholar]
- 5. Grundy G.J., Rulten S.L., Arribas-Bosacoma R., Davidson K., Kozik Z., Oliver A.W., Pearl L.H., Caldecott K.W.. The Ku-binding motif is a conserved module for recruitment and stimulation of non-homologous end-joining proteins. Nat. Commun. 2016; 7:11242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma Y., Lu H., Tippin B., Goodman M.F., Shimazaki N., Koiwai O., Hsieh C.L., Schwarz K., Lieber M.R.. A biochemically defined system for mammalian nonhomologous DNA end joining. Mol. Cell. 2004; 16:701–713. [DOI] [PubMed] [Google Scholar]
- 7. Ochi T., Blackford A.N., Coates J., Jhujh S., Mehmood S., Tamura N., Travers J., Wu Q., Draviam V.M., Robinson C.V. et al. . DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science. 2015; 347:185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xing M., Yang M., Huo W., Feng F., Wei L., Jiang W., Ning S., Yan Z., Li W., Wang Q. et al. . Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nat. Commun. 2015; 6:6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Henner W.D., Rodriguez L.O., Hecht S.M., Haseltine W.A.. gamma Ray induced deoxyribonucleic acid strand breaks. 3΄ Glycolate termini. J. Biol. Chem. 1983; 258:711–713. [PubMed] [Google Scholar]
- 10. Buchko G.W., Weinfeld M.. Influence of nitrogen, oxygen, and nitroimidazole radiosensitizers on DNA damage induced by ionizing radiation. Biochemistry. 1993; 32:2186–2193. [DOI] [PubMed] [Google Scholar]
- 11. Lennartz M., Coquerelle T., Bopp A., Hagen U.. Oxygen–effect on strand breaks and specific end-groups in DNA of irradiated thymocytes. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1975; 27:577–587. [DOI] [PubMed] [Google Scholar]
- 12. Menon V., Povirk L.F.. End-processing nucleases and phosphodiesterases: an elite supporting cast for the non-homologous end joining pathway of DNA double-strand break repair. DNA Repair (Amst.). 2016; 43:57–68. [DOI] [PubMed] [Google Scholar]
- 13. Andres S.N., Schellenberg M.J., Wallace B.D., Tumbale P., Williams R.S.. Recognition and repair of chemically heterogeneous structures at DNA ends. Environ. Mol. Mutagen. 2015; 56:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koch C.A., Agyei R., Galicia S., Metalnikov P., O’Donnell P., Starostine A., Weinfeld M., Durocher D.. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J. 2004; 23:3874–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clements P.M., Breslin C., Deeks E.D., Byrd P.J., Ju L., Bieganowski P., Brenner C., Moreira M.C., Taylor A.M., Caldecott K.W.. The ataxia-oculomotor apraxia 1 gene product has a role distinct from ATM and interacts with the DNA strand break repair proteins XRCC1 and XRCC4. DNA Repair (Amst.). 2004; 3:1493–1502. [DOI] [PubMed] [Google Scholar]
- 16. Iles N., Rulten S., El-Khamisy S.F., Caldecott K.W.. APLF (C2orf13) is a novel human protein involved in the cellular response to chromosomal DNA strand breaks. Mol. Cell. Biol. 2007; 27:3793–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kanno S., Kuzuoka H., Sasao S., Hong Z., Lan L., Nakajima S., Yasui A.. A novel human AP endonuclease with conserved zinc-finger-like motifs involved in DNA strand break responses. EMBO J. 2007; 26:2094–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bekker-Jensen S., Fugger K., Danielsen J.R., Gromova I., Sehested M., Celis J., Bartek J., Lukas J., Mailand N.. Human Xip1 (C2orf13) is a novel regulator of cellular responses to DNA strand breaks. J. Biol. Chem. 2007; 282:19638–19643. [DOI] [PubMed] [Google Scholar]
- 19. Macrae C.J., McCulloch R.D., Ylanko J., Durocher D., Koch C.A.. APLF (C2orf13) facilitates nonhomologous end-joining and undergoes ATM-dependent hyperphosphorylation following ionizing radiation. DNA Repair (Amst.). 2008; 7:292–302. [DOI] [PubMed] [Google Scholar]
- 20. Weinfeld M., Mani R.S., Abdou I., Aceytuno R.D., Glover J.N.. Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair. Trends Biochem. Sci. 2011; 36:262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schellenberg M.J., Tumbale P.P., Williams R.S.. Molecular underpinnings of Aprataxin RNA/DNA deadenylase function and dysfunction in neurological disease. Prog. Biophys. Mol. Biol. 2015; 117:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grundy G.J., Rulten S.L., Zeng Z., Arribas-Bosacoma R., Iles N., Manley K., Oliver A., Caldecott K.W.. APLF promotes the assembly and activity of non-homologous end joining protein complexes. EMBO J. 2013; 32:112–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ahel I., Ahel D., Matsusaka T., Clark A.J., Pines J., Boulton S.J., West S.C.. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature. 2008; 451:81–85. [DOI] [PubMed] [Google Scholar]
- 24. Rulten S.L., Cortes-Ledesma F., Guo L., Iles N.J., Caldecott K.W.. APLF (C2orf13) is a novel component of poly(ADP-ribose) signaling in mammalian cells. Mol. Cell. Biol. 2008; 28:4620–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li S., Kanno S., Watanabe R., Ogiwara H., Kohno T., Watanabe G., Yasui A., Lieber M.R.. Polynucleotide kinase and aprataxin-like forkhead-associated protein (PALF) acts as both a single-stranded DNA endonuclease and a single-stranded DNA 3΄ exonuclease and can participate in DNA end joining in a biochemical system. J. Biol. Chem. 2011; 286:36368–36377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cherry A.L., Nott T.J., Kelly G., Rulten S.L., Caldecott K.W., Smerdon S.J.. Versatility in phospho-dependent molecular recognition of the XRCC1 and XRCC4 DNA-damage scaffolds by aprataxin-family FHA domains. DNA Repair (Amst.). 2015; 35:116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bernstein N.K., Williams R.S., Rakovszky M.L., Cui D., Green R., Karimi-Busheri F., Mani R.S., Galicia S., Koch C.A., Cass C.E. et al. . The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase. Mol. Cell. 2005; 17:657–670. [DOI] [PubMed] [Google Scholar]
- 28. Loizou J.I., El-Khamisy S.F., Zlatanou A., Moore D.J., Chan D.W., Qin J., Sarno S., Meggio F., Pinna L.A., Caldecott K.W.. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. 2004; 117:17–28. [DOI] [PubMed] [Google Scholar]
- 29. Ali A.A., Jukes R.M., Pearl L.H., Oliver A.W.. Specific recognition of a multiply phosphorylated motif in the DNA repair scaffold XRCC1 by the FHA domain of human PNK. Nucleic Acids Res. 2009; 37:1701–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Breslin C., Caldecott K.W.. DNA 3΄-phosphatase activity is critical for rapid global rates of single-strand break repair following oxidative stress. Mol. Cell. Biol. 2009; 29:4653–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tahbaz N., Subedi S., Weinfeld M.. Role of polynucleotide kinase/phosphatase in mitochondrial DNA repair. Nucleic Acids Res. 2012; 40:3484–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mandal S.M., Hegde M.L., Chatterjee A., Hegde P.M., Szczesny B., Banerjee D., Boldogh I., Gao R., Falkenberg M., Gustafsson C.M. et al. . Role of human DNA glycosylase Nei-like 2 (NEIL2) and single strand break repair protein polynucleotide kinase 3΄-phosphatase in maintenance of mitochondrial genome. J. Biol. Chem. 2012; 287:2819–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rasouli-Nia A., Karimi-Busheri F., Weinfeld M.. Stable down-regulation of human polynucleotide kinase enhances spontaneous mutation frequency and sensitizes cells to genotoxic agents. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:6905–6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Freschauf G.K., Karimi-Busheri F., Ulaczyk-Lesanko A., Mereniuk T.R., Ahrens A., Koshy J.M., Rasouli-Nia A., Pasarj P., Holmes C.F., Rininsland F. et al. . Identification of a small molecule inhibitor of the human DNA repair enzyme polynucleotide kinase/phosphatase. Cancer Res. 2009; 69:7739–7746. [DOI] [PubMed] [Google Scholar]
- 35. Freschauf G.K., Mani R.S., Mereniuk T.R., Fanta M., Virgen C.A., Dianov G.L., Grassot J.M., Hall D.G., Weinfeld M.. Mechanism of action of an imidopiperidine inhibitor of human polynucleotide kinase/phosphatase. J. Biol. Chem. 2010; 285:2351–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zereshkian A., Leyton J.V., Cai Z., Bergstrom D., Weinfeld M., Reilly R.M.. The human polynucleotide kinase/phosphatase (hPNKP) inhibitor A12B4C3 radiosensitizes human myeloid leukemia cells to Auger electron-emitting anti-CD123 (1)(1)(1)In-NLS-7G3 radioimmunoconjugates. Nucl. Med. Biol. 2014; 41:377–383. [DOI] [PubMed] [Google Scholar]
- 37. Karimi-Busheri F., Lee J., Tomkinson A.E., Weinfeld M.. Repair of DNA strand gaps and nicks containing 3΄-phosphate and 5΄-hydroxyl termini by purified mammalian enzymes. Nucleic Acids Res. 1998; 26:4395–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bernstein N.K., Hammel M., Mani R.S., Weinfeld M., Pelikan M., Tainer J.A., Glover J.N.. Mechanism of DNA substrate recognition by the mammalian DNA repair enzyme, polynucleotide kinase. Nucleic Acids Res. 2009; 37:6161–6173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Coquelle N., Havali-Shahriari Z., Bernstein N., Green R., Glover J.N.. Structural basis for the phosphatase activity of polynucleotide kinase/phosphatase on single- and double-stranded DNA substrates. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:21022–21027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Garces F., Pearl L.H., Oliver A.W.. The structural basis for substrate recognition by mammalian polynucleotide kinase 3΄ phosphatase. Mol. Cell. 2011; 44:385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wilson S.H., Kunkel T.A.. Passing the baton in base excision repair. Nat. Struct. Biol. 2000; 7:176–178. [DOI] [PubMed] [Google Scholar]
- 42. Mani R.S., Karimi-Busheri F., Fanta M., Cass C.E., Weinfeld M.. Spectroscopic studies of DNA and ATP binding to human polynucleotide kinase: evidence for a ternary complex. Biochemistry. 2003; 42:12077–12084. [DOI] [PubMed] [Google Scholar]
- 43. Whitehouse C.J., Taylor R.M., Thistlethwaite A., Zhang H., Karimi-Busheri F., Lasko D.D., Weinfeld M., Caldecott K.W.. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001; 104:107–117. [DOI] [PubMed] [Google Scholar]
- 44. Chappell C., Hanakahi L.A., Karimi-Busheri F., Weinfeld M., West S.C.. Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J. 2002; 21:2827–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Della-Maria J., Hegde M.L., McNeill D.R., Matsumoto Y., Tsai M.S., Ellenberger T., Wilson D.M. 3rd, Mitra S., Tomkinson A.E.. The interaction between polynucleotide kinase phosphatase and the DNA repair protein XRCC1 is critical for repair of DNA alkylation damage and stable association at DNA damage sites. J. Biol. Chem. 2012; 287:39233–39244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu M., Mani R.S., Karimi-Busheri F., Fanta M., Wang H., Litchfeld D.W., Weinfeld M.. Independent mechanisms of stimulation of polynucleotide kinase/phosphatase by phosphorylated and non-phosphorylated XRCC1. Nucleic Acids Res. 2010; 38:510–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mani R.S., Fanta M., Karimi-Busheri F., Silver E., Virgen C.A., Caldecott K.W., Cass C.E., Weinfeld M.. XRCC1 stimulates polynucleotide kinase by enhancing its damage discrimination and displacement from DNA repair intermediates. J. Biol. Chem. 2007; 282:28004–28013. [DOI] [PubMed] [Google Scholar]
- 48. Mani R.S., Yu Y., Fang S., Lu M., Fanta M., Zolner A.E., Tahbaz N., Ramsden D.A., Litchfield D.W., Lees-Miller S.P. et al. . Dual modes of interaction between XRCC4 and polynucleotide kinase/phosphatase: implications for nonhomologous end joining. J. Biol. Chem. 2010; 285:37619–37629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shen J., Gilmore E.C., Marshall C.A., Haddadin M., Reynolds J.J., Eyaid W., Bodell A., Barry B., Gleason D., Allen K. et al. . Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat. Genet. 2010; 42:245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wales T.E., Engen J.R.. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 2006; 25:158–170. [DOI] [PubMed] [Google Scholar]
- 51. Rey M., Yang M., Burns K.M., Yu Y., Lees-Miller S.P., Schriemer D.C.. Nepenthesin from monkey cups for hydrogen/deuterium exchange mass spectrometry. Mol. Cell. Proteomics. 2013; 12:464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rey M., Sarpe V., Burns K.M., Buse J., Baker C.A., van Dijk M., Wordeman L., Bonvin A.M., Schriemer D.C.. Mass spec studio for integrative structural biology. Structure. 2014; 22:1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Modesti M., Junop M.S., Ghirlando R., van de Rakt M., Gellert M., Yang W., Kanaar R.. Tetramerization and DNA ligase IV interaction of the DNA double-strand break repair protein XRCC4 are mutually exclusive. J. Mol. Biol. 2003; 334:215–228. [DOI] [PubMed] [Google Scholar]
- 54. Putnam C.D., Hammel M., Hura G.L., Tainer J.A.. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007; 40:191–285. [DOI] [PubMed] [Google Scholar]
- 55. Hammel M., Rey M., Yu Y., Mani R.S., Classen S., Liu M., Pique M.E., Fang S., Mahaney B.L., Weinfeld M. et al. . XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. J. Biol. Chem. 2011; 286:32638–32650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Williams G.J., Hammel M., Radhakrishnan S.K., Ramsden D., Lees-Miller S.P., Tainer J.A.. Structural insights into NHEJ: building up an integrated picture of the dynamic DSB repair super complex, one component and interaction at a time. DNA Repair (Amst.). 2014; 17:110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rambo R.P., Tainer J.A.. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013; 496:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rambo R.P., Tainer J.A.. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers. 2011; 95:559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pelikan M., Hura G.L., Hammel M.. Structure and Flexibility within proteins as identified through small angle X-ray scattering. Gen. Physiol. Biophys. 2009; 28:174–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bernado P., Mylonas E., Petoukhov M.V., Blackledge M., Svergun D.I.. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007; 129:5656–5664. [DOI] [PubMed] [Google Scholar]
- 61. Reynolds J.J., Walker A.K., Gilmore E.C., Walsh C.A., Caldecott K.W.. Impact of PNKP mutations associated with microcephaly, seizures and developmental delay on enzyme activity and DNA strand break repair. Nucleic Acids Res. 2012; 40:6608–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Klement K., Luijsterburg M.S., Pinder J.B., Cena C.S., Del Nero V., Wintersinger C.M., Dellaire G., van Attikum H., Goodarzi A.A.. Opposing ISWI- and CHD-class chromatin remodeling activities orchestrate heterochromatic DNA repair. J. Cell Biol. 2014; 207:717–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li M., Lu L.Y., Yang C.Y., Wang S., Yu X.. The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes Dev. 2013; 27:1752–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ropars V., Drevet P., Legrand P., Baconnais S., Amram J., Faure G., Marquez J.A., Pietrement O., Guerois R., Callebaut I. et al. . Structural characterization of filaments formed by human Xrcc4-Cernunnos/XLF complex involved in nonhomologous DNA end-joining. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:12663–12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Andres S.N., Vergnes A., Ristic D., Wyman C., Modesti M., Junop M.. A human XRCC4-XLF complex bridges DNA. Nucleic Acids Res. 2012; 40:1868–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Roy S., Andres S.N., Vergnes A., Neal J.A., Xu Y., Yu Y., Lees-Miller S.P., Junop M., Modesti M., Meek K.. XRCC4's interaction with XLF is required for coding (but not signal) end joining. Nucleic Acids Res. 2012; 40:1684–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Graham T.G., Walter J.C., Loparo J.J.. Two-stage synapsis of DNA ends during non-homologous end joining. Mol. Cell. 2016; 61:850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li G.Y., McCulloch R.D., Fenton A.L., Cheung M., Meng L., Ikura M., Koch C.A.. Structure and identification of ADP-ribose recognition motifs of APLF and role in the DNA damage response. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:9129–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Eustermann S., Brockmann C., Mehrotra P.V., Yang J.C., Loakes D., West S.C., Ahel I., Neuhaus D.. Solution structures of the two PBZ domains from human APLF and their interaction with poly(ADP-ribose). Nat. Struct. Mol. Biol. 2010; 17:241–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shirodkar P., Fenton A.L., Meng L., Koch C.A.. Identification and functional characterization of a Ku-binding motif in aprataxin polynucleotide kinase/phosphatase-like factor (APLF). J. Biol. Chem. 2013; 288:19604–19613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hammel M., Yu Y., Radhakrishnan S.K., Chokshi C., Tsai M.S., Matsumoto Y., Kuzdovich M., Remesh S.G., Fang S., Tomkinson A.E. et al. . An intrinsically disordered APLF links Ku, DNA-PKcs and XRCC4-DNA ligase IV in an extended flexible non-homologous end joining complex. J. Biol. Chem. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang Q., Karnak D., Tan M., Lawrence T.S., Morgan M.A., Sun Y.. FBXW7 facilitates nonhomologous end-joining via K63-linked polyubiquitylation of XRCC4. Mol. Cell. 2016; 61:419–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Breslin C., Hornyak P., Ridley A., Rulten S.L., Hanzlikova H., Oliver A.W., Caldecott K.W.. The XRCC1 phosphate-binding pocket binds poly (ADP-ribose) and is required for XRCC1 function. Nucleic Acids Res. 2015; 43:6934–6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.