

Abstract
Lung cancer is the leading cause of cancer related death, and the past years’ improved insight into underlying molecular events has significantly improved outcome for specific subsets of patients. In particular, several new therapies that target protein kinases have been implemented, and many more are becoming available. We have investigated lung cancer specimens for somatic mutations in a targeted panel of 612 human genes, the majority being protein kinases. The somatic mutation profiles were correlated to profiles of immune cell infiltration as well as relapse‐free survival. Targeted deep sequencing was performed on 117 tumour/normal pairs using the SureSelect Human Kinome kit (Agilent Technologies), with capture probes targeting 3.2 Mb of the human genome, including exons and untranslated regions of all known kinases, kinase receptors and selected cancer‐related genes (612 genes in total). CD8 staining was determined using Ventana Benchmark. Survival analyses were performed using SPSS. The number of mutations per sample ranged from 0 to 50 (within the 612 genes tested), with a median of nine. The prognosis was worse for patients with more than the median number of mutations. A significant correlation was found between mutations in one of selected DNA‐repair genes and the total number of mutations in that tumour (p < 0.001). There was a significant inverse correlation between the number of infiltrating stromal CD8+ lymphocytes and the presence of EGFR mutations.
Keywords: lung cancer, mutations, kinome, sequencing, prognosis
Short abstract
What's new?
Lung carcinomas are among the tumours with highest mutation frequency. Here, the authors performed mutational analyses of 612 genes–including all known kinases and kinase receptors–in 117 non‐small cell lung cancer (NSCLC) tumours. They also investigated the relationship of mutation rate to number of infiltrating lymphocytes and to the clinical course of the disease. The number of mutations per sample varied, and the relapse‐free survival was worse for patients with more than the median number of mutations. Also, there was a significant inverse correlation between the number of infiltrating stromal CD8+ lymphocytes and the presence of EGFR mutations.
Abbreviations
- NSCLC
non‐small cell lung cancer
- SNV
single nucleotide variant
- TCGA
The Cancer Genome Atlas
- UTRs
untranslated regions
Lung cancer is a common cancer with a poor prognosis, and a five‐year survival rate of 15–20%. Most non‐small cell lung cancer (NSCLC) patients have locally advanced or metastatic disease (stage III/IV) at the time of presentation. Lung cancer is responsible for approximately the same number of life years lost as breast, prostate and colorectal cancers taken together.1
During the past 10 years, there has been a change in treatment algorithms for patients with NSCLC. For patients harbouring activating mutations in the EGFR‐gene, several clinical studies have demonstrated a better efficacy of treatment with EGFR tyrosine kinase inhibitors in terms of response rates, progression free survival and quality of life, when compared to standard chemotherapy.2 All patients with non‐squamous NSCLC in Norway are currently analysed for presence of EGFR‐activating mutations and ALK‐aberrations in tumour biopsies.
Mutagenesis drives transformation of a normal cell to a tumour. Mutations may be a result of endogenous processes or of exposure to exogenous mutagens, in combination with a failure of DNA repair pathways. Somatic mutations are abundant in most cancers. However, most mutations do not appear to play a role in the initiation of the cancer phenotype, but are rather indicative of a high mutation rate.3 The majority of mutations are thus most likely bystander events, that is, passenger mutations with no apparent fitness advantage for the tumour clone. A minor set of mutations is on the other hand thought to act as driver mutations, being directly involved in the development or progression of the tumour.4 The development of massively parallel sequencing technologies has now enabled us to analyse the tumour genome for substitutions, insertions and deletions with a high sensitivity. The Cancer Genome Atlas (TCGA) project has shown that lung carcinomas are among the tumours with highest mutation frequency. Most of these mutations are passenger mutations, contributing to a mutational signature characteristic of tobacco smoking.5 Drugs targeting selected mutations are currently being evaluated in clinical trials. Most of these drugs are targeting protein kinases, thus we have in this study focused on mutations in the “kinome,” that is, in genes coding for kinases.
Stromal T‐cell infiltration has prognostic impact in several cancers. In colorectal cancers, assessment of T cells infiltrating the cancer tissue is used to create an “Immunoscore,” an important supplement to the TNM‐classification, designated TNM‐Immune (TNM‐I). The prognostic value of this immune‐classification seems to be superior to the AJCC/UICC TNM‐classification alone.6 In NSCLC, the immune stroma is recognised as highly important, with stromal T‐cell infiltration having a prognostic impact.7 In addition, immunotherapy has introduced a paradigm shift in lung cancer treatment. The immune checkpoint inhibitors enable the patient's own T‐cells to kill tumour cells. PDL1‐expression is being used as a predictive biomarker for response, but PDL1‐negative tumours still can have long‐lasting effect of treatment. An association between the number of non‐synonymous mutations and immunogenicity in lung carcinomas has been observed.8
Here, we present mutational analyses of 612 genes, including all known kinases and kinase receptors in addition to selected cancer‐related genes, in 117 NSCLC tumours. We have investigated the relationship of mutation rate to immunoscore and to the clinical course of the disease.
Material and Methods
Blood samples and tumour tissues were obtained from 117 operated lung cancer patients admitted to Oslo University Hospital‐Rikshospitalet in the period 2006–2011. The collected tumor samples consisted of 93 adenocarcinomas, 2 large cell carcinomas and 22 squamous cell carcinomas. Sixty patients were female. One patient was diagnosed with metastases based on pathology report, and was omitted from survival analyses.
Tumour tissue was snap frozen in liquid nitrogen and stored at −80°C until DNA isolation. DNA was extracted using Maxwell®16 DNA Purification Kits and a Maxwell®16 instrument according to technical manual (Literature # TM284)(http://www.promega.com). DNA from blood was isolated using the Master Pure DNA purification Kit for blood according to the DNA Purification Protocol (http://www.epicentre.com).
Targeted resequencing was performed using the SureSelect Human Kinome kit (Agilent Technologies), with capture probes targeting 3.2 Mb of the human genome, including exons and untranslated regions (UTRs) of all known kinases and selected cancer‐related genes (612). Library construction and in solution capturing was performed following Agilent's SureSelectXT library construction kit and SureSelect Target enrichment protocol, respectively. Sequencing was performed on an Illumina Genome Analyzer using TruSeq SBS Kit v5 generating paired‐end reads of 75 bp in length. Base calling, demultiplexing and quality filtering was performed using Illumina's software packages SCS2.8/RTA1.8 and Off‐line Basecaller‐v1.8. The raw sequencing reads derived from tumour and control samples were subsequently processed in a computational pipeline to identify and functionally annotate somatic DNA changes.
A variant calling pipeline was established to identify somatic mutations in each tumour/control sample pair. This included several steps: (i) alignment of sequence reads toward the reference genome (GRCh37 ‐ including unlocalised/unplaced contigs and decoy sequences) using BWA‐mem,9 (ii) marking of potential PCR‐derived duplicate reads using Picard (http://broadinstitute.github.io/picard/), (iii) local realignment around indel sites using GATK RealignerTargetCreator and IndelRealigner,10 (iv) base quality recalibration using GATK BaseRecalibrator, and (v) somatic single nucleotide variant (SNV) and short insertion/deletion (InDel) calling using Strelka and MuTect algorithms.11, 12 For SNVs, we only considered calls designated as somatic by both MuTect and Strelka (InDels were called by Strelka only). Our pipeline was recently part of a whole‐genome benchmarking exercise organized through ICGC, in which our performance was assessed for SNV (precision = 0.90, recall = 0.73) and InDel calling (precision = 0.71, recall = 0.70).13
To understand the functional role of identified variants, all variants were subject to a computational annotation workflow including a modified version of ANNOVAR (release 2015Dec14, using Ensembl as the gene model), COSMIC (known somatic cancer variants and Cancer Gene Census, v76 February 2016), PFAM (protein domain information, v27.0), UniProt KB (functional protein properties, release 2016_02), the Drug Gene Interaction Database (druggable targets, version 2.0), Database of Curated Mutations (release January 2016), and database of computational predictions of effects of non‐synonymous variants, v2.1.13, 14, 15, 16, 17, 18
We used IntOGen (Integrative Onco Genomics) to identify cancer driver genes in our sample cohort. Considering that a frequency‐based approach to discover significantly mutated genes is suboptimal in samples with low mutation counts, IntOGen uses an approach by which a clustering and functional bias among gene mutations (across the sample cohort) indicates driver candidates.19
We have previously reported on the survival impact of stromal CD8 expression (cytotoxic T‐cells) including most of the patients in this study (n = 107).7 In short, tissue micro‐arrays were used for determination of an immunoscore by immunohistochemistry CD8 staining by Ventana Benchmark, XT automated slide stainer (Ventana Medical System). By light microscopy, the tissue cores were scored for the degree of infiltration of CD8+ lymphocytes. The percentages of CD8+ lymphocytes among all nucleated cells in the stromal compartments were assessed. Scoring cut‐off points at 5%, 25%, or 50% were used for each core. All samples were anonymized and independently scored by two pathologists. The mean score for duplicate cores from each individual was calculated and the median stromal CD‐8 score was used as cut‐off.
Time to relapse was calculated from the date of diagnosis to the date of diagnosed local relapse, distant metastasis, or lung cancer death with a maximum follow‐up time of 80 months after surgery. Patients that died of other causes were treated as censored. A p values < 0.05 was considered as statistically significant. The survival analyses were performed using SPSS (PASW Statistics for Windows, version 18.0, Chicago: SPSS). Survival curves and estimation of statistical significance between the survival curves utilized the Kaplan‐Meier method and log rank test, respectively. A Cox regression model was applied for the multivariate survival analysis using SPSS (v. 18). Factors included in the multivariate model were stage, sex and histology. A p values < 0.05 was considered statistically significant.
Results
Probes that targeted the human kinome were captured and sequenced at 50–60× coverage in 117 tumour‐control sample pairs. The medians for allelic fraction and read depth at confident variant sites were 0.2 and 84, respectively. The best covered variant sites (read depth of 150) enabled detection of variants with allelic fraction as low as 3–4%. In addition to coding exonic sequences, the targeted regions included canonical splice sites in introns, as well as UTRs. Intronic sequences, except those included in splice sites, were excluded from analysis. Each tumour sample had an accompanying control sample (blood), which allowed the detection of point mutations and insertions/deletions of somatic origin. Our pipeline for somatic mutation detection relied on a consensus between two different algorithms (MuTect and Strelka), a strategy that performed well in a previous benchmarking exercise.20 Only mutations called by both the algorithms were thus included in the further analysis. The number of coding mutations varied extensively between tumours, ranging from 0 to 50 (mean = 11, median = 9). Calculation of coding mutations in the same genes (i.e., kinome) of TCGA lung adenocarcinoma samples revealed similar numbers (mean 13.2, median 9).21 There was no significant difference in number of mutations between men and women or between tumours with different histological subtypes. Number of mutations was significantly associated with packyears of tobacco smoking (p = 0.009).
The TP53 gene and the TTN gene were the most frequently mutated genes, the latter being a known artefact due to its large size (Fig. 1 a). The TP53 mutations were all validated by Sanger sequencing where the sequences were aligned and analysed using SeqScape v.2.5 according to the project template [TP53 accession nr. NM 000546 (TP53refNC000017.9NT010718.15)] (http://www.appliedbiosystems.com.).
Figure 1.
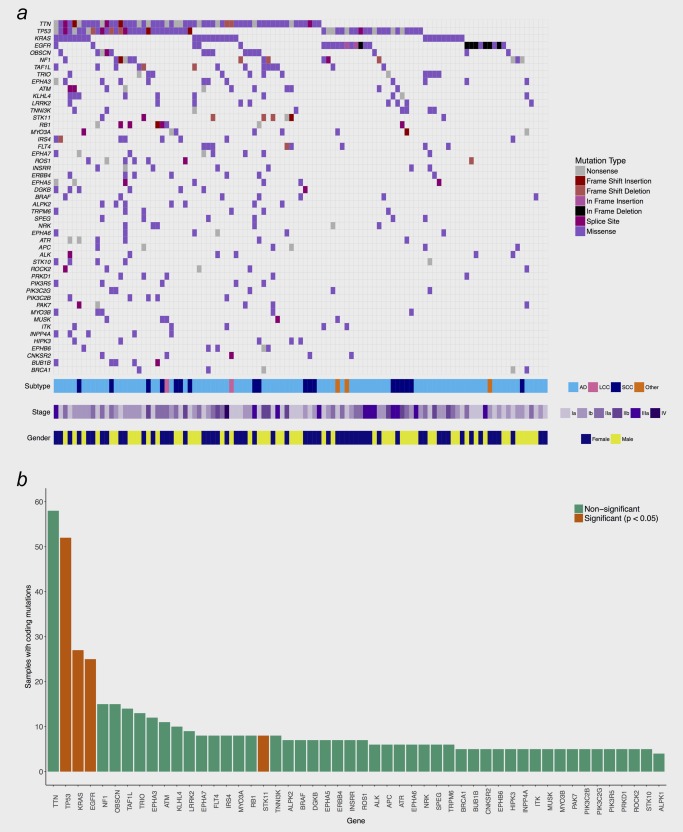
(a) Recurrence of somatically mutated genes. Shown are all mutated genes with a recurrence frequency >= 4% among all 117 samples. AD = Adenocarcinoma, SCC = Squamous Cell Carcinoma, LCC = Large Cell Carcinoma. (b) Frequency of somatic mutations for individual genes. Genes defined by IntoGen as significant cancer drivers are indicated in red.
Through use of IntOGen, KRAS, EGFR, TP53 and STK11 were identified as significant cancer driver genes (Fig. 1 b). TP53 mutations were identified in 52 samples, of which two had double mutations. KRAS was mutated in 28 samples (27 adenocarcinomas, one large cell carcinoma). Eight samples (seven adenocarcinomas) were mutated in the STK11 gene. Only adenocarcinomas were EGFR‐mutated, with mainly L858R mutations and exon 19 deletions.
Mutations in DNA repair genes
Twenty‐seven samples had mutations in known DNA repair genes,1 including PALB2, ATM, ATR, BRCA1, BRCA2, FANCL, CHEK1, CHEK2 and CLK2. There was a significant correlation between mutations in one of these genes and the total number of mutations (p < 0.001). Tumours with a mutation in TP53, ATM or ATR had an increased number of mutations when analysed separately (Fig. 2), whereas tumours with mutations in the other repair genes did not show a significant increase by themselves; however, the number of samples in these groups were small.
Figure 2.
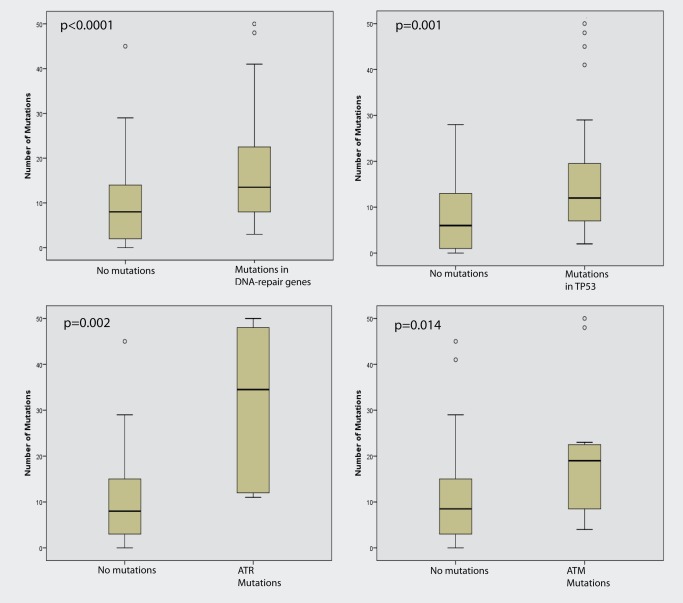
Mutations in one of the DNA repair genes were significantly associated with a higher number of mutations. Mutations in TP53, ATR and ATM alone were associated with a higher number of mutations.
Immunoscore
The tumours were divided in three separate groups based on an immunoscore reflecting the number of infiltrating CD8+ T‐lymphocytes, as defined by Donnem et al.7 There was no significant association between immunoscore and the number of point mutations. However, when using the median stromal CD8‐score as cut‐off (high score n = 58, low score n = 49), there was a significant inverse correlation between the number of infiltrating stromal CD8+ lymphocytes and presence of EGFR mutations (r = −0.21, p = 0.029) (Pearson χ2).
Survival
Patients with more than the median number of mutations in their tumours had shorter relapse‐free survival than patients with sub‐median mutation counts (Fig. 3, p = 0.056).
Figure 3.
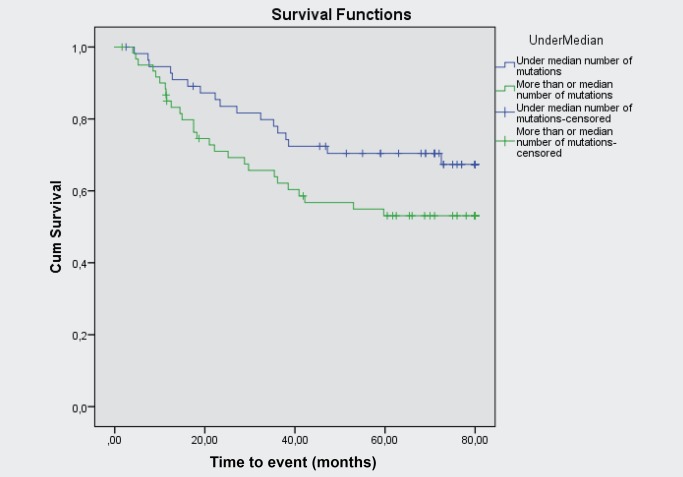
Relapse‐free survival in patients with more or less than the median number of mutations.
Multivariate analysis of relapse‐free survival was performed using Cox regression to study the influence of known prognostic factors (histology, stage, sex). The number of mutations was not significantly associated with survival in a Cox‐analysis with number of mutations as a continuous variable (p = 0.60). However, the number of mutations (over median) was found to be significantly associated with time to progression after adjusting for clinical variables (p = 0.021).
There was a tendency toward increased relapse‐free survival for patients with high CD8‐score (p = 0.10).
Discussion
By second‐generation sequencing, we have confirmed that lung cancers are highly heterogeneous in terms of somatic mutation profiles, and that the number of mutations is associated with patient survival.
An improved understanding of tumour emergence relies on the identification of genes that drive carcinogenesis. Several bioinformatics approaches have been proposed to identify driver genes, all with shortcomings and particular biases.19 Given the limited number of mutations per tumour kinome, we chose an approach (IntOGen) that emphasizes enrichment of clustered and functionally biased mutations. This algorithm has previously demonstrated its capability of detecting cancer drivers.19
When applying the IntOGen algorithm on our dataset, TP53, EGFR, KRAS, and STK11 were identified as significant driver genes, as previously described.21 Despite extensive efforts, no effective treatments targeting TP53 and KRAS have been presented up to now. Conversely, targeted treatment of mutated EGFR has changed the prospects for the subset of patients with lung EGFR‐mutation‐positive lung cancers. STK11 is a serine/threonine kinase and a tumour suppressor and has been ranked as the third most frequently mutated gene in lung cancers.22 It is implicated in cell polarity, energy metabolism, apoptosis, cell cycle arrest and proliferation. The mutations observed were in the protein kinase domain, but are probably inactivating as STK11 is a tumour suppressor. Other known genes involved in lung carcinogenesis, like ALK and ROS1, were also mutated in a small subset of the tumours. However, these genes are usually changed by inversions and translocations, not detected by our sequencing analysis.
T‐cells play pivotal effector‐like roles in the human immune system. They hamper tumour development, but unfortunately tumours can prevent sustained T‐cell responses via immune checkpoints. The immune checkpoint‐inhibitors can restore T‐cell involvement and lead to prolonged treatment responses. Unfortunately, good predictive biomarkers for response to checkpoint inhibitors are hitherto lacking.23
There have been several reports indicating that the effect of induced immune‐response correlates with the number of mutations, with the novel immunotherapeutic check‐point inhibitors having better efficacy on tumours with a high number of mutations.8 EGFR‐mutated tumours generally have lower number of mutations.24 In our study, we found significantly more infiltrating stromal CD8+ T‐cells in tumours without EGFR mutations. This would be in accordance with a higher number of immunogenic antigens in tumours with a high number of mutations. In addition, this is in accordance with the lack of CD8+ lymphocyte response in EGFR‐mutant mice models and human tumours.25
Mutations in DNA repair genes would most often lead to repair defects, which would in turn allow certain types of mutations to accumulate. In our material, we find a significant association between number of mutations and mutated DNA repair genes. Whether the significantly higher number of mutations is a consequence of dysfunctional DNA repair genes in these cases has not been determined. Clearly, our study is limited in size, so ruling out which genes in the DNA‐repair pathway are the most important should be studied more extensively, perhaps revealing a correlation of mutation signature to specific defects. A total of only 28 samples were mutated in the DNA repair genes.
Despite the modest number of samples in our cohort, we report a correlation between number of mutations and course of disease. In the present era of evolving immune therapies, we believe this to be an important insight, which needs more attention in larger cohorts, and possibly with a further potential in the clinical setting.
The authors have no conflict of interest.
Footnotes
References
- 1. Brustugun OT, Moller B, Helland A. Years of life lost as a measure of cancer burden on a national level. Br J Cancer 2014;111:1014–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin‐paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57. [DOI] [PubMed] [Google Scholar]
- 3. Pleasance ED, Cheetham RK, Stephens PJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010;463:191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pon JR, Marra MA. Driver and passenger mutations in cancer. Annu Rev Pathol 2015;10:25–50. [DOI] [PubMed] [Google Scholar]
- 5. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Galon J, Mlecnik B, Bindea G, et al. Towards the introduction of the 'Immunoscore' in the classification of malignant tumours. J Pathol 2014;232:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Donnem T, Hald SM, Paulsen EE, et al. Stromal CD8+ T‐cell density‐a promising supplement to TNM staging in non‐small cell lung cancer. Clin Cancer Res 2015;21:2635–43. [DOI] [PubMed] [Google Scholar]
- 8. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li H, Durbin R. Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saunders CT, Wong WS, Swamy S, et al. Strelka: accurate somatic small‐variant calling from sequenced tumor‐normal sample pairs. Bioinformatics 2012;28:1811–7. [DOI] [PubMed] [Google Scholar]
- 12. Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Forbes SA, Bindal N, Bamford S, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011;39:D945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Finn RD, Bateman A, Clements J, et al. Pfam: the protein families database. Nucleic Acids Res 2014;42:D222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Griffith M, Griffith OL, Coffman AC, et al. DGIdb: mining the druggable genome. Nat Methods 2013;10:1209–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non‐synonymous SNVs and their functional predictions and annotations. Hum Mutat 2013;34:E2393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Apweiler R, Bairoch A, Wu CH, et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res 2004;32:D115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gonzalez‐Perez A, Perez‐Llamas C, Deu‐Pons J, et al. IntOGen‐mutations identifies cancer drivers across tumor types. Nat Methods 2013;10:1081–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alioto TS, Buchhalter I, Derdak S, et al. A comprehensive assessment of somatic mutation detection in cancer using whole‐genome sequencing. Nat Commun 2015;6:10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014;505:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008;455:1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Borghaei H, Paz‐Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N Engl J Med 2015;373:1627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012;150:1107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Busch SE, Hanke ML, Kargl J, et al. Lung cancer subtypes generate unique immune responses. J Immunol 2016;197:4493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]