

Abstract
Diabetic retinopathy (DR) is one of the leading causes of blindness in the developed world. Characteristic features of DR are retinal neurodegeneration, pathological angiogenesis and breakdown of both the inner and outer retinal barriers of the retinal vasculature and retinal pigmented epithelial (RPE)–choroid respectively. Vascular endothelial growth factor (VEGF-A), a key regulator of angiogenesis and permeability, is the target of most pharmacological interventions of DR. VEGF-A can be alternatively spliced at exon 8 to form two families of isoforms, pro- and anti-angiogenic. VEGF-A165a is the most abundant pro-angiogenic isoform, is pro-inflammatory and a potent inducer of permeability. VEGF-A165b is anti-angiogenic, anti-inflammatory, cytoprotective and neuroprotective. In the diabetic eye, pro-angiogenic VEGF-A isoforms are up-regulated such that they overpower VEGF-A165b. We hypothesized that this imbalance may contribute to increased breakdown of the retinal barriers and by redressing this imbalance, the pathological angiogenesis, fluid extravasation and retinal neurodegeneration could be ameliorated. VEGF-A165b prevented VEGF-A165a and hyperglycaemia-induced tight junction (TJ) breakdown and subsequent increase in solute flux in RPE cells. In streptozotocin (STZ)-induced diabetes, there was an increase in Evans Blue extravasation after both 1 and 8 weeks of diabetes, which was reduced upon intravitreal and systemic delivery of recombinant human (rh)VEGF-A165b. Eight-week diabetic rats also showed an increase in retinal vessel density, which was prevented by VEGF-A165b. These results show rhVEGF-A165b reduces DR-associated blood–retina barrier (BRB) dysfunction, angiogenesis and neurodegeneration and may be a suitable therapeutic in treating DR.
Keywords: angiogenesis, diabetic retinopathy, vascular endothelial growth factor
Clinical perspectives
-
•
Diabetes results in leakage of fluid into the retina through the BRB. This permeability increase is modulated by VEGF-A. VEGF-A165b is a splice form of VEGF-A, which is anti-angiogenic. We, therefore, tested to see whether it could also be anti-permeability.
-
•
VEGF-A165b inhibited permeability of the outer BRB in vitro and retinal permeability in vivo.
-
•
This suggests that increasing VEGF-A165b expression could prevent increased permeability in DR.
Introduction
Diabetic retinopathy (DR) affects both Type 1 and Type 2 diabetic patients [1] and is the leading cause of blindness in the working population of the western world. Proliferative diabetic retinopathy (PDR), like many other neovascular retinopathies, occurs as a result of an ischaemic retina. Persistent hyperglycaemia causes structural, functional and haemodynamic changes in the vasculature [2] that can occur early on in diabetes before noticeable signs of retinopathy, eventually resulting in focal ischaemia [3]. As a consequence of hypoxia, angiogenesis is triggered by increased vascular endothelial growth factor (VEGF-A) expression [4]. Newly formed vessels rarely recruit pericytes [5], lack vessel integrity and are prone to rupture, as evidenced by microaneurysms and haemorrhage [6]. Diabetic macular oedema (DME) occurs from blood–retina barrier (BRB) breakdown causing accumulation of fluid and plasma proteins from leaky blood vessels [7]. Both DME and PDR worsen with persistent hyperglycaemia and VEGF-A up-regulation [8].
Treatments for DR can involve surgical and/or pharmacological intervention. Laser photocoagulation for both PDR and DME aims to increase the amount of oxygen that is available to the retina [9]. Since the discovery of VEGF-A in the pathogenesis of DR [4], the use of anti-VEGF-A agents such as ranibizumab (Lucentis, Genentech, San Francisco, U.S.A.), bevacizumab (Avastin, Genentech, San Francisco, U.S.A.) and aflibercept (Eyelea, Regeneron, New York, U.S.A.) have been widely investigated in their treatment of DR [10]. These anti-VEGF-A treatment strategies are mainly targeted at the inner BRB. Anti-VEGF-A therapy is effective in treating DR in approximately 50% of patients [11]. Conversely, anti-VEGF-A therapy in treating wet AMD is much more effective with up to 87% of patients responding positively [12]. Neutralizing or inhibiting an endogenous cytokine such as VEGF-A could prove to be deleterious. Within the eye itself, suppression of VEGF-A may lead to widespread geographic atrophy [13].
The BRB is a selectively permeable region formed by tight junctions (TJs) on retinal endothelial cells (RECs) [14] and retinal pigmented epithelial (RPE) cells [15], which form the inner and outer BRB respectively. The BRB protects the neural retina from contents of the retinal and choroidal circulations, has a role in homoeostatic maintenance of the neural retina and controls fluid and molecular flux between cells.
Unlike the retinal vasculature, the choroidal vasculature is fenestrated to allow transport of nutrients from the systemic circulation to the RPE, which in turn transports them to the outer retina [16,17]. This transcellular movement is also used to transport outer retinal waste products back to the choroidal circulation [16]. Due to its close association with the choroidal circulation, TJ integrity of the RPE is critical for the maintenance of the outer BRB. Moreover, the fenestrae of the choroidal plexus may allow macromolecular leakage from the choroid that may otherwise be deleterious in the retina. As well as providing a barrier function and a transporter of nutrients and waste, the RPE layer also protects against oxidative stress by expressing superoxide dismutase, lipofuscin and melanin [18].
VEGF-A pre-mRNA can be alternatively spliced at exon 8 [19] to generate two families of isoforms. Selection of either a proximal splice site or a distal splice site results in two different families of isoforms respectively [19]. The VEGF-Axxxb (x=amino acid number) family has a different terminal six amino acid sequence (SLTRKD) to the pro-angiogenic family, VEGF-Axxxa (CDKPRR) [20]. Of the many isoforms of the VEGF-Axxxb family (x=amino acid number), VEGF-A165b is the most well characterized and understood, and has properties that differ from VEGF-A165a, its pro-angiogenic sister isoform. VEGF-A165b binds to VEGFR2, however unlike VEGF-A165a, it does not fully stablize neuropilin 1 binding, and only weakly phosphorylates VEGFR2 [21]. VEGF-A165a and VEGF-A165b competitively bind to VEGFR2, so VEGF-A165b prevents pro-angiogenic activity in vivo.
VEGF-Axxxb isoforms are found in the adult human vitreous indicating that both families of isoforms are required for adequate ocular function [22]. However, in diabetic patients, this balance shifts in favour of VEGF-Axxxa, with the percentage of VEGF-A in the vitreous that is anti-angiogenic decreasing (12.5±3%) compared with that of normal eyes (65.3±7.2%) [22]. This indicates that progression of DR may be influenced by a switch in VEGF-A splicing. Intravitreal administration of VEGF-A165b reduces neovascularization in oxygen-induced retinopathy (OIR) [23] and in choroidal neovascularization [24], and has been shown to increase RPE cytoprotection [25]. VEGF-A165b is also neuroprotective and anti-apoptotic [26].
It, therefore, seems possible that VEGF-A165b could ameliorate the vascular aspect of DR, and as it appears to be cytoprotective to RPE cells, could act on the outer BRB, which would be an advantageous addition to ‘anti-VEGF-A’ therapies. We, therefore, tested the hypothesis that the cytoprotective, anti-angiogenic and anti-permeability properties of VEGF-A165b enable it to prevent BRB breakdown in vitro and in vivo in an animal model of DR.
Materials and methods
Animal experiments
All animal experiments were undertaken under an U.K. Home Office project licence, and approved by the local ethics review board.
Streptozotocin-induced diabetes–8 weeks
A total of 20 female Sprague–Dawley rats (200–300g, Charles River, U.K.) were weighed and fasted overnight, prior to diabetes induction. Rats were given a single intraperitoneal (i.p.) injection of streptozotocin (STZ, 50 mg/kg, Sigma–Aldrich, MO, U.S.A.). A total of ten control rats were injected with 300 μl saline i.p. A third of an insulin capsule (LinShin, ON, Canada) was implanted subcutaneously to maintain body weight over the following 8 weeks in the diabetic rats. On day 4 post-induction, blood glucose was tested from the tail vein. Rats with blood glucose of 15 mmol/l and above were deemed diabetic. STZ-injected rats that did not become hyperglycaemic on day 4 were re-fasted overnight and then re-injected with STZ the following morning. For the chronic experiment, diabetic rats were treated with 20 ng/g recombinant human (rh)VEGF-A165b (i.p., R&D Systems, MN, U.S.A.) or saline biweekly for the experimental period. Control groups remained untreated for the duration of the experiment.
Streptozotocin-induced diabetes–1 week
A total of ten rats were used for the 1 week induction of diabetes. STZ treatment was as described above on five rats but without insulin treatment. On day 6 post-induction, rats with blood glucose ≥15 mmol/l received an intravitreal injection of vehicle (5 μl of PBS) in one eye and 50 ng of VEGF-A165b in the contralateral eye as described below. Five control animals had vehicle in one eye and no injection in the contralateral eye.
Intravitreal injections
Rats were anaesthetized with a single 10 mg/ml i.p. injection of Domitor (medetomidine hydrochloride, Pfizer, U.K.) and Ketaset (ketamine hydrochloride, Zoetis, NJ, U.S.A.). A 1.5 cm 34-gauge hypodermic needle (Hamilton, NV, U.S.A.) attached to a 5 μl syringe (World Precision Instruments, FL, U.S.A.) was inserted into the posterior chamber of the eye at a 45° angle. Either 5 μl of sterile PBS or 50 ng rhVEGF-A165b was injected into the vitreous. The following day animals were perfused with Evans Blue dye as described below.
Evans Blue dye perfusion
The Evans Blue dye perfusion technique was used as described [27] but with minor alterations. Evans Blue dye (Sigma–Aldrich, MO, U.S.A.) was prepared by dissolving in normal saline for a final concentration of 45 mg/ml. Rats were anaesthetized (i.p.) with 10 mg/ml Vetalar (ketamine hydrochloride, Boehringer Ingelheim, Germany) and Domitor (medetomidine hydrochloride, Pfizer, U.K.) with additional anaesthesia provided as needed. The left jugular vein and femoral artery were cannulated with 0.6 mm OD tubing and Evans Blue (45 mg/ml) injected via the left jugular vein over 10 s. Two minutes post-infusion, 200 μl of blood was removed from the femoral artery to establish the initial vascular Evans Blue concentration. Fifteen minutes post-infusion, 100 μl of blood was removed, and this volume was withdrawn every 15 min for 2 h. Eyes were kept moist using Viscotears (Novartis, Switzerland). Two hours post-infusion, the chest cavity was opened and 200 μl of blood was withdrawn from the left ventricle to determine the final plasma Evans Blue concentration. Rats were then perfused with 50 ml of saline through the left ventricle at a physiological pressure of 120 mmHg, and exsanguinated. Both eyes were enucleated and bisected at the equator. The retinas were dissected and weighed (wet weight) and then dried at 70°C overnight, and weighed again (dry weight). Formamide (120 μl) was added to each sample and incubated at 70°C overnight. Following dye extraction, the samples were centrifuged at 12000 rpm at 4°C for 45 min. The supernatant was used to determine the Evans Blue concentration. The blood samples were centrifuged at 4°C at 12000 rpm for 45 min and diluted 1/100 in formamide (Sigma–Aldrich, MO, U.S.A.). The absorbance of all samples was then measured at 620 nm, and the concentration of Evans Blue dye in formamide was calculated using a standard curve of Evans Blue in formamide. Formulae for solute flux and permeability–surface area product (PA) used are:
-
(a) Evans Blue (EB) absorbance in wet weight (μg/g):
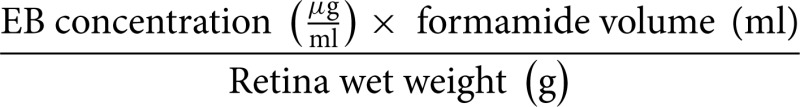
EB absorbance in dry weight (μg/g): same as (a), substituting wet weight for dry weight
-
(b) EB accumulation in tissue fluid (μg/ml):
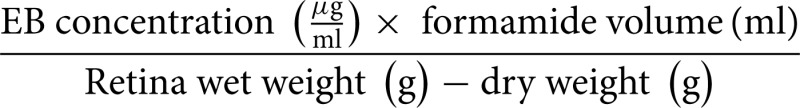
-
(c) EB wet weight solute flux (μg/min per g):
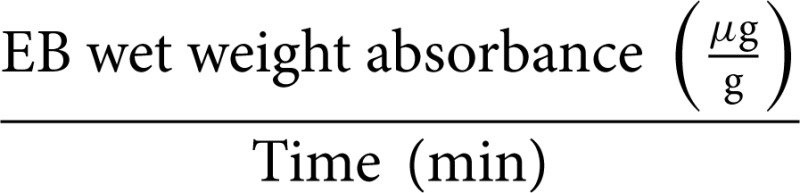
-
(d) EB dry weight permeability − surface area product (ml/min per g):

The control permeability and solute flux were different between the 1-week and 8-week group. To compare the effect of 8-week and 1-week, the values were normalized to their control cohort by subtracting the relevant control cohort values from the STZ-treated (or STZ + VEGF-A165b-treated) values.
Isolation of human primary retinal pigmented epithelial cells
All RPE isolations were performed as described [28]. Upon reaching 80% confluence, cells were split and either transferred to a T75 flask (Greiner Bio One, Austria) or used for experimental purposes. RPE cells were used until passage 8 and cultured in DMEM:F12 + GlutaMAX (Invitrogen, CA, U.S.A.) supplemented with 10% FBS (Invitrogen, CA, U.S.A.) and 1% penicillin/streptomycin (Invitrogen, CA, U.S.A.).
Trans-epithelial electrode resistance
Trans-epithelial electrode resistance (TEER) was measured using electric cell-substrate impedance sensing (ECIS, Applied Biophysics, NY, U.S.A.) using a 1600R ECIS model. Cells were plated on eight-well plates (8W10E+ plates, Applied Biophysics). Wells were pre-treated with 10 mM L-cysteine (Sigma–Aldrich, MO, U.S.A.) for 30 min at 37°C to neutralize the electrodes. Wells were then washed with distilled water and stabilized in complete medium. Sixty thousand cells were plated per well and were deemed confluent when the resistance was greater than 1000 Ω. Measurements were collected at multiple frequencies (125, 250, 500, 1000, 2000, 4000, 8000 and 16000 Hz) for 1 h to establish baseline data. Once baseline was established, growth factor treatment was prepared in 50 μl of medium. Fifty microlitres of medium was carefully aspirated from each well of the plate and replaced with 50 μl of growth factor treatment, and was allowed to equilibrate for 30 min before recommencing TEER evaluation. Plates were then assayed for changes in TEER for at least 15 h. Data were analysed at 500 Hz to give an indication of TJ integrity and paracellular flux.
Immunoblotting
Excised retinae or sub-confluent cells were lysed in RIPA lysis buffer supplemented with protease inhibitor cocktail (Sigma–Aldrich, MO, U.S.A.). A total of 50 μg of total protein was re-suspended in sample buffer, heated at 95°C for 5 min and subjected to SDS/PAGE under reducing conditions. Subsequently, proteins were electrotransferred for 2 h at 4°C to polyvinylidene fluoride membranes (PVDF, Bio-Rad, CA, U.S.A.). The membranes were then exposed to primary antibodies (rabbit anti-ZO1 [Invitrogen], mouse anti-occludin [Invitrogen], rabbit anti-VEGFR2 Ty115 [Cell Signaling Technology], all 1:500 5% BSA and mouse anti-α tubulin [Santa Cruz], 1:1000 5% BSA). PVDF was washed in Tris-buffered saline/0.3% Tween-20 (Sigma–Aldrich, MO, U.S.A.; TBS-T) and incubated with a secondary peroxidase-conjugated antibody at a 1:10000 dilution (Thermo Fisher Scientific, MA, U.S.A.). Signals were detected by enhanced chemiluminescence (ECL) substrate (GE Healthcare, U.K.). Western blots were also detected using a fluorescent-labelled secondary antibody (donkey anti-mouse IR dye 1:7000, LI-COR Biosciences, NE, U.S.A.), incubated at room temperature for 1 h, followed by detection using LI-COR Odyssey imaging system.
Immunofluorescence and quantification
To stain for TJs, RPE cells grown on 13 mm glass coverslips (Thermo Fisher Scientific, MA, U.S.A.) were treated with either VEGF-A165a, VEGF-A165b, anti-VEGFxxxb, (clone 56/8) or glucose (Sigma–Aldrich, MO, U.S.A.). Cells were fixed at room temperature with 4% paraformaldehyde (Sigma–Aldrich, MO, U.S.A.) 24-h post treating, permeabilized in 0.05% Triton-X (Sigma–Aldrich, MO, U.S.A.) in PBS and then blocked in 5% normal goat serum (Sigma–Aldrich, MO, U.S.A.) in PBS, all for 30 min at room temperature. All cells were incubated in primary antibody in PBS at 4°C overnight (mouse anti-occludin and rabbit anti-ZO1 [both Invitrogen at 4 μg/ml]). Cells were then incubated with Alexa-Fluor 488 conjugated secondary antibody (1:500 Invitrogen, CA, U.S.A.) in PBS, for 45 min at room temperature, followed by incubation with Hoechst (1:5000, Sigma–Aldrich, MO, U.S.A.) for 10 min at room temperature. Coverslips were mounted on glass slides with Vectashield (both Thermo Fisher Scientific, MA, U.S.A.).
To stain retinal flat mounts, eyes were enucleated from culled rats and fixed in 4% PFA for 1 h. Eyes were then hemisected and retinae were excised and transferred to blocking solution (5% NGS, 3% Triton-X100 and 1% BSA) for 2 h. Retinae were incubated at 4°C overnight in mouse anti-NeuN (Merck Millipore, MA, U.S.A., 2 μg/ml) and isolectin B4 (biotin conjugated from Bandeiraea simplicifolia, Sigma–Aldrich, MO, U.S.A., 5 μg/ml) prepared in blocking solution followed by three PBS washes and then incubation with Alexa-Fluor 488 (1:500, diluted in blocking solution) at room temperature for 2 h. After the last wash, samples were carefully flat-mounted onto a microscope slide (VWR International, PA, U.S.A.) and mounted with Vectashield containing DAPI (Thermo Fisher Scientific, MA, U.S.A.).
TJ staining in cells were imaged using an epifluorescence microscope (Nikon Eclipse E400) and analysed using a custom made macro on Fiji [29]. Flat-mounted retinae were imaged using confocal microscopy (Leica SPE). Tortuosity and vascular density was determined using Imaris software (Bitplane, U.K.) and confirmed using Fiji.
Statistical analyses
Unless otherwise stated, all data are shown as mean ± SEM. All data, graphs and statistical analyses were calculated with Microsoft Excel (Microsoft Office Software), GraphPad Prism v6 (GraphPad Software Inc., CA, U.S.A.), Fiji and Imaris parametric and non-parametric statistical tests were chosen upon the results of the D'Agostino–Pearson normality test in GraphPad Prism. Curve fitting was carried out using Prism. All results were considered statistically significant at p<0.05 (*), p<0.01 (**) and p<0.001 (***).
Results
VEGF-A165a causes RPE tight junction breakdown
To assess the effect of VEGF-A165a on the outer BRB in vitro, RPE cells were treated with increasing concentrations of rhVEGF-A165a and expression of the TJ marker occludin measured by immunofluorescence. As VEGF-A165a concentration increased, occludin fluorescence intensity decreased. There was a significant reduction upon 1.0 nM VEGF-A165a treatment (Figure 1A), 2.5 nM and 5.0 nM when compared with untreated controls (Figure 1B). Immunoblotting confirmed that occludin expression dose dependently decreased with VEGF-A165a treatment (Figure 1C and D). Conversely, VEGFR2 expression increased with increasing VEGF-A165a concentration (Figure 1C and D). The changes in occludin expression were matched by an increase in trans-epithelial conductance, a measure of permeability to small solutes, upon increased VEGF-A165a concentration (Figure 1E). Permeability increased in all groups treated with VEGF-A165a, being significantly different from control at 2.5 and 5nM (Figure 1E). Total solute flux over a 24-h time period (area under the curve) demonstrated a dose dependent increase in permeability with an EC50 of 832 pM (Figure 1F).
Figure 1. VEGF-A165a causes RPE tight junction breakdown.

RPE cells were treated with increasing doses of rhVEGF-A165a for 24 h (n=3, mean ± SEM). (A) Cells were fixed and stained for TJ marker, occludin (green) and nuclei co-stained with Hoechst (Blue). (B) Intensity was measured using ImageJ and normalized to untreated. (C) Protein lysate was extracted and immunoblotted for occludin and VEGFR2 expression (n=2, mean ± SD). Membranes were stripped and re-probed for α-tubulin expression to confirm equal loading of protein and densitometry (D). (E) RPE cells were plated on ECIS plates (n=3, mean ± SEM) to assess paracellular flux treated with a range of VEGF-A165a concentrations (0–5 nM). (F) Area under the curve was calculated over the 25-h period and plotted against concentration. The concentration that gave a 50% increase in permeability was determined by curve fitting using a four-parameter variable slope log(agonist) versus response curve in GraphPad Prism7; *p<0.05, **p<0.01, ***p<0.001, Figure A–C: one-way ANOVA with Dunnett's post-test; Figure E: two-way ANOVA with Bonferroni post-hoc, significance shown at 5-h intervals.
VEGF-A165b prevents VEGF-A165a -induced changes in tight junctions
To assess whether VEGF-A165b could be used to treat DR, its effect on RPE integrity was investigated by measuring TEER of RPE monolayers. There was no significant increase in paracellular flux upon VEGF-A165b treatment up to the highest concentration tested (5 nM) (Figure 2A). To determine whether VEGF-A165b could abrogate VEGF-A165a-induced changes in TJ expression and permeability, we treated RPE cells with a single concentration of VEGF-A165a (1 nM, EC70) and increasing concentrations of VEGF-A165b. When compared with untreated wells (Figure 2B), VEGF-A165b inhibited the VEGF-A165a-mediated increase in permeability at 0.2 nM and above. Calculation of the total flux over 24 h indicated that VEGF-A165b inhibited the permeability induced by 1 nM VEGF-A165a with an IC50 of 0.1 nM, and was completely abolished with equimolar treatment (Figure 2C).
Figure 2. VEGF-A165b prevents VEGF-A165a -induced changes in tight junctions.

RPE cells were plated on ECIS plates (n=3, mean ± SEM) and treated with increasing concentrations (A) of VEGF-A165b (0–5 nM) or with different proportions (B) of VEGF-A165b: VEGF-A165a and TEER were measured over 24 h (n=3, mean ± SEM) and impedance was measured over 24 h; ***p<0.001, two-way ANOVA with Bonferroni post-hoc. (C) Inhibition of the RPE total solute flux induced by 1 nM VEGF-A165a calculated from the area under the curve; IC50=1 nM. Cells were stained (D) for ZO1 (green) and fluorescence intensity was calculated (E). Protein lysate was immunoblotted for ZO1 and occludin expression (F). Membranes were stripped and re-probed for α-tubulin expression and expression was quantified (G); n=3, one-way ANOVA, Tukey's post-hoc test, *p<0.05, **p<0.01.
To determine the effect on tight junctional proteins, RPE cells were treated with 2.5 nM VEGF-A165A, 2.5 nM VEGF-A165b and 2.5 nM VEGF-A165a co-treated with 2.5 nM VEGF-A165b for 24 h. Cells were then fixed and stained for ZO1, a marker of TJ function (Figure 2D), and membrane fluorescence intensity was measured for each cell per field of view. Staining intensity was calculated relative to control cells.
Figure 2E shows that VEGF-A165b by itself had no significant effect on ZO1 expression or location (86.3% ± 25.0 of control). VEGF-A165a significantly reduced ZO1 expression (43.7% ± 5.6), similar to its effect on occludin expression (Figure 1). Co-treatment of VEGF-A165a and VEGF-A165b (‘both’) showed no significant change in ZO1 expression (84.3% ± 5.6), compared with untreated, but showed a significant increase relative to VEGF-A165a-treated wells. This was also seen by immunoblotting (Figure 2F). VEGF-A165b has no detrimental effect on either ZO1 or occludin expression VEGF-A165a-reduced ZO1 and occludin expression (12.6±9% and 22.7±15% of control respectively). VEGF-A165a and VEGF-A165b co-treatment showed no significant TJ loss (Figure 2G).
VEGF-A165b prevents hyperglycaemia-induced changes in TJ integrity
DR is associated with persistent hyperglycaemia. To determine whether outer BRB breakdown was also associated with hyperglycaemia, RPE were cultured in normal or high glucose and (Figure 3) and assayed for occludin expression. Cells cultured in low glucose conditions alone showed no difference in occludin expression relative to untreated wells (Figure 3A) and the occludin was predominantly associated with cell junctions. When cultured in 35 mM glucose, occludin expression was not clearly delineated at the cell borders, and Western blotting showed that there was less occludin expression overall at 35 mM glucose (Figure 3B and C). This was not an osmotic effect, as treatment with 35 mM mannitol did not affect occludin expression. When co-treated with 2.5 nM VEGF-A165b, the occludin expression was restored and was present at the junctions (Figure 3A and B) in the presence of 35 mM glucose.
Figure 3. VEGF165b prevents hyperglycaemia-induced changes in TJ integrity.

(A) RPE cells (n=3, mean ± SEM) were cultured in 5 mM glucose, 5 mM glucose + 2.5 nM VEGF-A165a, 5 mM glucose + 1 μM 56/8, 35 mM glucose and 35 mM glucose + 2.5 nM VEGF-A165b for 24 h and stained for occludin (green) and nuclei were stained for Hoechst (blue). (B) Protein lysate was also extracted under these conditions (n=2, mean ± SD) and immunoblotted against occludin. Blots were then stripped and re-probed for β-actin and quantified (C). (D) TEER was assayed and area under the curve (E) was quantified (A–D and F, one-way ANOVA with Tukey's post-hoc test, **P<0.0). (E) Two-way ANOVA with Tukey's post-hoc, **P<0.01, ***P<0.001.
To determine whether this effect was functional, we measured TEER under the same conditions. Hyperglycaemia significantly increased permeability relative to normoglycaemic cells. In contrast with the effect on occludin expression, this increase was also seen with co-treatment with 2.5 nM VEGF-A165b (Figure 3D). Measurement of total solute flux indicated that VEGF-A165b co-treatment did partially reduce permeability, but did not restore it to the level of normoglycaemic cells.
VEGF-A165b prevents diabetes-induced increase in retinal vascular density
To determine whether the effects of VEGF-A165b seen in vitro were also seen in animal models of DR, we investigated the effect of long-term systemic treatment of diabetic rats with VEGF-A165b. Treatment of two groups of five rats with STZ resulted in hyperglycaemia after 1 week, which was maintained for 8 weeks (Figure 4A). Half of the diabetic animals were given biweekly injection (i.p.) of VEGF-A165b (20 ng/g). The half-life of VEGF-A165b is 4.2 h when given i.p. [30] and this dose and timing have previously been shown to exert anti-nociceptive [31] and anti-nephropathic [32] effects in rats and mice. A control group did not receive STZ. After 8 weeks, the retinae were excised, flat mounted and stained for blood vessels with isolectin B4 (IB4) (Figure 4B). Interestingly, there was a significantly increased vascular area in the diabetic retinae (Figure 4C) when compared with control retinae (1.15±0.08 mm2 and 0.85±0.007 mm2 respectively, p<0.01 one-way ANOVA). VEGF-A165b-treated diabetic retinae showed a reduction in vascular area compared with both control retinae and diabetic + vehicle-treated retinae. To confirm this, we calculated the volume occupied by vasculature. Diabetic + vehicle-treated retinae expressed a greater vascular volume (Figure 4D) relative to control retinae (5.6×106 μm3 ± 4.8×105 and 3.8×106 μm3 ± 3.8×105 respectively). VEGF-A165b treatment prevented diabetes-induced vascular remodelling (2.3×106 μm3 ± 2.9×105), but also showed a significant reduction in volume relative to control retinae. Calculation of integrated density also demonstrated an increase in IB4 staining in the diabetic animals, which was reduced by VEGF-A165b treatment (Figure 4E). Vessel tortuosity is an another pathological feature of PDR [7] where vessels dilate in response to ischaemia and become more tortuous. This aspect is thought to precede neovascularization [33] and is often used as an indicator of severity of disease in other ocular diseases as well as DR. Tortuosity was defined as ‘change in straightness’ and analysis showed there was a trend in diabetic + vehicle animals where individual rats showed an increase in tortuosity (Figure 4F) relative to control rats and diabetic + VEGF-A165b-treated rats. Systemic VEGF-A165b intervention showed a statistically significant reduction in average vessel tortuosity (0.074±0.004). There was no difference between untreated and VEGF-A165b-treated groups.
Figure 4. VEGF-A165b prevents diabetes-induced increase in retinal vascular density.

(A) STZ (50 mg/kg, i.p.) was used to induce diabetes in Sprague–Dawley rats (n=10). Saline was injected in vehicle controls (i.p., n=4). Diabetic rats were either treated with VEGF-A165b (20 ng/g, biweekly i.p., n=5) or saline (biweekly i.p., n=5). All groups were weighed weekly and rats with a blood glucose ≥15 mmol/l were deemed diabetic. (B) Rats were killed at 8 weeks and retinae were stained for blood vessels using IB4 and DAPI. Vascular density was calculated from three parts of the retina in each animal (denoted by shading) using Imaris software, measured as both area (C) and volume (D) occupied by vasculature in a 2D and 3D plane respectively. This was corroborated by measuring integrated density on Fiji software (E). Retinae were also assessed for vessel straightness using Imaris software as a marker for vessel tortuosity (F); one-way ANOVA, Tukey's post-hoc test, *p<0.05, **p<0.01 and ***p<0.001).
Systemic VEGF-A165b prevents diabetes-induced increase in solute flux
In an additional cohort of five animals per group, Evans Blue was used to assay retinal permeability in diabetic and control rats. Again, STZ-treated rats became hyperglycaemic within 1 week and maintained hyperglycaemia for 8 weeks (Figure 5A). Excised STZ + vehicle-treated retinae were noticeably more blue (Figure 5B) than both control- and STZ + VEGF-A165b-treated retinae. When the amount of Evans Blue per g of tissue that had leaked during the 2 h of the experiment (solute flux) was calculated (Figure 5C), it was significantly increased in diabetic retinae (0.148±0.047 μg/min per g) relative to control retinae (0.012±0.028 μg/min per g, n=10 retinae). VEGF-A165b-treated retinae had a significantly reduced EB solute flux (0.0002±0.021 μg/min per g) relative to diabetic + vehicle retinae. There was no statistically significant difference between control- and diabetic + VEGF-A165b-treated retinae. Solute flux was used to calculate the permeability surface area product (PA) based on the concentration of plasma Evans Blue during the experiment (Figure 5D). There was a significantly increased EB PA in diabetic retinae (0.007±0.002 μl/g per h) relative to control retinae (1.12×10−5 ± 4.08×10−4 μl/g per h). There was a significant decrease in EB PA in VEGF-A165b-treated groups (6.63×10−5 ± 8.96×10−4 μl/g per h), relative to diabetic + vehicle-treated groups. There was no significant difference between control- and VEGF-A165b-treated groups.
Figure 5. Systemic VEGF-A165b prevents diabetes-induced increase in solute flux.

(A) Sprague–Dawley female rats were induced with diabetes using STZ (50 mg/kg i.p., n=10) and control rats (n=5) were injected with saline (i.p.) on day 0. After 4 days, blood glucose was tested and blood glucose ≥ 15mmol/l were deemed diabetic. (B) Diabetic rats were treated with either vehicle (saline, biweekly i.p., n=5) or VEGF-A165b (20 ng/g, biweekly i.p., n=5). At 8 weeks post-STZ induction, Evans Blue (EB, 45 mg/kg) was injected i.v. into terminally anaesthetized rats. Plasma was collected every 15 minutes for 2 h, after which, animals were killed and retinae were excised. (C) Retinae were weighed and EB was extracted using formamide, allowing EB solute flux (C) and permeability surface area product (D) to be calculated; Kruskal–Wallis test with Dunn's post-hoc test, **p<0.01, ***p<0.001.
Intravitreal VEGF-A165b prevents diabetes-induced increase in solute flux
Most treatments for DR and AMD are administered locally, either intravitreally (IVT) or subretinally, as most drugs are hypothesized to not cross the BRB effectively. VEGF-A165b was given IVT to see if local administration could prevent diabetes-mediated TJ dysfunction. However, given the short half-life of VEGF-A165b [30], the 8-week STZ model would be unsuitable as the rat would require several intraocular injections a week. We, therefore, investigated the effect of intraocular injection of VEGF-A165b 24 h before assessment of Evans Blue extravasation in diabetes after 1-week based on a 7-day protocol as established in Xu et al [27]. Again, upon excision, diabetic + vehicle-treated retinae were considerably more blue than contralateral, VEGF-A165b-injected eyes (Figure 6A). EB solute flux (Figure 6B) increased in diabetic retinae (0.244±0.05 μg/min per g) relative to control + vehicle-injected retinae (0.089±0.024 μg/min per g). VEGF-A165b treatment induced no change in EB solute flux (0.131±0.038μg/min per g) compared with control, but was lower, but not significantly so, than diabetic animals. There was no significant difference between vehicle-injected control retinae and untreated retinae (0.067±0.016 μg/min per g) indicating that the IVT injection did not exert a significant, inflammatory effect. When measuring PA (Figure 6C), diabetes alone caused a significant increase in EB extravasation (0.012±0.004 μl/g per h) relative to vehicle-treated and untreated controls (0.002±0.008 μl/g per h and 0.002±4.0×10−4 μl/g per h respectively). VEGF-A165b treatment significantly reduced EB extravasation (0.005±0.001 μl/g per h) relative to contralateral diabetic retinae. There was no significant difference between STZ + VEGF-A165b-treated, control + vehicle and untreated groups. When these data were compared with data obtained at 8 weeks (Figure 5), relative solute flux (Figure 6D) increased robustly between 1 and 8 weeks in diabetic animals (by 0.18±0.05 μg/min per g and 0.16±0.047 μg/min per g respectively), whereas relative solute flux in VEGF-A165b-injected animals remained relatively unchanged between 1 and 8 weeks of treatment (0.064 ± −0.038 μg/min per g and 0.012±0.022 μg/min per g greater than their respective controls). The difference between STZ + vehicle-treated solute flux and STZ + VEGF-A165b-treated solute flux at 1 and 8 weeks was different at both time points (p=0.056 and p<0.01 for 1 and 8 weeks respectively). This trend was similar for PA (Figure 6E). PA was raised above control at both 1 and 8 weeks in diabetic + vehicle retinae (11.2±4.42×10−3 μl/g per h and 5.94±2.2×10−3 μl/g per h greater than their respective controls). In VEGF-A165b-treated retinae, while there was less of an increase in PA at 1 week (4.0±1.0 μl/g per h above control) compared with STZ-treated animals (p<0.01) at 8 weeks, the increase in PA was completely abolished (−0.8±0.89 μl/g per h compared with control, Figure 6E).
Figure 6. Intravitreal VEGF-A165b prevents diabetes-induced increase in solute flux.

(A) Sprague–Dawley rats were induced with diabetes using STZ (50 mg/kg). After 6 days, 5 μl of saline was injected into one eye of each diabetic rat, and 5 μl of 10 ng/μl rhVEGF-A165b injected into the contralateral eye (n=5). Control rats had no injection in one eye, and 5 μl of saline in the contralateral eye (n=5). (B) On day 7, Evans Blue (EB, 45 mg/kg) was injected into anaesthetized rats (i.v.). Plasma was collected every 15 min for 2 h, after which, animals were killed and retinae excised. (C) Evans Blue solute flux was calculated from total EB absorbance after 2 h and (D). Permeability surface area product (PA) was calculated using solute flux as a measure of plasma EB absorbance. The increase in solute flux (D) and permeability (E) after 1 week and 8 weeks in STZ + vehicle-treated retinae compared with their respective controls was compared that with VEGF-A165b treatment; B and C=one-way ANOVA with Bonferroni post-hoc test, E=two-way ANOVA with Tukey's post-hoc test, *p<0.05, **p<0.01.
VEGF-A165b prevents retinal ganglion cell shrinkage
Retinae from three animals were stained with NeuN to assess retinal ganglion cells (RGC) number (Figure 7A). Whilst there was no change in number of RGCs (Figure 7B) expressed in diabetic + vehicle retinae (67±15.8) relative to control retinae (94±9.07), there was a slight non-significant increase in RGC number in VEGF-A165b-treated diabetic retinae (76.9±2.97). When assessing NeuN positive area (Figure 7C), a similar trend was observed. The distribution of cells was further analysed by measuring the area of each NeuN positive cell within each single high-power field (40× magnification). Cells were then binned according to size, into bins of 40 μm2, and displayed as the average number of cells per high-power field in each bin from 40 to 320 μm2. The data were fitted with a Gaussian curve (Figure 7D). Both the control- and VEGF-A165b-treated groups demonstrated a normal distribution of areas of cells with NeuN positive staining (Figure 7D, grey and black filled curves), whereas the STZ group had a significant left skew to the distribution (Figure 7D, red filled curve, p<0.01 F-test for comparison of distribution). This indicates that diabetic NeuN positive stained foci in diabetic rats were smaller than control, with most RGC staining being 0–39 and 80–19 μm2 respectively. Diabetic rats treated with VEGF-A165b also had peak expression within 80–119 μm2. Interestingly, at 0–30 μm2, there was significantly more RGC staining within that range in diabetic retinae (4.19±0.908) relative to both control (0.583±0.583) and diabetic + VEGF-A165b (0.021±0.917).
Figure 7. VEGF-A165b prevents retinal ganglion cell shrinkage.

(A) Sprague–Dawley rats induced with diabetes were treated with vehicle (saline i.p., n=4) or VEGF-A165b (20 ng/g i.p., n=4) biweekly for 8 weeks. Animals were then killed and retinae excised and (A) stained for RGC marker NeuN. RGC NeuN+ staining was counted (B) and areas measured (C). (D) The frequency of NeuN+ areas (50 μm bins) was plotted against area, and Lorentzian curve fitted, and curve fit compared with an F-test; P<0.01.
Discussion
VEGF-A165b prevents VEGF-A165a-mediated changes in barrier properties
Studies looking at the inner BRB, specifically RECs, have shown that increasing levels of VEGF-A both in vitro and in vivo induce ‘rapid phosphorylation’ of occludin and ZO1 resulting in increased barrier permeability [34]. We show here that VEGF-A165a dose-dependently reduced occludin expression (Figure 1A and C) in RPE cells, which form the outer BRB. This decrease in occludin expression coincided with an increase in paracellular conductance (Figure 1E), confirming that occludin expression is tightly linked with paracellular flux. As VEGF-A165a concentration increased from 0 to 5 nM, VEGFR2 expression increased in RPE cells. An increase in VEGFR2 stimulation results in increased VEGFR2 phosphorylation and increased activity of PKC [35]. Increased PKC activity is hypothesized to induce changes in the actin cytoskeleton, which would in turn induce changes in the ZO1–occludin complex, resulting in TJ degradation/internalization and subsequent increase in permeability. VEGF-A165b, whilst it binds to VEGFR2 with equal affinity as VEGF-A165a [20], does not activate the receptor completely. When it binds, it only partially phosphorylates VEGFR2, and therefore elicits a different downstream effect of reducing permeability, promoting cell survival and reducing angiogenesis [36].
It has previously been acknowledged that there is a switch in isoform splicing in the diabetic eye in favour of pro-angiogenic isoforms such that VEGF-A165a levels overwhelm VEGF-A165b levels [22]. This means that competitive binding of both isoforms to VEGFR2 will also be in favour of VEGF-A165a, resulting in increased angiogenesis and increased permeability. VEGF-A165b alone does not elicit an increased paracellular flux in RPE (Figure 2A) when treated at the same concentrations. Figure 2D and F show that 2.5 nM VEGF-A165b treatment alone has no significant effect on location of ZO1 or on ZO1 and occludin expression.
By co-treating VEGF-A165a with VEGF-A165b, there was a restoration in TJ expression and a reduction in permeability of the RPE to baseline (Figure 2). Similar but less robust effects have also been shown using typical and atypical PKC inhibitors in endothelial cells, where VEGF-A-induced increased TEER and occludin phosphorylation were reduced [35,37]. As mentioned earlier, VEGF-A165b binding to VEGFR2 results in incomplete auto-phosphorylation of the receptor in endothelial cells. As a result, downstream signalling events such as activation of PKC is reduced [36] thereby reducing TJ phosphorylation.
VEGF-A165b prevents diabetes-induced retinal vascular remodelling
The use of STZ-induced hyperglycaemia is a widely used and accepted model of Type 1 diabetes due to its rapid, reliable and robust phenotypic onset. STZ combined with the addition of long-term insulin enables a more accurate model of Type 1 diabetes. It has been hypothesized that PDR is the predominant ‘sight-threatening lesion’ in Type 1 diabetic patients [38]; however, comorbidities associated with diabetes typically take many years of poor glycaemic control to develop. To compensate for this, the experimental protocol duration was set at 7–8 weeks in rats. This time frame has been shown to produce reproducible features of diabetic neuropathy [31], and as diabetic neuropathy and retinopathy occur over a similar time frame, we hypothesized that this would also be a suitable model of PDR.
It was evident that there was indeed an increase in retinal vascular density in diabetic retinae compared with untreated animals, after 8 weeks of diabetes. Increased area and volume occupied by vasculature both increased in vehicle-treated diabetic retinae (Figure 4C and D). This is the first report of an increase/change in the retinal vasculature in STZ diabetes. This model of insulin-supplemented chronic diabetes in rats could lead to vascular remodelling, and would thus be a valuable new model of PDR. It would be useful to understand whether this is associated with increased endothelial cell division earlier in the model, and whether true angiogenesis is happening. The increased vessel tortuosity seen in diabetic retinae suggests a model that is more representative of human DR.
Vessel tortuosity typically precedes angiogenesis and is indicative of a hypoxic environment. Instances where there are changes in NO expression in the retina, will also result in changes in vasodilatation both longitudinally and radially, both occurring at different rates and therefore resulting in a meandering structure [33], as observed in the diabetic + vehicle-treated rat retinae (Figure 4F). Interestingly, in VEGF-A165b-treated groups there was less vessel tortuosity. This may indicate that VEGF-A165b, when applied systemically, can cross the BRB and potentially interferes with eNOS signalling by preventing HIF transcription or by interfering with NO production. This would prevent the need for the vessels to constantly dilate and reperfuse, as the vasculature may not be occluded at all. This demonstrates the importance in maintaining the correct balance between both VEGF-A isoforms. The increased vascular density observed in Figure 4 is likely to be mediated by delta-like ligand 4 (DLL4). In hypoxic microenvironments where there is increased VEGF-A expression and VEGFR2 activation, endothelial cells in these regions may express more DLL4. DLL4–Notch-mediated tip and stalk cell formation respectively result in tubule formation and, eventually, widespread angiogenesis [39,40]. In models of CNV and choroidal hypoxia, angiogenesis has been linked with increased VEGF-A-induced DLL4 expression [41]. This has been consistently shown to be the key aspect of retinal angiogenesis [40]. Interestingly, in a model of in vitro haemangioma, VEGF-A165b treatment prevented DLL4 expression by VEGF-A165a in haemangioma endothelial cells [42]. Thus, in the diabetic + VEGF-A165b-treated retinae competitive binding of both isoforms to VEGFR2 could be preventing excessive VEGF-A165a-induced VEGFR2 activation resulting in more VEGF-A165b binding to VEGFR2 and a resultant weakened activation of the receptor.
STZ-induced diabetes as a model of DME and the effect of rhVEGF-A165b on diabetic hyperpermeability
There was an increase in extravasation of Evans Blue dye in the retina after both 1 and 8 weeks of diabetes (Figure 6E and 6F), the former similar to that previously shown by Qaum et al. [43] to be VEGF-A dependent. Development of increased retinal permeability at 8 weeks has not previously been described in STZ diabetes, but it appears that 8 weeks of diabetes provides a more robust phenotype, relative to 1-week diabetes as there is an increase in both EB solute flux and PA in the 8-week model relative to the 1-week model.
There are many pathways by which extravasation could occur in this model, but it is perhaps most likely to be TJ phosphorylation. This relies on the assumption that the increased solute flux is due to increased vascular permeability, and not due to other changes in vasoactivity. Similar models using the EB technique are in agreement that diabetes increases EB leakage and is likely mediated by PKC [35,37,44]. PKC has been linked directly to occludin phosphorylation in vitro and in vivo predominantly at the serine and threonine residues, and potentially these residues on ZO1 [35,37], resulting in their internalization to an intracellular pool. Inhibition of atypical PKC isoforms, such as PKCζ, have shown promising results with regard to VEGF-induced but not diabetes-induced permeability in vivo [37].
Activation of DAG and PKC results in an up-regulation of VEGF-A165a (or the mouse equivalent, VEGF-A164a) and NF-κB, both of which are pro-inflammatory and up-regulate expression of intracellular adhesion molecule (ICAM-1) and TNF-α, which result in increased retinal permeability [45,46–48]. VEGF-A165b has been shown to reduce TNF-α mediated ICAM activation in RPE cells, and therefore is potentially anti-inflammatory [49]. VEGF-A165b competitively binds to VEGFR2, in line with the partial activation of VEGFR2 by VEGF-A165b, this ligand only partially activates DAG and therefore partially activates typical PKC isoforms; therefore, there is little or no phosphorylation of TJs. VEGF-A165b also reduces TNF-α expression in the outer BRB, perhaps by blocking atypical PKC activation in the RPE, also preventing TJ phosphorylation and subsequent hyperpermeability.
However, it is possible that the increase in extravasation is due to an increase in blood flow to the retina, which would result in increased delivery of Evans Blue to the tissue [50] .While this cannot be ruled out it is unlikely as blood flow is generally reduced in DR [2].
VEGF-A165b protects against the reduction in NeuN staining induced by diabetes
RGC loss is a prominent feature of the neurodegenerative aspect of DR and is significantly part of vision loss. We did not see any significant reduction in NeuN cell number, consistent with previous work that shows SD rats develop a non-significant reduction in RGCs after 4 months of insulin-supplemented diabetes [51]. However, it was evident that diabetic + vehicle-treated rats showed a change in the distribution of the size of NeuN staining. NeuN is an antibody that stains Fox3, an RNA-binding protein that is normally found in the nucleus. A reduction in the size of the NeuN staining area could therefore be due to a reduction in expression of Fox3, or a small number of RGC undergoing apoptosis resulting in nuclear condensation (pyknosis) or fragmentation (karyorrhexis). However, we were unable to confirm this due to the nature of the NeuN staining and the lack of nuclear marker in those retinas. Further work could be undertaken to investigate whether this 8-week model of insulin supplementation in diabetes does lead to RGC undergoing cell death.
VEGF-A is up-regulated in the diabetic retina, particularly in insulin-treated diabetes in SD rats [52] and both the VEGF-A165a and VEGF-A165b isoforms have neuroprotective functions in the retina [26,53,54]. Furthermore, insulin has been hypothesized to prevent RGC apoptosis in rat models of DR [55], conflicting with what is observed in patients where insulin therapy causes a transient worsening of retinopathy [56,57]. VEGF-A and insulin may be protecting the RGC layer from the vascular dysfunction in the earlier stages of DR. It is possible that over time, once the vascular dysfunction develops and there is an increase pro-inflammatory markers, RGCs are no longer sufficiently protected by VEGF-A and insulin. If we extend this model to a significantly longer time point, there may be RGC dysfunction. An additional caveat to this experiment is that some NeuN antibodies have been known to also stain cholinergic amacrine cells that have been displaced to the GCL. However, this is usually only 15–20% of cells stained by NeuN, and it is therefore likely that the majority of these results are from RGCs [58]. Nevertheless, VEGF-A165b does appear to protect against RGC nuclear shrinkage and perhaps amacrine cell nuclear shrinkage, both cell types associated with visual dysfunction in DR.
Summary
VEGF-A165b, systemic or locally provided, was able to reduce signs of DR, particularly increased vascular permeability, vessel remodelling and neuronal dysfunction in the diabetic eye. This appears to be one of, if not the, only endogenous agent able to reduce all three cardinal signs of retinopathy in an animal model of diabetes. VEGF-A165b is generated by alternative splicing of the VEGF-A gene at the last exon. The mechanisms underlying this splice site control have recently been elucidated, and it is clear that the activation of the protein kinase SRPK1 is one mechanism through which VEGF-A165a is up-regulated and VEGF-A165b is down-regulated [59]. Inhibitors of SRPK1 have been shown to be anti-angiogenic in animal models of choroidal neovascularization [28], prostate [60] and colorectal cancer [61] and malignant melanoma [62]. These inhibitors are being developed as a new class of anti-angiogenic agents for age-related macular degeneration [63], and if they can up-regulate VEGF-A165b in DR, then the results shown here indicate that they could be potential novel therapeutics, having the advantage over existing anti-angiogenic therapies of being neuroprotective for RGC cells and protective for RPE cells, at the same time as reducing extravasation and preventing new vessel growth.
Abbreviations
- AMD
age related macular degeneration
- BRB
blood–retina barrier
- CNV
choroidal neovascularization
- DAG
Diacylglycerol
- DLL4
delta-like ligand 4
- DME
diabetic macular oedema
- DMEM
Dulbecco's Modified Eagle's Medium
- DR
diabetic retinopathy
- ECIS
electric cell-substrate impedance sensing
- eNOS
endothelial nitric oxide synthase
- GCL
ganglion cell layer
- IB4
isolectin B4
- ICAM
intracellular adhesion molecule
- HIF
hypoxia inducible factor
- i.p.
intraperitoneal
- i.v.
intravenous
- IVT
intravitreally
- NF-κB
nuclear factor -κB
- NGS
normal goat serum
- NO
nitric oxide
- PDR
proliferative diabetic retinopathy
- PKC
protein kinase C
- REC
retinal endothelial cell
- RGC
retinal ganglion cells
- rh
recombinant human
- RIPA
Radioimmunoprecipitation assay
- RPE
retinal pigmented epithelial
- SD
standard deviation
- SEM
standard error mean
- SRPK1
Serine-arginine protein kinase 1
- STZ
streptozotocin
- TEER
trans-epithelial electrode resistance
- TJ
tight junction
- VEGF-A
vascular endothelial growth factor
Author contribution
The work was conceived by D.O.B., J.W.B. and L.F.D. Experiments were designed by D.O.B., L.F.D. and R.P.H. Results were collected by N.V., S.M.B. and R.P.H. and analysed by D.O.B. and N.V. The article was drafted by N.V. and D.O.B. and revised by all authors. Final approval of the manuscript was provided by all authors.
Funding
This work was supported by Diabetes U.K. grants [grant numbers 10/0004152 (to D.O.B. and J.C.B.) and 11/0004192 (to L.F.D. and D.O.B.)]; and an Medical Research Council grant [MR/K020366/1 (to D.O.B.)].
Competing interests
Professor David Bates and Professor Lucy Donaldson are founders and shareholders of Exonate Ltd., a company that is pursuing SRPK1 inhibitors for age-related macular degeneration and other indications.
References
- 1.Klein R., Klein B.E. and Moss S.E. (1984) Visual impairment in diabetes. Ophthalmology 91, 1–9 10.1016/S0161-6420(84)34337-8 [DOI] [PubMed] [Google Scholar]
- 2.McGahon M.K., Dash D.P., Arora A., Wall N., Dawicki J., Simpson D.A.. et al. (2007) Diabetes downregulates large-conductance Ca2+-activated potassium beta 1 channel subunit in retinal arteriolar smooth muscle. Circ. Res. 100, 703–711 10.1161/01.RES.0000260182.36481.c9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bursell S.E., Clermont A.C., Kinsley B.T., Simonson D.C., Aiello L.M. and Wolpert H.A. (1996) Retinal blood flow changes in patients with insulin-dependent diabetes mellitus and no diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 37, 886–897 [PubMed] [Google Scholar]
- 4.Aiello L.P., Avery R.L., Arrigg P.G., Keyt B.A., Jampel H.D., Shah S.T.. et al. (1994) Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 331, 1480–1487 10.1056/NEJM199412013312203 [DOI] [PubMed] [Google Scholar]
- 5.Hammes H.P. (2005) Pericytes and the pathogenesis of diabetic retinopathy. Horm. Metab. Res. 37, Suppl 139–43 10.1055/s-2005-861361 [DOI] [PubMed] [Google Scholar]
- 6.Cogan D.G., Toussaint D. and Kuwabara T. (1961) Retinal vascular patterns. IV. Diabetic retinopathy. Arch. Ophthalmol. 66, 366–378 10.1001/archopht.1961.00960010368014 [DOI] [PubMed] [Google Scholar]
- 7.Gardner T.W., Antonetti D.A., Barber A.J., LaNoue K.F. and Levison S.W. (2002) Diabetic retinopathy: more than meets the eye. Surv. Ophthalmol. 47, Suppl 2S253–S262 10.1016/S0039-6257(02)00387-9 [DOI] [PubMed] [Google Scholar]
- 8.Simo R., Sundstrom J.M. and Antonetti D.A. (2014) Ocular anti-VEGF therapy for diabetic retinopathy: the role of VEGF in the pathogenesis of diabetic retinopathy. Diabetes Care 37, 893–899 10.2337/dc13-2002 [DOI] [PubMed] [Google Scholar]
- 9.Photocoagulation therapy for diabetic eye disease. (1985) Early Treatment Diabetic Retinopathy Study Research Group. J. Am. Med. Assoc. 254, 3086 10.1001/jama.1985.03360210102042 [DOI] [PubMed]
- 10.Cheung N., Wong I.Y. and Wong T.Y. (2014) Ocular anti-VEGF therapy for diabetic retinopathy: overview of clinical efficacy and evolving applications. Diabetes Care 37, 900–905 10.2337/dc13-1990 [DOI] [PubMed] [Google Scholar]
- 11.Stitt A.W., Curtis T.M., Chen M., Medina R.J., McKay G.J., Jenkins A.. et al. (2016) The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 51, 156–186 10.1016/j.preteyeres.2015.08.001 [DOI] [PubMed] [Google Scholar]
- 12.Falavarjani K.G. and Nguyen Q.D. (2013) Adverse events and complications associated with intravitreal injection of anti-VEGF agents: a review of literature. Eye (Lond.) 27, 787–794 10.1038/eye.2013.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holz F.G., Bellman C., Staudt S., Schutt F. and Volcker H.E. (2001) Fundus autofluorescence and development of geographic atrophy in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 42, 1051–1056 [PubMed] [Google Scholar]
- 14.Shakib M. and Cunha-Vaz J.G. (1966) Studies on the permeability of the blood-retinal barrier. IV. Junctional complexes of the retinal vessels and their role in the permeability of the blood-retinal barrier. Exp. Eye Res. 5, 229–234 10.1016/S0014-4835(66)80011-8 [DOI] [PubMed] [Google Scholar]
- 15.Cunha-Vaz J.G. (1976) The blood-retinal barriers. Documenta ophthalmologica. Adv. Ophthalmol. 41, 287–327 [DOI] [PubMed] [Google Scholar]
- 16.Weinberger D., Fink-Cohen S., Gaton D.D., Priel E. and Yassur Y. (1995) Non-retinovascular leakage in diabetic maculopathy. Br. J. Ophthalmol. 79, 728–731 10.1136/bjo.79.8.728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saint-Geniez M., Kurihara T., Sekiyama E., Maldonado A.E. and D'Amore P.A. (2009) An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc. Natl. Acad. Sci. U.S.A. 106, 18751–18756 10.1073/pnas.0905010106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simo R., Lecube A., Segura R.M., Garcia Arumi J. and Hernandez C. (2002) Free insulin growth factor-I and vascular endothelial growth factor in the vitreous fluid of patients with proliferative diabetic retinopathy. Am. J. Ophthalmol. 134, 376–382 10.1016/S0002-9394(02)01538-6 [DOI] [PubMed] [Google Scholar]
- 19.Bates D.O., Cui T.G., Doughty J.M., Winkler M., Sugiono M., Shields J.D.. et al. (2002) VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 62, 4123–4131 [PubMed] [Google Scholar]
- 20.Woolard J., Wang W.Y., Bevan H.S., Qiu Y., Morbidelli L., Pritchard-Jones R.O.. et al. (2004) VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 64, 7822–7835 10.1158/0008-5472.CAN-04-0934 [DOI] [PubMed] [Google Scholar]
- 21.Kawamura H., Li X., Goishi K., van Meeteren L.A., Jakobsson L., Cebe-Suarez S.. et al. (2008) Neuropilin-1 in regulation of VEGF-induced activation of p38MAPK and endothelial cell organization. Blood 112, 36–38 10.1182/blood-2007-12-125856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perrin R.M., Konopatskaya O., Qiu Y., Harper S., Bates D.O. and Churchill A.J. (2005) Diabetic retinopathy is associated with a switch in splicing from anti- to pro-angiogenic isoforms of vascular endothelial growth factor. Diabetologia 48, 2422–2427 10.1007/s00125-005-1951-8 [DOI] [PubMed] [Google Scholar]
- 23.Konopatskaya O., Churchill A.J., Harper S.J., Bates D.O. and Gardiner T.A. (2006) VEGF165b, an endogenous C-terminal splice variant of VEGF, inhibits retinal neovascularization in mice. Mol. Vis. 12, 626–632 [PubMed] [Google Scholar]
- 24.Hua J., Spee C., Kase S., Rennel E.S., Magnussen A.L., Qiu Y.. et al. (2010) Recombinant human VEGF165b inhibits experimental choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 51, 4282–4288 10.1167/iovs.09-4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Magnussen A.L., Rennel E.S., Hua J., Bevan H.S., Beazley Long N., Lehrling C.. et al. (2010) VEGF-A165b is cytoprotective and antiangiogenic in the retina. Invest. Ophthalmol. Vis. Sci. 51, 4273–4281 10.1167/iovs.09-4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beazley-Long N., Hua J., Jehle T., Hulse R.P., Dersch R., Lehrling C.. et al. (2013) VEGF-A165b is an endogenous neuroprotective splice isoform of vascular endothelial growth factor A in Vivo and in Vitro. Am. J. Pathol. 183, 918–929 10.1016/j.ajpath.2013.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Q., Qaum T. and Adamis A. (2001) Sensitive blood-retinal barrier breakdown quantitation using Evans blue. Invest. Ophthalmol. Vis. Sci. 42, 789–794 [PubMed] [Google Scholar]
- 28.Gammons M.V., Dick A.D., Harper S.J. and Bates D.O. (2013) SRPK1 inhibition modulates VEGF splicing to reduce pathological neovascularization in a rat model of retinopathy of prematurity. Invest. Ophthalmol. Vis. Sci. 54, 5797–5806 10.1167/iovs.13-11634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T.. et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rennel E.S., Hamdollah-Zadeh M.A., Wheatley E.R., Magnussen A., Schuler Y., Kelly S.P.. et al. (2008) Recombinant human VEGF165b protein is an effective anti-cancer agent in mice. Eur. J. Cancer 44, 1883–1894 10.1016/j.ejca.2008.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hulse R.P., Beazley-Long N., Ved N., Bestall S.M., Riaz H., Singhal P.. et al. (2015) Vascular endothelial growth factor-A165b prevents diabetic neuropathic pain and sensory neuronal degeneration. Clin. Sci. (Lond. 129, 741–756 10.1042/CS20150124 [DOI] [PubMed] [Google Scholar]
- 32.Oltean S., Qiu Y., Ferguson J.K., Stevens M., Neal C., Russell A.. et al. (2014) Vascular endothelial growth factor-A165b is protective and restores endothelial glycocalyx in diabetic nephropathy. J. Am. Soc. Nephrol. 26, 1889–1904 10.1681/ASN.2014040350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hart W.E., Goldbaum M., Cote B., Kube P. and Nelson M.R. (1999) Measurement and classification of retinal vascular tortuosity. Int. J. Med. inform. 53, 239–252 10.1016/S1386-5056(98)00163-4 [DOI] [PubMed] [Google Scholar]
- 34.Antonetti D.A., Barber A.J., Hollinger L.A., Wolpert E.B. and Gardner T.W. (1999) Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J. Biol. Chem. 274, 23463–23467 10.1074/jbc.274.33.23463 [DOI] [PubMed] [Google Scholar]
- 35.Harhaj N.S., Felinski E.A., Wolpert E.B., Sundstrom J.M., Gardner T.W. and Antonetti D.A. (2006) VEGF activation of protein kinase C stimulates occludin phosphorylation and contributes to endothelial permeability. Invest. Ophthalmol. Vis. Sci. 47, 5106–5115 10.1167/iovs.06-0322 [DOI] [PubMed] [Google Scholar]
- 36.Harper, S.J. and Bates, D.O. (2008) VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat. Rev. Cancer 8, 880–887 10.1038/nrc2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Titchenell P.M., Lin C.M., Keil J.M., Sundstrom J.M., Smith C.D. and Antonetti D.A. (2012) Novel atypical PKC inhibitors prevent vascular endothelial growth factor-induced blood-retinal barrier dysfunction. Biochem. J. 446, 455–467 10.1042/BJ20111961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simo R., Villarroel M., Corraliza L., Hernandez C. and Garcia-Ramirez M. (2010) The retinal pigment epithelium: something more than a constituent of the blood-retinal barrier–implications for the pathogenesis of diabetic retinopathy. J. Biomed. Biotechnol. 2010, 190724. 10.1155/2010/190724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hellstrom M., Phng L.K., Hofmann J.J., Wallgard E., Coultas L., Lindblom P.. et al. (2007) Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445, 776–780 10.1038/nature05571 [DOI] [PubMed] [Google Scholar]
- 40.Suchting S., Freitas C., le Noble F., Benedito R., Breant C., Duarte A.. et al. (2007) The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc. Natl. Acad. Sci. U.S.A. 104, 3225–3230 10.1073/pnas.0611177104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong X., Wang Y.S., Dou G.R., Hou H.Y., Shi Y.Y., Zhang R.. et al. (2011) Influence of Dll4 via HIF-1alpha-VEGF signaling on the angiogenesis of choroidal neovascularization under hypoxic conditions. PLoS One 6, e18481. 10.1371/journal.pone.0018481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye X., Abou-Rayyah Y., Bischoff J., Ritchie A., Sebire N.J., Watts P.. et al. (2016) Altered ratios of pro- and anti-angiogenic VEGF-A variants and pericyte expression of DLL4 disrupt vascular maturation in infantile haemangioma. J. Pathol. 239, 139–151 10.1002/path.4715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qaum T., Xu Q., Joussen A.M., Clemens M.W., Qin W., Miyamoto K.. et al. (2001) VEGF-initiated blood-retinal barrier breakdown in early diabetes. Invest. Ophthalmol. Vis. Sci. 42, 2408–2413 [PubMed] [Google Scholar]
- 44.Aiello L.P., George D.J., Cahill M.T., Wong J.S., Cavallerano J., Hannah A.L.. et al. (2002) Rapid and durable recovery of visual function in a patient with von hippel-lindau syndrome after systemic therapy with vascular endothelial growth factor receptor inhibitor su5416. Ophthalmology 109, 1745–1751 10.1016/S0161-6420(02)01159-4 [DOI] [PubMed] [Google Scholar]
- 45.Ishida S., Usui T., Yamashiro K., Kaji Y., Amano S., Ogura Y.. et al. (2003) VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J. Exp. Med. 198, 483–489 10.1084/jem.20022027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ishida S., Usui T., Yamashiro K., Kaji Y., Ahmed E., Carrasquillo K.G.. et al. (2003) VEGF164 is proinflammatory in the diabetic retina. Invest. Ophthalmol. Vis. Sci. 44, 2155–2162 10.1167/iovs.02-0807 [DOI] [PubMed] [Google Scholar]
- 47.Joussen A.M., Poulaki V., Le M.L., Koizumi K., Esser C., Janicki H.. et al. (2004) A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 18, 1450–1452 [DOI] [PubMed] [Google Scholar]
- 48.Tang J. and Kern T.S. (2011) Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 30, 343–358 10.1016/j.preteyeres.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thichanpiang P., Harper S.J., Wongprasert K. and Bates D.O. (2014) TNF-alpha-induced ICAM-1 expression and monocyte adhesion in human RPE cells is mediated in part through autocrine VEGF stimulation. Mol. Vis. 20, 781–789 [PMC free article] [PubMed] [Google Scholar]
- 50.Bates D.O. (2010) Vascular endothelial growth factors and vascular permeability. Cardiovasc. Res. 87, 262–271 10.1093/cvr/cvq105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kern T.S., Miller C.M., Tang J., Du Y., Ball S.L. and Berti-Matera L. (2010) Comparison of three strains of diabetic rats with respect to the rate at which retinopathy and tactile allodynia develop. Mol. Vis. 16, 1629–1639 [PMC free article] [PubMed] [Google Scholar]
- 52.Poulaki V., Qin W., Joussen A.M., Hurlbut P., Wiegand S.J., Rudge J.. et al. (2002) Acute intensive insulin therapy exacerbates diabetic blood-retinal barrier breakdown via hypoxia-inducible factor-1alpha and VEGF. J. Clin. Invest. 109, 805–815 10.1172/JCI0213776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin K.L., Mao X.O. and Greenberg D.A. (2000) Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc. Natl. Acad. Sci. U.S.A. 97, 10242–10247 10.1073/pnas.97.18.10242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shima D.T., Adamis A.P., Ferrara N., Yeo K.T., Yeo T.K., Allende R.. et al. (1995) Hypoxic induction of endothelial cell growth factors in retinal cells: identification and characterization of vascular endothelial growth factor (VEGF) as the mitogen. Mol. Med. 1, 182–193 [PMC free article] [PubMed] [Google Scholar]
- 55.Kern T.S. and Barber A.J. (2008) Retinal ganglion cells in diabetes. J. Physiol. (Lond.) 586, 4401–4408 10.1113/jphysiol.2008.156695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chantelau E. and Kohner E.M. (1997) Why some cases of retinopathy worsen when diabetic control improves. BMJ 315, 1105–1106 10.1136/bmj.315.7116.1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dahl-Jorgensen K., Brinchmann-Hansen O., Hanssen K.F., Sandvik L. and Aagenaes O. (1985) Rapid tightening of blood glucose control leads to transient deterioration of retinopathy in insulin dependent diabetes mellitus: the Oslo study. Br. Med. J. (Clin. Res. Ed.) 290, 811–815 10.1136/bmj.290.6471.811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schlamp C.L., Montgomery A.D., Mac Nair C.E., Schuart C., Willmer D.J. and Nickells R.W. (2013) Evaluation of the percentage of ganglion cells in the ganglion cell layer of the rodent retina. Mol. Vis. 19, 1387–1396 [PMC free article] [PubMed] [Google Scholar]
- 59.Nowak D.G., Woolard J., Amin E.M., Konopatskaya O., Saleem M.A., Churchill A.J.. et al. (2008) Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by known splicing and growth factors. J. Cell Sci. 121, 3487–3495 10.1242/jcs.016410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mavrou A., Brakspear K., Hamdollah-Zadeh M., Damodaran G., Babaei-Jadidi R., Oxley J.. et al. (2014) Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 34, 4311–4319 10.1038/onc.2014.360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Amin E.M., Oltean S., Hua J., Gammons M.V., Hamdollah-Zadeh M., Welsh G.I.. et al. (2011) WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell. 20, 768–780 10.1016/j.ccr.2011.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gammons M.V., Lucas R., Dean R., Coupland S.E., Oltean S. and Bates D.O. (2014) Targeting SRPK1 to control VEGF-mediated tumour angiogenesis in metastatic melanoma. Br. J. Cancer 111, 477–485 10.1038/bjc.2014.342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Batson J., Toop H.D., Redondo C., Babaei-Jadidi R., Chaikuad A., Wearmouth S.F.. et al. (2017) Development of potent, selective SRPK1 inhibitors as potential topical therapeutics for neovascular eye disease. ACS Chem. Biol. 12, 825–832 10.1021/acschembio.6b01048 [DOI] [PubMed] [Google Scholar]