


Abstract
Genetic mutations are important biomarkers for cancer diagnostics and surveillance. Preferably, the methods for mutation detection should be straightforward, highly specific and sensitive to low-level mutations within various sequence contexts, fast and applicable at room-temperature. Though some of the currently available methods have shown very encouraging results, their discrimination efficiency is still very low. Herein, we demonstrate a branch-migration based fluorescent probe (BM probe) which is able to identify the presence of known or unknown single-base variations at abundances down to 0.3%-1% within 5 min, even in highly GC-rich sequence regions. The discrimination factors between the perfect-match target and single-base mismatched target are determined to be 89–311 by measurement of their respective branch-migration products via polymerase elongation reactions. The BM probe not only enabled sensitive detection of two types of EGFR-associated point mutations located in GC-rich regions, but also successfully identified the BRAF V600E mutation in the serum from a thyroid cancer patient which could not be detected by the conventional sequencing method. The new method would be an ideal choice for high-throughput in vitro diagnostics and precise clinical treatment.
INTRODUCTION
Small genetic variations such as DNA point mutations serve as important drivers for disease development and indicators for diagnosis (1,2). They are often present at very low abundances (<1%) in human body because of local environment, cell heterogenity and migration (3,4). Integrated technologies are in urgent need for point-of-care test, field measurement, or high-throughput analysis of low-abundance genetic variations (5). Currently, the widely adopted procedure for detection of low-abundance point mutations includes: PCR amplification of the sample genomic DNA, treatment of PCR products and post-PCR detection of the mutation abundance. Many efforts have been made to improve the selectivity of PCR toward the mutant alleles, such as allele-specific amplification PCR (6), digital PCR (7), wild-type blocking PCR (8) and coamplification at lower denaturation temperature PCR (9). However, these approaches rely on precise temperature control and primer design. Since PCR has become a common tool to amplify the target DNA fragments, highly sensitive and specific methods for post-PCR discrimination of the low-abundance mutation are greatly appreciated. Ideally, these post-PCR detection methods should have the following features: convenience (fast, room-temperature, capable of identification of unknown mutations within a short region), high specificity, and importantly, with consistent performance on various targets. However, the sequence contexts surrounding the mutation site often have significant influences on the detection of the mismatched base pairs, especially within a GC-rich region, where a point mutation might only cause a very slight change to the thermodynamics and local conformation of the duplex, making it very difficult to be detected. Therefore, unstable performances are frequently encountered in the detection of low-abundance mutations in complex sequence contexts by using the currently available methods.
In recent years, we and other groups have developed a variety of enzyme-mediated oligonucleotide-based fluorescent sensing methods for post-PCR mutation detection (10–14). These methods are highly sensitive and specific, which are able to identify DNA mutations at very low abundances (0.01–0.1%). However, these methods are limited to the detection of minority known mutations at fixed positions. Besides, the recognition properties of enzymes for the mismatched base pairs are sometimes influenced by the surrounding sequences and thereby leads to unstable performance on different DNA targets. Thus more flexible and widely applicable methodologies for the detection of both known and unknown mutations with high specificity are needed in the clinical molecular diagnostics.
Many simpler methods that only employ nucleic acid probes such as molecular beacons (15,16), binary probes (17,18), yin-yang probes (19), base-discriminating fluorescent (BDF)probes (20), triple-stem probes (21) and toehold exchange probes (22) have also been developed. Molecular beacons require temperature optimization and typically do not perform well at room temperature. By using more complicated structures, some other probes are able to discriminate single base mismatch within a short region at room temperature. However, they still suffer from the unstable performance on different targets because of the complex surrounding sequences. Their discrimination factors (the ratio of signal induced by perfect-match target to that induced by single-base mismatched target) are less than 30, which are not high enough for the detection of very low-abundance mutations (<1%). Moreover, the kinetics of these probes are slow, thus requiring long incubation time to obtain observable signals. Based on toehold exchange, Pierce and colleagues constructed shielded covalent probes which covalently captured complementary DNA or RNA oligo targets and rejected single-base mismatched targets with near-quantitative yields at room temperature (23). Covalent linkage was able to eliminate the longstanding trade-off between selectivity and durable target capture, and thereby achieved higher discrimination factors than conventional toehold exchange probes. In the subsequent work, they further developed a small conditional RNA based probe (24). After the toehold exchange process induced by cognate RNA strand, a programmable hybridization cascades would take place, which significantly enhanced the signals and increased the discrimination factors. However, the measurement procedures of these methods were still complex. More robust and simpler approaches were highly desired.
Herein, we propose a novel branch-migration based probe (BM probe) that combines simplicity and exquisite selectivity. Shown in Figure 1A, the BM probe comprises of two complementary strands. The longer strand (L-strand, shown in dark gray) is labeled with Dabcyl, and the shorter strand (S-strand, shown in blue) is labeled with FAM. The distance between FAM and Dabcyl is designed to be close so that in the initial state the fluorescence of FAM is quenched by Dabcyl due to FRET. The BM probe can also be designed as a hairpin (shown in purple dash) to ensure the stability of the duplex in complex environments. In the presence of perfectly matched target (PM target, shown in red), the single-stranded region (17-nt) of the BM probe would immediately hybridize with part of the PM target. This process is very fast and the reverse reaction is negligible due to the ultra-high affinity of 17-base pairing. The rest 10 nucleotides of the PM target is also complementary to the L-strand, so branch migration would take place at the branch point. Theoretically, the structure of the three-strand complex would oscillate from two directions of the branch migration, and the average fluorescence signal would increase due to the distance change. For single-base mismatched target (1-MM target, shown in red with an arch), the process of initial 17-nt hybridization is also very fast. However, the branch migration would be greatly obstructed due to the mismatch between the L-strand and 1-MM target. Therefore, only slight increase of fluorescence intensity would be observed.
Figure 1.

Schematic illustration of the branch migration based probe (BM probe, A) and normal probe (B) for discrimination of perfect match target (PM target) and single-base mismatch target (1-MM target). The BM probe could be designed as double stranded or hairpin. (C) The curve of the conversion ratio of a reaction versus free energy change (ΔG) at 37°C.
As we know, the curve of conversion ratio versus free energy change of a reaction drops sharply near the point that the corresponding free energy change is zero (shown in Figure 1C). For normal nucleic acid probes at room temperature (shown in Figure 1B), the free energy change for the hybridization of PM target (ΔGa) and 1-MM target (ΔGb) are both far below zero and fall into the zone of high conversion ratio, rendering low discrimination efficiency. In the case of BM probe, the free energy change of the branch migration process is zero (ΔG1 = 0) for PM target and above zero for 1-MM target (ΔG2 > 0), rendering high discrimination efficiency. Importantly, the entropy change of the branch migration process is nearly zero, so the free energy change is not influenced by the reaction temperature. Due to the sharp drop around ΔG1 and the short length of the branch migration domain, a slight change of the base located within the domain is able to cause significant deviation of the conversion ratio, enabling BM probe to perform consistently without being affected by the surrounding sequences, and to discriminate unknown mutations within the branch migration domain.
The reaction pathways of the branch migration processes induced by perfect-match target (PM target) and single-base mismatch target (1-MM target) are identical. Then, it is reasonable to assume that the difference in activation energy equals to the difference in free energy change (23, 26). Therefore, the theoretical discrimination factor in the kinetic mode would be identical to that in the thermodynamic mode:
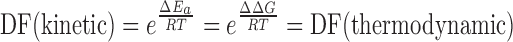 |
(1) |
However, detection through kinetic curves was considered to be much less affected by the instrumental variability during the measurement processes than detection through the change of thermodynamic plateaus. Therefore, we adopted detection in the kinetic mode in this work. Compared with the toehold exchange process, the dynamic equilibrium of the initial hybridization step (docking) and the final dissociation step of toehold would actually have a leveling effect on the discrimination efficiency of the branch migration step. Based on the kinetic modeling of strand displacement established by Winfree (25), we calculated the ratio of the discrimination factors (DF) toward perfect-match target and single-base mismatched target between the branch migration process and the toehold exchange process (see supporting information for detailed calculation). The discrimination capability of BM probe was estimated to be about twice as large as that of the toehold exchange probe. Furthermore, removing the docking step and the last dissociation step offers two distinct advantages: First, the free energy change (ΔG = 0) was unrelated to the sequences of the docking domain and the dissociation domain, making the probe design very convenient. Second, the reaction rate was significantly increased so that ultra-fast detection could be achieved.
MATERIALS AND METHODS
Materials
DNA strands were synthesized and purified by HPLC (Sangon Co., China). The sequences of all the probes and targets that have been studied in this work are summarized in Supplementary Table S1. Lambda exonuclease (λ exo), Exonuclease I (exo I), Vent exo− polymerase, Lambda Exonuclease Buffer (67 mM glycine–KOH (pH:9.4), 2.5 mM MgCl2, 50 μg/ml BSA) and ThermoPol Reaction Buffer (20 mM Tris–HCl, 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4 and 0.1% Triton X-100, pH 8.8) were all purchased from New England Biolabs (MA, USA). Taq DNA polymerase and deoxyribonucleoside triphosphates (dNTPs) were purchased from Tiangen Biotech Co. (Beijing, China). DNase/RNase free deionized water from Tiangen Biotech Co. was used in all the experiments.
Determination of the discrimination capability of the BM probe between PM target and 1-MM target
For experiments in Figure 2A, to a 200 μl PCR tube, 200 nM of BM probes was mixed with 200 nM of PM target and 1-MM-1 target, respectively. The buffer was ThermoPol reaction buffer. The solutions were immediately put into a Rotor-Gene Q 5plex Instrument (QIAGEN, Hilden, Germany) for fluorescence measurement at 37°C. Fluorescence intensity was recorded once a cycle (5 s per cycle) for 240 cycles with gain level of 7.33. The excitation and emission wavelengths were set to 470 and 510 nm, respectively.
Figure 2.

(A) The fluorescence intensity responses of BM-1 probe toward PM-1 target and 1-MM-1 target. (B) Illustration of the reaction pathways of BM-1 probe toward PM-1 target and 1-MM-1 target with and without the assistance of polymerase extension. (C) The fluorescence response curves of BM-1 probe toward PM-1 target and 1-MM-1 target in a concurrent reaction of branch migration and polymerase elongation. (D) Gel electrophoresis results of the branch migration reaction products shown in (A). Lane 1: BM-1 probe + PM-1 target. Lane 2: BM-1 probe + 1-MM-1 target. (E) The discrimination factors (DF) of BM probe toward different types of mismatches. The data were obtained based on the rate of fluorescence increase of the BM probe with the assistance of polymerase elongation. The six types of mismatches indicated by the blue bracket can cover all the 12 types of mismatches as the real DNA samples come as double stranded.
For polymerase elongation experiments, to a 200 μl PCR tube, 100 nM of BM probes and 2 units of Vent exo− polymerase, and 5 mM of dNTPs was mixed with 100 nM of PM target and 1-MM-1 target, respectively. The buffer was ThermoPol reaction buffer. The fluorescence intensity responses were immediately recorded at a gain level of 9 at 37°C. The rate of fluorescence increase was calculated based on the slope of the linear portion of the time curve. Discrimination factors (DFs) shown in Figure 2E were determined by calculating the ratio of the increase rate of fluorescence intensity induced by perfect-match target to that induced by single-base mismatched target.
Detection of low-abundance PM targets
To a 200 μl PCR tube, 500 nM of BM probes was mixed with 400 nM of target strands with the PM target at different abundances. The solutions were immediately put into a Rotor-Gene Q 5plex Instrument for fluorescence measurement. Fluorescence intensity was recorded once a cycle (5 s per cycle) for 240 cycles with gain level of 9 at 37°C (for experiments in Supplementary Figures S4–S6, the temperatures were set at 33, 28, 25°C, respectively). The excitation and emission wavelengths were set at 470 and 510 nm, respectively. The rate of fluorescence increase was determined by the slope of the linear portion of the time curve.
Agarose gel electrophoresis analysis
Agarose gel electrophoresis was carried out using 2.5% agarose gel at 120 V for 30 min in a 0.5× TBE buffer (90 mM Tris, 90 mM boric acid, 2 mM EDTA; pH 8.0). 9 μl of samples solution and 1.8 μl of loading buffer were added to each well. After separation, the gels containing DNA were stained using GelSafe Dye and visualized at a wavelength of 595 nm using a Tanon 1600 gel imaging system (Tanon, China).
Post-PCR detection of low-abundanceEGFR associated point mutations
To a 200 μl PCR tube, 25.5 μl of water, 5 μl of 10× ThermolPol Reaction Buffer, 4 μl of dNTPs (10 nmol), 1 μl of forward primers (12 pmol), 1 μl of reverse primers (12 pmol), 1 μl of mixed ssDNA templates (total amount 1 fmol, 32 pg) (see Supplementary Table S1 for sequence), 0.5 μl of Taq (1.25 U) were added and mixed well. PCR procedure (94°C for 30 s, 60°C for 30 s, 72°C for 20 s, 25 cycles) was performed on a Rotor-Gene Q 5plex Instrument. After the PCR amplification, 1 μl of exo I (5 U) was added to the amplicons to remove the unreacted primers, followed by inactivation at 85°C for 10 min. Then 3 μl of λ exo (5 U) was added to digest the strand containing 5΄-PO4 in the duplex products for 20 min at 37°C. After inactivation of λ exo at 85°C for 10 min, 5 μl of probe (500 nM) were added and mixed well. The detection was performed at 37°C in the same manner as described above (gain = 9).
Detection of BRAF V600E in circulating free DNA (cfDNA) extracted from the serum of cancer patients
The cfDNA was extracted from 200 μl of serum using the Qiagen DNeasy Kit. The amount of the extracted cfDNA was determined by Nanodrop 2000 (Thermo, Germany). To a 200 μl PCR tube, 23.5 μl of water, 5 μl of 10× ThermolPol Reaction Buffer, 4 μl of dNTPs (10 nmol), 1 μl of forward primer-BRAF (12 pmol), 1 μl of reverse primer-BRAF (12 pmol), 3 μl of extracted cfDNA (see Supplementary Table S1 for sequence), 0.5 μl of Taq (1.25 U) were added and mixed well. PCR procedure (94°C for 30 s, 60°C for 30 s, 72°C for 20 s, 45 cycles) was performed on a Rotor-Gene Q 5plex Instrument. After the PCR amplification, the following enzymatic treatment procedures and the fluorescence detection were performed in the same manner as described above (gain = 9, 37°C).
RESULTS
The discrimination capability of the BM probe between PM target and 1-MM target
We firstly investigated the discrimination capability of the BM probe between PM target and 1-MM target. As shown in Figure 2A, the fluorescence intensity increased very fast when 200 nM of BM-1 probe was reacted with 200 nM of PM-1 target, while only a slight increase of the fluorescence signal was observed when the probe was incubated with 200 nM of 1-MM-1 target. However, the signal generated by the 1-MM-1 target was still higher than the expected background signals. We attributed this slight signal increase to the intermediate branch migration that terminated at the mismatch site, Since this intermediate branch migration was a common process for both the PM-1 target and 1-MM-1 target, it could not directly reflect the amount of the final branch migration products of the 1-MM-1 target. Actually, the discrimination between the two targets was initiated in the following stage of further branch migration (shown in grey dash frame in Figure 2B).
Due to the existence of intermediate branch migration process, it is hard to obtain the real signals of 1-MM target by direct measurement of the fluorescence increase of the resultant three-strand complex. To eliminate the influences of the intermediate branch migration on the measurement of the actual discrimination factors, we added vent exo− polymerase to the solution at the beginning of the branch migration reaction. When the branch migration reached the end, the resulting structure could be immediately elongated by the polymerase and produce very strong fluorescence signals due to the dissociation of the S-strand from the L-strand. By contrast, most of the 1-MM-1 targets paused at the intermediate branch migration state which could not be elongated. Compared to the signals produced after polymerase elongation, the signals induced by the intermediate branch migration process were negligible. Thus, the experimental DF can be measured more accurately. Since the vent polymerase was added at the beginning of the reaction, the obtained experimental DFs were kinetic DF values. As shown in Figure 2C, the increase rate of fluorescence intensity for PM target was ∼300-fold higher than that for 1-MM target. We then used gel electrophoresis to further confirm the existence of intermediate branch migration. After branch migration, the products were loaded into an agarose gel and electrophoresis was performed. From Figure 2D, we could see that the band in lane 1 (BM-1 probe + PM-1 target) was much broader than the band in lane 2 (BM-1 probe + 1-MM-1 target), indicating that the branch migration of BM-1 probe reached much farther on the PM-1 target than on the 1-MM-1 target. The above two experiments substantially demonstrated the slight signal produced by 1-MM target was due to the intermediate branch migration, and the actual discrimination factor was ∼300.
Next, we optimized the length of the branch migration region and the position of the fluorophore. According to Supplementary Figures S1 and S2, the optimum length of the branch migration region was 10 nt, and the best position of the fluorophore was at the 5΄ end of the S-strand. Using the optimized structure, we determined the actual discrimination factor of BM probe toward different types of mismatches. Shown in Figure 2E, the DFs varied from 10 to 311, and the median DF was 127. For detection of low-abundance point mutations, the first six types of mismatches (from C:C to A:A, as indicated by the blue bracket) can cover all 12 types of mismatches. Therefore, the developed BM probe is able to detect all types of low-abundance point mutations with DFs higher than 89. We then changed the position of the C:C mismatch. From Supplementary Figure S3, we could see that C:C mismatch at the three tested positions were all effectively detected, demonstrating that the BM probe was able to detect unknown mismatches within a region of at least 6-nucleotide long.
Detection of low-abundance point mutations by BM probe
To evaluate the performance of BM-1 probe in the detection of low-abundance mutations, we prepared a series of mixed samples with the abundances of PM-1 target ranging from 10% to 0%, and the detection was performed at 37°C. The mismatch between BM-1 probe and 1-MM-1 target was C:C. As the DF for C:C mismatch was determined to be 311, we anticipated the limit of detection (LOD) for low-abundance PM-1 target to be ∼0.3%. Results shown in Figure 3A confirmed our anticipation. If we focused on the kinetic region of the curves to determine the LOD, the entire detection process would only need 5 min. To the best of our knowledge, this is the fastest hybridization-based probe for the detection of point mutations at such low abundances, which is very suitable for applications that require quick analysis. We then performed the same detection process at different temperatures (25, 28, 33°C). As shown in Supplementary Figures S4–S6, the BM-1 probe performed very well over a wide temperature range, from 25 to 37°C. For other types of mismatch, we selected another 1-MM-1A target that would form an A:A mismatch with BM-1A probe to further evaluate the performance of BM probe. Shown in Supplementary Figure S7, PM-1A target with abundance down to 1% was clearly differentiated from 0%, in accordance with the DF of 89 in Figure 2E.
Figure 3.

Fluorescence curves show the capability of the BM probe in discrimination of low-abundance mismatched base pairs in a target strand with different length and surrounding sequences. (A) The fluorescence intensity responses of BM-1 probe toward PM-1 target in the presence of different amount of 1-MM-1 target. (B) The fluorescence intensity responses of BM-1 probe toward PM-1L target in the presence of different amount of 1-MM-1L target. The overhangs in the PM target and 1-MM target are shown in green. (C) Performance of BM-1 probe in a turn-off mode for the discrimination of low-abundance 1-MM-1L-T target by employing PM-1L as the wild-type DNA. (D) The fluorescence intensity responses of BM-2 probe toward PM-2 target at different abundances when the mutation site is surrounded by guanines and cytosines. Such point mutations are difficult for enzyme-dependent probes to identify. We chose an EGFR associated point mutation (NM_0005228.3:c.2573T>G) as our model target here.
In above experiments, the target strand was 27-nt long. Nevertheless, the real DNA samples may be longer, so we synthesized longer PM-1L target and 1-MM-1L target to investigate whether the rest nucleotides would interfere the branch migration process. Results shown in Figure 3B demonstrated that the BM-1 probe worked very well on long target strands.
Detection of unknown point mutations within the branch migration domain under the turn-off mode
For detection of unknown point mutations within the branch migration region, the probe was designed to be complementary with the wild-type DNA, and thereby mis-matched with the mutant-type DNA at an unknown position. In this scenario, the signal would decrease with the increase of the abundance of mutant-type DNA, which is often termed as turn-off detection mode. So we further investigated the performance of BM probe under the turn-off detection mode. PM-1L target was regarded as wild-type DNA and 1-MM-1L-T target as mutant-type DNA. So the mutant-type DNA would form a C:T mismatch with BM probe. Shown in Figure 3C, the BM probe was able to differentiate 0.5% mutant-type DNA under the turn-off mode within 3 min, consistent with the discrimination factor for C:T mismatch (235). Above results demonstrated that the proposed BM probe functioned in the turn-off mode as well as in the turn-on mode, and therefore it could be further applied in the detection of low-abundance unknown point mutations within a short region.
Detection of an EGFR associated point mutations (NM_0005228.3:c.2573T>G) located in GC-rich areas
To show the capability of our method in the detection of single-point mutation within or near GC-rich regions which are more difficult to be distinguished by other methods, we chose an EGFR-associated point mutation (NM_0005228.3:c.2573T>G) which locates in a region with high GC content. The wild-type target (1-MM-2) has a C:T mismatch with the BM-2 probe. As we can see from Figure 3D, the BM-2 probe successfully detected the mutant-type target (PM-2) with an abundance down to 0.5%. The excellent performance of BM probe toward the point mutation within GC-rich sequences may be attributed to the short length and constant free energy change of the branch migration region, in which a slight change of single base may produce significant effects.
Post-PCR detection of another EGFR associated point mutations (NM_000222.2:c.1676T>G) located in GC-rich areas
We further applied our method to post-PCR detection of another EGFR-associated point mutation (NM_000222.2:c.1676T>G) which is also located in GC-rich region. It was a T to G mutation, so the wild-type sequence would form a C:T mismatch with the BM-3 probe. We synthesized 74-nt wild-type strands and 74-nt mutant-type strands, and prepared a series of mixed strands with the abundances of mutant-type sequences ranging from 0% to 10%. These mixed samples were then measured following the procedures described in Figure 4, and the results showed that the BM-3 probe detected the mutant-type target with an abundance down to 0.5%. Above experiments demonstrated that our method can be conveniently coupled with PCR for quick analysis of real clinical samples.
Figure 4.

Post-PCR detection of low-abundance point mutations. Another EGFR associated point mutation (NM_000222.2:c.1676T>G) located in GC-rich region is used here to show the generality of the method. The entire procedures of the detection were illustrated at the right. Inset shows the increase rates of fluorescence intensity (FL) for the tested EGFR associated point mutation at different abundance levels. All the experiments were repeated for three times, and standard deviations were shown in error bars.
Detection of BRAF V600E mutation in circulating free DNA (cfDNA) in the serum of cancer patients
BRAF V600E mutation was frequently found in thyroid cancer cells. In contrast, the BRAF V600E mutation frequency in liver cancer cells is much lower. To test the capability of the BM probe in discrimination of very low-abundance mutations, we tried to detect BRAF V600E mutation in circulating free DNA (cfDNA) extracted from the serum of a thyroid cancer patient. We first measured the enzymatic activity of human apurinic/apyrimidinic endonuclease I (APE-1) in the serum, which was observed to be 37 U/ml, about ten-fold higher than the normal level of healthy persons. Such high activity of APE-1 in serum significantly reflected the active tumor behavior in the patient (26), implying that the amount of circulating tumor DNA released from the tumor cells could be considerable. We extracted the cfDNA from 200 μl of the serum samples and then conducted PCR. The PCR products were first sequenced and the sequencing results were shown in Supplementary Figure S8, which proved that the expected fragment of DNA was successfully amplified. However, the abundance of BRAF V600E mutation in the serum was lower than the detection limit of Sanger Sequencing. Then we measured the PCR products with BM-4 probe following the procedures described in Figure 4. From the detection results, the fluorescence signal of the serum of the thyroid cancer patient was higher than those of the negative sample and the serum of the liver cancer patient, but lower than that of 1% abundance mutation. Therefore, we performed student's t test to determine whether the increase of the signal was of statistical significance. As shown in Figure 5, the level of BRAF V600E mutation in the cfDNA from the serum of the thyroid cancer patient was significantly higher than the control sample from the healthy person (t > tα(0.01)); While the signal of the cfDNA from the serum of the liver cancer patient was only slightly higher than that from the healthy person (tα(0.01) < t < tα(0.05)). These results clearly demonstrated the high discrimination capability of the developed BM probe in identification of low-level DNA mutations in clinical samples. As the amount of PCR products may vary between different samples, the method for accurate quantification of the exact abundance of specific mutation in real samples will be investigated in detail and reported later.
Figure 5.

(A) The fluorescence intensity responses of BM-4 probe toward BRAF mutant-type targets at different abundance levels. Inset shows the increase rates of fluorescence intensity (FL) for the BRAF mutant-type targets at different abundance levels. All the experiments were repeated for three times, and standard deviations were shown in error bars. (B) Detection of BRAF V600E mutation in circulating free DNA (cfDNA) extracted from the serum of a thyroid cancer patient. Serum samples obtained from a healthy person and a liver cancer patient were also measured as controls.
DISCUSSIONS
The discrimination capability of BM probe is fundamentally determined by the thermodynamic favourability, i.e. the difference in binding energy between perfect and single-base mismatch targets. As above-mentioned in the introduction, the theoretical DF in the kinetic mode is identical to that in the thermodynamic mode. For the bimolecular reaction model of toehold exchange (25), the kinetics of strand displacement processes can be accurately predicted only when the concentrations of invading strand and substrate complex are sufficiently low (about nM level). At higher concentrations of invading strand (>300 nM) and substrate complex (>100 nM), the experimentally observed reaction kinetics were significantly slower than those predicted by the model. This may partially explain why the experimentally observed DFs were usually lower than the theoretical predicted DFs. In a more recent work by Zhang et al., the theoretical DF for C:C mismatch was calculated to be >10 000 (23). However, the reported experimental results were far below this value. The large discrepancy may be ascribed to the quantifiable limit (the highest DF that can be measured accurately and precisely) of the detection methods. If the signals of PM target and 1-MM target can both be measured accurately without suffering from the limited sensitivity and precision of the instruments, the experimentally observed DF would be much closer to the theoretical DF.
The experimental DF is usually calculated based on the following equation:
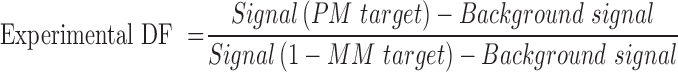 |
(2) |
Thus, much effort has been focused on enhancing the signals of PM target and lowering down the signals of 1-MM target. However, when the signals of 1-MM target decrease to very close to the background signals, the accuracy of the difference between the signal of 1-MM target and the background signal will be greatly affected by the precision of the instrument used for the measurement. Therefore, the actual DF of a DNA probe was determined by both the theoretical DF and the quantifiable limit of the detection method. For the proposed BM probe, the theoretical DFs in thermodynamic mode and kinetic mode are identical. Since kinetic curves was much less affected by the variability of the instruments and measurement processes, we used the data of increase rate of fluorescence intensity induced by PM target and 1-MM target to calculate the DF of the BM probe.
Due to the existence of intermediate branch migration process, it's hard to obtain the real signals of 1-MM target by direct measurement of the fluorescence increase of the resultant three-strand complex. As shown in Figure 2A, the increase of fluorescence intensity induced by 1-MM target was mostly owed to the intermediate branch migration. Ideally, the background signal could be measured by using a control probe with a 4-nt branch migration domain to mimic the intermediate branch migration process. Then the experimental DF can be calculated by
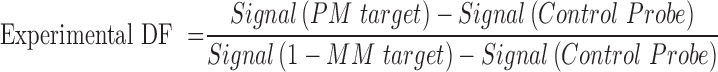 |
(3) |
However, the signal produced by the control probe would be very weak and significantly affected by the variability of the instruments. To avoid this, we employed an alternative strategy to measure the actual DF of BM probe. By incorporating the polymerase elongation reaction into the branch migration system, the fully migrated products could generate much higher fluorescent signals. Compared to the signals produced after polymerase elongation, the signals induced by the intermediate branch migration process were negligible. Thus the experimental DF can be measured more accurately. Since the vent polymerase was added at the beginning of the reaction, the obtained experimental DFs were kinetic DF values.
It is worth to note that the polymerase elongation method has a higher quantifiable limit than the method of using a control probe with a 4-nt branch migration domain. But in the subsequent detection of low-abundance mutations, we did not use polymerase elongation to enhance the quantifiable limit. In such cases, since we mainly focused on obtaining the signals of a small amount of PM targets in a background of large amounts of 1-MM targets, it was not necessary to measure the large fluorescence signals of 100% PM targets. Therefore, we increased the amount of probes and enlarged the gain level of the instrument, which actually increased the signal intensity and thereby enhanced the quantifiable limit. In this case, the absolute signals of the small amount of PM targets were higher than those in Figure 1a, and the discrimination effect of low-abundance mutations were comparable to those with the polymerase elongation (Figures 3 and 4). These results substantially demonstrated the capability of the BM-probe in post-PCR detection of low-abundance mutations in a simple, enzyme free and straightforward way. But it is worth to note that combining enzymes with branch migration process is potentially powerful in further development of more sensitive and specific assays. Further investigation is undergoing.
Compared with the kinetics of toehold exchange process (23), the BM probe has three advantages: (i) Due to the leveling effect of the docking step and dissociation step in the toehold exchange process, the kinetic theoretical DF of toehold exchange probe was only half of the kinetic theoretical DF of the BM probe. (ii) The reaction rate of BM probe was higher than that of toehold exchange probe. As mentioned above, higher reaction rate means higher sensitivity and thereby higher quantifiable limit of DF. (iii) The docking step and dissociation step contribute to the deviations of signals, lowering down the quantifiable limit of DF.
The BM probe was designed to be used as a convenient tool to discriminate low-abundance DNA mutations after a regular PCR process. Usually the PCR products were sequenced to find out if there were mutations. However, first-generation sequencing is not sensitive enough to distinguish mutations at abundances lower than 10%, and next-generation sequencing is too expensive for detection of low-abundance hotspot mutations at known positions. Compared with other probes for post-PCR mutation detection, the BM probe performed much better with the GC-rich sequences such as the EGFR mutation. Besides, it can detect unknown mutations within a short region, while the enzyme-dependent probes can only measure known mutations at fixed positions. With the same steps of regular PCR and treatment of the PCR products as those for the enzyme-dependent probes, the BM probe is much faster and simpler in the subsequent detection step. It is more suitable for the detection that requires multiplicity and fast speed. We must admit that the detection limit of BM probe is slightly compromised for the convenience we are trying to achieve; While other enzyme-dependent probes can differentiate point mutations at much lower abundance levels. But as we have discussed above, the branch migration process is versatile. By coupling with other approaches such as polymerase elongation, the BM probe holds great potential for further development of more powerful tools for DNA mutation detection.
CONCLUSIONS
We have successfully constructed a novel type of branch-migration based fluorescent probe (BM probe) for fast, robust, and sensitive detection of known or unknown single-base variations at abundances down to 0.3–1% within 5 min, even in highly GC-rich sequence regions. The short length of the branch migration domain and the inherent significant difference between the free energy change of the branch migration process for the single-mismatch target and that for the perfect-match target ensures the BM probe to be sensitive to even very slight changes in the target strands without the need of precise temperature control. The outstanding discrimination capability of the BM probe was demonstrated not only by the fast detection of an EGFR-associated point mutation (NM_005228.3T>G) located in GC-rich regions with an abundance down to 0.5%, but also by the successful discrimination of very low level BRAF V600E mutation in the serum sample from a thyroid cancer patient which could not be detected by the conventional sequencing method. In consideration of all the features provided, we anticipate the BM probe would find wide applications in point-of-care test, high-throughput human in vitro diagnostics and precise clinical treatment.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [81571130100, 21575008, 21375004]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bronner C.E., Baker S.M., Morrison P.T., Warren G., Smith L.G., Lescoe M.K., Kane M., Earabino C., Lipford J., Lindblom A. et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994; 368:258–261. [DOI] [PubMed] [Google Scholar]
- 2. Lievre A., Bachet J.B., Le Corre D., Boige V., Landi B., Emile J.F., Cote J.F., Tomasic G., Penna C., Ducreux M. et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006; 66:3992–3995. [DOI] [PubMed] [Google Scholar]
- 3. Bielas J.H., Loeb K.R., Rubin B.P., True L.D., Loeb L.A.. Human cancers express a mutator phenotype. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:18238–18242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shah S.P., Morin R.D., Khattra J., Prentice L., Pugh T., Burleigh A., Delaney A., Gelmon K., Guliany R., Senz J. et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009; 461:809–813. [DOI] [PubMed] [Google Scholar]
- 5. Fan J.B., Chen X., Halushka M.K., Berno A., Huang X., Ryder T., Lipshutz R.J., Lockhart D.J., Chakravarti A.. Parallel genotyping of human SNPs using generic high-density oligonucleotide tag arrays. Genome Res. 2000; 10:853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu D.Y., Ugozzoli L., Pal B.K., Wallace R.B.. Allele-specific enzymatic amplification of beta-globin genomic DNA for diagnosis of sickle cell anemia. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:2757–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vogelstein B., Kinzler K.W.. Digital PCR. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:9236–9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dominguez P.L., Kolodney M.S.. Wild-type blocking polymerase chain reaction for detection of single nucleotide minority mutations from clinical specimens. Oncogene. 2005; 24:6830–6834. [DOI] [PubMed] [Google Scholar]
- 9. Li J., Wang L., Mamon H., Kulke M.H., Berbeco R., Makrigiorgos G.M.. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat. Med. 2008; 14:579–584. [DOI] [PubMed] [Google Scholar]
- 10. Xiao X., Song C., Zhang C., Su X., Zhao M.. Ultra-selective and sensitive DNA detection by a universal apurinic/apyrimidinic probe-based endonuclease IV signal amplification system. Chem. Commun. 2012; 48:1964–1966. [DOI] [PubMed] [Google Scholar]
- 11. Xiao X.J., Zhang C., Su X., Song C., Zhao M.P.. A universal mismatch-directed signal amplification platform for ultra-selective and sensitive DNA detection under mild isothermal conditions. Chem. Sci. 2012; 3:2257–2261. [Google Scholar]
- 12. Xiao X., Liu Y., Zhao M.. Endonuclease IV discriminates mismatches next to the apurinic/apyrimidinic site in DNA strands: constructing DNA sensing platforms with extremely high selectivity. Chem. Commun. 2013; 49:2819–2821. [DOI] [PubMed] [Google Scholar]
- 13. Gerasimova Y.V., Kolpashchikov D.M.. Enzyme-assisted target recycling (EATR) for nucleic acid detection. Chem. Soc. Rev. 2014; 43:6405–6438. [DOI] [PubMed] [Google Scholar]
- 14. Wu T.B., Xiao X.J., Zhang Z., Zhao M.P.. Enzyme-mediated single-nucleotide variation detection at room temperature with high discrimination factor. Chem. Sci. 2015; 6:1206–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tyagi S., Kramer F.R.. Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol. 1996; 14:303–308. [DOI] [PubMed] [Google Scholar]
- 16. Piatek A.S., Tyagi S., Pol A.C., Telenti A., Miller L.P., Kramer F.R., Alland D.. Molecular beacon sequence analysis for detecting drug resistance in Mycobacterium tuberculosis. Nat. Biotechnol. 1998; 16:359–363. [DOI] [PubMed] [Google Scholar]
- 17. Kolpashchikov D.M. Split DNA enzyme for visual single nucleotide polymorphism typing. J. Am. Chem. Soc. 2008; 130:2934–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kolpashchikov D.M. A binary DNA probe for highly specific nucleic Acid recognition. J. Am. Chem. Soc. 2006; 128:10625–10628. [DOI] [PubMed] [Google Scholar]
- 19. Tyagi S., Bratu D.P., Kramer F.R.. Multicolor molecular beacons for allele discrimination. Nat. Biotechnol. 1998; 16:49–53. [DOI] [PubMed] [Google Scholar]
- 20. Okamoto A., Kanatani K., Saito I.. Pyrene-labeled base-discriminating fluorescent DNA probes for homogeneous SNP typing. J. Am. Chem. Soc. 2004; 126:4820–4827. [DOI] [PubMed] [Google Scholar]
- 21. Xiao Y., Plakos K.J., Lou X., White R.J., Qian J., Plaxco K.W., Soh H.T.. Fluorescence detection of single-nucleotide polymorphisms with a single, self-complementary, triple-stem DNA probe. Angew. Chem. Int. Ed. 2009; 48:4354–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang D.Y., Chen S.X., Yin P.. Optimizing the specificity of nucleic acid hybridization. Nat. Chem. 2012; 4:208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vieregg J.R., Nelson H.M., Stoltz B.M., Pierce N.A.. Selective nucleic acid capture with shielded covalent probes. J. Am. Chem. Soc. 2013; 135:9691–9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sternberg J.B., Pierce N.A.. Exquisite sequence selectivity with small conditional RNAs. Nano Lett. 2014; 14:4568–4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang D.Y., Winfree E.. Control of DNA strand displacement kinetics using toehold exchange. J. Am. Chem. Soc. 2009; 131:17303–17314. [DOI] [PubMed] [Google Scholar]
- 26. Futagami S., Hiratsuka T., Shindo T., Horie A., Hamamoto T., Suzuki K., Kusunoki M., Miyake K., Gudis K., Crowe S.E. et al. Expression of apurinic/apyrimidinic endonuclease-1 (APE-1) in H-Pylori-associated gastritis, gastric adenoma, and gastric cancer. Helicobacter. 2008; 13:209–218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.