

Abstract
Background
Improving the salt tolerance of direct-seeding rice at the seed germination stage is a major breeding goal in many Asian rice-growing countries, where seedlings must often establish in soils with a high salt content. Thus, it is important to understand the genetic mechanisms of salt tolerance in rice and to screen for germplasm with salt tolerance at the seed germination stage. Here, we investigated seven seed germination-related traits under control and salt-stress conditions and conducted a genome-wide association study based on the re-sequencing of 478 diverse rice accessions.
Results
The analysis used a mixed linear model and was based on 6,361,920 single nucleotide polymorphisms in 478 rice accessions grouped into whole, indica, and non-indica panels. Eleven loci containing 22 significant salt tolerance-associated single nucleotide polymorphisms were identified based on the stress-susceptibility indices (SSIs) of vigor index (VI) and mean germination time (MGT). From the SSI of VI, six major loci were identified, explaining 20.2% of the phenotypic variation. From the SSI of MGT, five major loci were detected, explaining 26.4% of the phenotypic variation. Of these, seven loci on chromosomes 1, 5, 6, 11, and 12 were close to six previously identified quantitative gene loci/genes related to tolerance to salinity or other abiotic stresses. The strongest association region for the SSI of MGT was identified in a ~ 13.3 kb interval (15450039–15,463,330) on chromosome 1, near salt-tolerance quantitative trait loci controlling the Na+: K+ ratio, total Na+ uptake, and total K+ concentration. The strongest association region for the SSI of VI was detected in a ~ 164.2 kb interval (526662–690,854) on chromosome 2 harboring two nitrate transporter family genes (OsNRT2.1 and OsNRT2.2), which affect gene expression under salt stress. The haplotype analysis indicated that OsNRT2.2 was associated with subpopulation differentiation and its minor/rare tolerant haplotype was detected.
Conclusions
These results provide valuable information for salt tolerance-related gene cloning and for understanding the genetic mechanisms of salt tolerance at the seed germination stage. This information will be useful to improve the salt tolerance of direct-seeding rice varieties by genomic selection or marker-assisted selection.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-017-1044-0) contains supplementary material, which is available to authorized users.
Keywords: Rice, Salt tolerance, Germination, Genome-wide association study
Background
Rice (Oryza sativa L.), as the most important cereal crop, is a staple food for more than half the world’s population, especially in developing countries. Because labor costs and water crises have increased in recent years, there is a growing demand for low-input direct-seeded rice in many growing regions [1]. Soil salinization is a serious problem worldwide and a key abiotic stress in agriculture [2]. Salinity affects rice growth during all developmental stages from seed germination to reproduction [3]. With the development and spread of direct-seeding technology, which requires high levels of seedling establishment in rice paddy fields that are salinized to some degree, salt tolerance at the seed germination stage has become a major rice breeding goal in many Asian countries. Therefore, it is necessary to understand the genetic mechanisms of salt tolerance at the seed germination stage in rice.
Salt tolerance is a quantitative genetic characteristic that is controlled by multiple genes in rice [4]. In recent decades, with the rapid development of molecular marker technology, a number of quantitative trait loci (QTLs) for salt tolerance in rice have been identified using bi-parental linkage mapping of plants at different developmental growth stages, such as at the seedling and mature stages [5]. More than 70 QTLs controlling salt-related traits including the Na+/K+ ratio and survival time have been reported for rice [5]. Salt tolerance at the germination stage is not significantly correlated with salt tolerance at other stages [4]. To date, few studies have focused on evaluating the salt tolerance of rice at the germination stage to identify QTLs related to salt tolerance at this stage [6, 7]. A previous study detected only 16 QTLs associated with seed germination traits under salt stress and control conditions in a recombinant inbred rice population [7].
Linkage analysis of QTLs uses bi-parental genetic mapping populations. To date, most studies on QTLs associated with salt tolerance have examined individual mapping populations [5, 8]. However, this method is insufficient to reveal the genetic variation in salt tolerance among the rice germplasm. Over the past several years, genome-wide association studies (GWASs) using high-density genome-wide single nucleotide polymorphisms (SNPs) detected by next-generation sequencing have provided a powerful strategy to detect variants that can be used directly to improve rice varieties [9–12]. Recently, the 3000 rice genomes project (3 K RGP) [13] used Illumina next generation sequencing of a core collection of 3024 rice accessions from 89 countries to generate sequence data with high coverage (~94%) and mapping rate (~92.5%) and to construct a high-density SNP database [14] providing genotype data for GWAS of agronomic traits in rice. Several GWASs of salt tolerance in rice at the early tillering stage [15, 16] and reproductive stage [17] have been reported. However, no studies have evaluated the diverse rice germplasm to identify potentially novel loci for salt tolerance at the seed germination stage.
In this study, therefore, 478 rice accessions with an appropriate growth period and without distinct unfavorable traits in southern China selected from 3024 rice genomes sequenced by 3 K RGP [13], were used to conduct an association analysis of salt tolerance at the germination stage. For this analysis, we used 6,361,920 SNPs filtered from the 3 K RG 6.5mio SNP dataset in the Rice SNP-Seek Database [14]. The goal of this study was to understand the genetic basis and differentiation of salt tolerance at the germination stage in rice. This information will be useful for marker-assisted selection of direct-seeding rice varieties suitable for cultivation in salinized paddies.
Results
Seed germination of rice accessions under salt stress
The germination index (GI), vigor index (VI), germination rate (GR), mean germination time (MGT), and imbibition rate (IR) were determined for 35 rice accessions randomly selected from the whole panel under three salt treatments (60, 80 and 100 mM NaCl) (Additional file 1: Table S1). The GI, VI, and GR at 5 d (GR-5d) and GR at 10 d (GR-10d) significantly decreased as the NaCl concentration increased, and MGT was significantly higher in the 100 mM NaCl treatment than in the 60 and 80 mM NaCl treatments. These findings suggested that the seed germination capability was positively correlated with GI, VI, GR-5d, and GR-10d, and negatively correlated with MGT. The IR values at 24 h (IR-24 h) and 48 h (IR-48 h) were not significantly different among the three NaCl treatments. The seeds of 23 accessions germinated in the 60 mM NaCl treatment, but the seeds of only 11 and 3 accessions germinated in the 80 and 100 mM NaCl treatments, respectively (Additional file 2: Table S2). Thus, the 60 mM NaCl treatment was used in further experiments to compare germination traits among the 478 accessions (Additional file 2: Table S2).
The distributions of the values of seed germination-related traits under stress and control conditions, and the stress susceptibility index (SSI) to salt across the whole panel, are shown in Additional file 3: Figure S1. The SSI has been used to define the varieties or genotypes that show superior performance under stress and non-stress conditions in wheat [18] and rice [17, 19]. Lower SSI values indicate higher tolerance to stress. All the traits showed a continuous distribution, and many accessions were susceptible to salt stress. The traits could be roughly classified into two categories; one with a symmetrical distribution and one with a skewed distribution. Three traits (IR-24 h, IR-48 h, and GR-10d) under salt stress, one trait (VI) in the control, and one SSI trait (GR-10d) showed approximately symmetrical distributions. The other 16 traits (GI, VI, MGT, and GR-5d under salt stress; GI, MGT, IR-24 h, IR-48 h, GR-5d, and GR-10d in the control; and the SSIs of GI, VI, MGT, IR-24 h, IR-48 h, and GR-5d) showed highly or moderately skewed distributions (Additional file 3: Figure S1). Compared with their respective values in the control, the average values of GI, VI, GR-5d, and GR-10d under salt stress were decreased by 58.5%, 75.7%, 57.0% and 54.8%, respectively, while the MGT was increased by 14.6% across the whole population. However, the IR values at 24 h and 48 h under salt-stress conditions were roughly the same as that in the control.
The results of a correlation analysis among the seven seed germination traits across the whole panel are shown in Table 1. There were significant positive correlations between VI and GR-related traits (such as GI, GR-5d, and GR-10d) (Table 1), and the correlations were stronger (higher correlation coefficient values) under salt-stress conditions. There were significant negative correlations between MGT and the other traits in the control, and the correlation coefficient values were lower under salt-stress conditions. There was a strong correlation between IR-24 h and IR-48 h, but not between IR (IR-24 h and IR-48 h) and the other germination-related traits in both the control and the salt treatment. The correlation analysis indicated that all traits, except for IR, exhibited a salt-stress response in this study. Uncovering novel loci for these traits at the seed germination stage has great significance in rice breeding for salt tolerance.
Table 1.
Correlation coefficients of paired traits evaluated under control and salt-stress conditions
| GI | VI | MGT | IR-24 h | IR-48 h | GR-5d | GR-10d | |
|---|---|---|---|---|---|---|---|
| GI | 0.198 | 0.971 | −0.538 | −0.009 | 0.068 | 0.980 | 0.966 |
| VI | 0.823 | 0.218 | −0.494 | −0.002 | 0.088 | 0.959 | 0.948 |
| MGT | -0.929 | −0.756 | 0.259 | −0.079 | −0.118 | −0.537 | -0.402 |
| IR-24 h | 0.083 | 0.053 | −0.123 | 0.836 | 0.870 | 0.006 | -0.013 |
| IR-48 h | 0.185 | 0.160 | −0.213 | 0.868 | 0.827 | 0.070 | 0.053 |
| GR-5d | 0.830 | 0.686 | −0.831 | 0.073 | 0.156 | 0.206 | 0.960 |
| GR-10d | 0.672 | 0.562 | −0.413 | −0.025 | 0.044 | 0.647 | 0.121 |
Upper and lower triangles indicate correlation coefficients under salt stress and control conditions, respectively. Diagonal values indicate correlation coefficients between control and salt stress conditions. All correlation coefficients are significant at p < 0.01 except for values in italics; values in bold indicate strong correlation (r > 0.8). GI, germination index; VI, vigor index; MGT, Mean germination time; IR, imbibition rate; GR, germination rate
There was highly significant genetic variation in all seed germination traits among rice germplasm accessions nested within the rice subgroups, together with less significant variance in GI, IR-24 h, IR-48 h, and GR-48d within subgroups (indica, japonica, aus and other) under salt stress (60 mM NaCl). There was no significant variance between replications for all seed germination traits (Table 2). The box plots of SSI indicated a substantial level of genotypic variation in salt-stress responses among different accessions (Additional file 4: Figure S2). An ANOVA demonstrated that the SSI of VI and GR at 10 d were significantly lower in the subgroup japonica than in the subgroups indica and aus, and the SSI of MGT was significantly lower in the aus subgroup than in the indica and japonica subgroups (Fig. 1). The average VI values for the japonica, indica, and aus subgroups were 6.48, 7.48, and 5.71, respectively, in the control, and 2.04, 1.68 and 1.35, respectively, under salt stress. This result suggested that the accessions in the japonica subgroup had higher salt tolerance than did the accessions in the other subgroups.
Table 2.
ANOVA (F-values) of seven seed germination-related traits in 478 rice accessions under salt stress (60 mM NaCl)
| Sources | Df | Germination index | Vigor index | Mean germination time | Imbibition rate | Germination rate | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 24 h | 48 h | 5d | 10d | ||||||||||||
| Rep | 1 | 0.6 | ns | 0.1 | ns | 2.6 | ns | 0.0 | b | 0.1 | ns | 0.7 | ns | 1.9 | ns |
| Subgroup | 3 | 3.3 | a | 2.2 | ns | 1.5 | ns | 4.0 | b | 6.0 | b | 2.6 | ns | 3.3 | a |
| Accession(Subgroup) | 474 | 436.4 | b | 485.7 | b | 385.2 | b | 392.0 | b | 390.9 | b | 707.5 | b | 571.3 | b |
a and b indicate significance at 0.05 and 0.01 levels, respectively. Ns, not significant
Fig. 1.
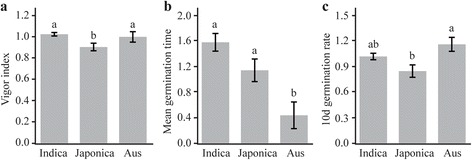
Comparison of stress susceptibility indices of seed germination traits with significant differences among accessions in different subgroups. Vigor index (a). Mean germination time (b). 10d germination rate (c). Figure shows average values ± SE. Multiple comparison tests were based on Duncan’s test at P < 0.01. Different letters above bars indicate significant differences among subgroups (indica, japonica, and aus)
Loci associated with salt tolerance at the germination stage identified by GWAS
We randomly selected 20% of SNPs with minor allele frequency (MAF) >10% and missing rate < 20% for the linkage disequilibrium (LD) decay analysis of each panel. The LD decayed quickly within ~8 kb for all panels, and decayed to its half-maximum within 95 kb for the whole panel, 52 kb for indica, and 230 kb for non-indica (Additional file 5: Figure S3). We selected 6,361,920 SNPs across the entire rice genome for the GWAS of seed germination traits under salt-stress and control conditions, and the related SSI. The LD blocks in all GWAS panels were defined by a fixed cutoff of <0.2 for the LD statistic r 2 on the same chromosome. Multiple significant SNPs in a LD block were clustered as one association locus in each panel, and the SNP with the minimum P value in a cluster was considered as the lead SNP. After Bonferroni multiple test correction, 11 loci (22 SNPs) were identified as being associated with the SSIs of the two seed germination traits (VI and MGT) at a significance level of 0.05 (Additional file 6: Table S3) in the whole, indica, and non-indica panels (Table 3 and Additional file 7: Table S4). The quantile–quantile plots for the GWAS results shown in Fig. 2 indicate that the model was well fitted to the data. Among the 11 loci, 7 were located in intervals containing previously mapped QTLs or genes associated with tolerance to salinity or other abiotic stresses (Table 3). Additionally, for GI, VI, MGT, GR-5d, and GR-10d in the control, 41, 136, and 7 significant SNPs were detected in the whole, indica, and non-indica panels, respectively. This result indicated that there is wide genetic variation in seed germination-related traits in the rice germplasm (Additional file 7: Table S4). Here, we focused on the significant associations of the SSIs of VI and MGT, the two parameters that underlie salt tolerance at the germination stage in rice.
Table 3.
Summary of loci significantly associated with stress susceptibility indices of seed germination-related traits
| Traita | Chr.b | Number of significant signalsc | Lead SNP position (bp)d | P value | Gene harbored lead SNP | Panele | Allelef | Mean SSI | Known QTL/Trait | |
|---|---|---|---|---|---|---|---|---|---|---|
| Major | Minor | |||||||||
| MGT | 1 | 3 | 15,450,039 | 6.22E-10 | LOC_Os01g27710-LOC_Os01g27720 | Whole | G/A | 1.09 | 2.92 | RM1-R886 [31]/Na+ uptake; K+ concentration; Na+: K+ ratio |
| MGT | 1 | 1 | 17,736,489 | 9.93E-09 | LOC_Os01g32330 | Whole | C/T | 1.23 | 2.57 | |
| VI | 2 | 7 | 666,962 | 1.15E-09 | LOC_Os02g02190 | Whole, non-Indica | A/G | 0.98 | 0.31 | |
| MGT | 4 | 1 | 16,501,431 | 2.32E-08 | LOC_Os04g27950-LOC_Os04g27960 | Indica | C/T | 1.57 | 1.47 | |
| MGT | 5 | 2 | 29,704,394 | 5.10E-09 | LOC_Os05g51770 | Whole | T/C | 1.35 | 1.32 | qRGR-5 [43] /relative germination rate with ABA; qGR5–1 [44] /Germination rate |
| VI | 6 | 1 | 23,605,165 | 2.93E-09 | LOC_Os06g39756-LOC_Os06g39750 | non-Indica | G/A | 1.08 | 0.27 | RG716 [45]/Salinity tolerance |
| VI | 9 | 2 | 2,214,950 | 9.71E-10 | LOC_Os09g04210 | non-Indica | A/T | 0.94 | 0.52 | |
| VI | 10 | 1 | 10,723,028 | 3.54E-10 | LOC_Os10g21100-LOC_Os10g21110 | Whole | G/A | 1.02 | 0.36 | |
| VI | 11 | 1 | 27,305,217 | 4.64E-09 | LOC_Os11g45110-LOC_Os11g45120 | non-Indica | T/C | 1.01 | 0.37 | RM206 [46]/Relative shoot elongation under submergence |
| MGT | 12 | 2 | 5,000,802 | 2.88E-08 | LOC_Os12g09510-LOC_Os12g09520 | Whole | A/C | 1.16 | 3.93 | qCTS12 [47]/Seedling cold tolerance; rv12–2 [48]/Root volume at seedling stage |
| VI | 12 | 1 | 14,215,505 | 2.41E-08 | LOC_Os12g24780 | Indica | C/T | 1.06 | 0.92 | qtl12.1 [49] /Drought resistance |
a Traits for which significant signals were associated with stress susceptibility index of that trait. MGT, mean germination time; VI, vigor index
b Chromosome number
c Number of significant association signals detected in region
d Representative SNP position on rice genome assembly MSU version 7.0
e Panel containing significant GWAS sites
f Major allele/minor allele
Fig. 2.
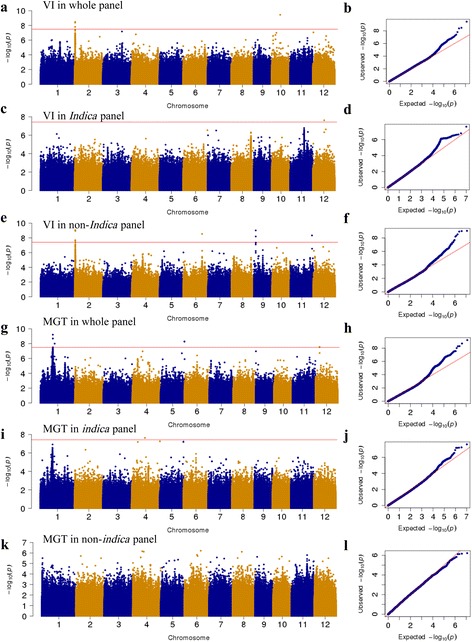
Genome-wide association mapping of rice salt tolerance at the germination stage. Manhattan and quantile–quantile plots for stress susceptibility indices (SSI) of vigor index in the whole (a, b), indica (c, d) and non-indica panels (e, f). Manhattan and quantile–quantile plots for SSIs of mean germination time in whole (g, h), indica (i, j) and non-indica panels (k, l). Red horizontal line indicates genome-wide significance threshold
For the SSI of MGT, five significant associated loci were identified on chromosomes 1, 4, 5, and 12 in the whole, indica, and non-indica panels. These loci explained 26.4% of the phenotypic variation in the whole panel. Four loci on chromosomes 1, 5, and 12 were close to QTLs/genes identified previously as being related to tolerance to salinity or other abiotic stresses (Table 3 and Fig. 2). The genome-wide peak locus, which contained the lead SNP chr1_15,450,039 (P = 6.2 × 10−10) in the whole panel, was identified in a ~ 13.3 kb interval (15450039–15,463,330) on chromosome 1. This locus included two associated SNPs located in the intergenic region of LOC_Os01g27710 (encoding an expressed protein) and LOC_Os01g27720 (encoding an E3 ubiquitin-protein ligase, MGRN1) and one associated SNP in the coding sequence-missense variant of LOC_Os01g27730 (encoding a GTPase with an unknown functional domain-containing protein) (Fig. 2 and Additional file 7: Table S4). The MGT of the minor genotype at the peak SNP chr1_15,450,039, AA (2.92 ± 2.87), was significantly greater than that of the major genotype, GG (1.09 ± 1.84). One significant locus on chromosome 4 containing SNP chr4_16,501,431 (P = 6.2 × 10−8) was detected only in the indica panel and was located in the intergenic region of LOC_Os04g27950 (encoding an expressed protein) and LOC_Os04g27960 (encoding a B3 DNA-binding domain-containing protein) (Additional file 7: Table S4).
For the SSI of VI, six significantly associated loci were identified on chromosomes 2, 6, 9, 10, 11, and 12 in the whole, indica, and non-indica panels. These loci explained 20.2% of the phenotypic variation in the whole panel. Three loci on chromosomes 6, 11, and 12 were close to previously identified QTLs related to tolerance to salinity or other abiotic stresses (Table 3 and Fig. 2). The strongest association in the hot region was observed on chromosome 2 in a ~ 164.2 kb interval (526662–690,854) that included three and four significant SNPs in the whole and non-indica panels, respectively (Additional file 7: Table S4). In this region, the lead SNP in the GWAS, chr2_666,962 (P = 1.2 × 10−9), was identified in the intergenic region of LOC_Os02g02180 (retrotransposon protein) and LOC_Os02g02190 (OsNRT2.2) (Fig. 3 and Additional file 7: Table S4). The SSI for the VI of the major susceptible genotype at the lead SNP (chr2_679199), AA (0.98 ± 0.32), was significantly greater than that of the minor resistant genotype, GG (0.31 ± 0.48), in the non-indica population. The haplotypes of LOC_Os02g02190 (OsNRT2.2) were built based on 30 SNPs in the 2-kb upstream promoter region and 5′ untranslated region and synonymous SNPs in the coding region (Fig. 3 and Additional file 8: Table S5). In total, we detected nine haplotypes (Hap1–5 mainly in the indica panel, Hap6 mainly in the japonica panel, and Hap7–9 mainly in the aus panel) shared by more than 5 of the 372 accessions. Multiple comparison tests showed that accessions with Hap7 had lower SSIs of VI than did accessions with other haplotypes (Fig. 3). The 16 accessions with the tolerant haplotype (Hap7) of OsNRT2.2 (LOC_Os02g02190) are mainly from Bangladesh and India and belong to the aus, basmati, indica and intermediate subgroups, but not the japonica subgroup. However, we did not find a significant difference in the SSIs of VIs among the accessions with different haplotypes of LOC_Os02g02170 (OsNRT2.1), which also belongs to the NRT2 family and is close to OsNRT2.2.
Fig. 3.
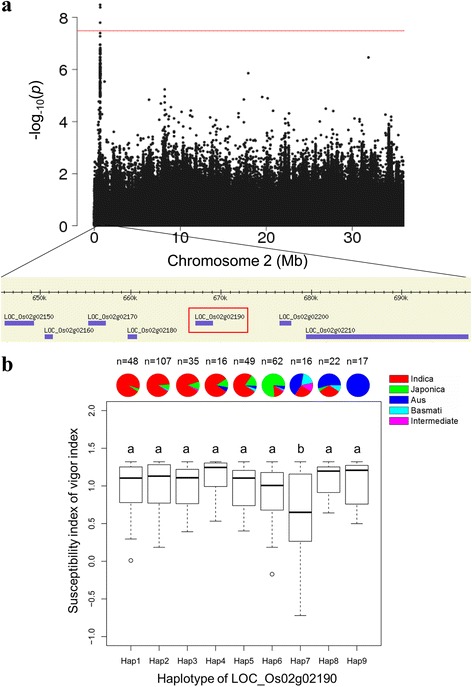
Strong association on rice chromosome 2 between stress susceptibility index and seed vigor index (a), and haplotype analysis of candidate gene LOC_Os02g02190 (b)
Another significant peak region with the lead SNP chr9_2,214,950 (P = 9.7 × 10−10) in the non-indica panel was located in the intron region of LOC_Os09g04210, which encodes a zinc ion-binding protein (Table 3). The SSI of the VI of the major genotype at the lead SNP (chr9_2,214,950), AA (0.94 ± 0.38), was significantly greater than that of the major genotype, TT (0.52 ± 0.49).
Discussion
Variations in the salt tolerance of rice at the germination stage
Accurate phenotyping is the most important part of GWASs on rice [10]. Here, we measured seven seed germination-related traits (GI, VI, MGT, IR-24 h, IR-48 h, GR-5d, and GR-10d) under salt stress and control conditions. These traits have been used successfully in previous studies for the QTL mapping of salt tolerance at the rice germination stage [6, 7]. The intensity of the salt treatment is an important factor in the effective evaluation of salt tolerance during rice germination. We compared seven seed germination-related traits for 35 randomly selected rice accessions under three different NaCl concentrations (60, 80, and 100 mM NaCl) (Additional file 1: Table S1) and found that 60 mM NaCl was the appropriate treatment to detect sufficient levels of genetic variation among the 478 rice accessions used in this study. These accessions showed continuous segregation of seed germination-related trait distribution frequencies (Additional file 3: Figure S1).
Mondal and Pramanik evaluated the response of 35 rice varieties to salt stress during seed germination, and found that japonica varieties had higher tolerance than indica varieties to high-salt conditions [20]. Our results indicated that the SSIs of VI and GR at 10d were significantly lower in the japonica subgroup than in the indica and aus subgroups (Fig. 1), indicating that japonica varieties had higher tolerance to salt stress than did the other subgroups at the germination stage. Different genetic and physiological mechanisms of salt tolerance exist in the diverse germplasm of rice, and salt tolerance mechanisms can vary among varieties even within a particular subgroup [21, 22]. It is generally thought that higher uptake of K+ in indica varieties results their higher tolerance to salinity than japonica rice at the seedling stage [23, 24]. However, the mechanism underlying salt tolerance at the seed germination stage in rice is still obscure. Because seed germination is a complex trait [25], and, like salt tolerance, governed by multiple genes [5], it is difficult to improve seed germination under salt stress. Thus, to improve salt tolerance at the germination stage in rice, breeders should pyramid the tolerant SNP genotype at the loci associated with salt tolerance using the potentially tolerant varieties identified in this study. For successful, knowledge-based crop improvement, further studies should focus on understanding the correlation between salt tolerance at the seed germination stage and that at the seedling stage, and on elucidating the molecular mechanisms underlying salt tolerance at different growth stages in rice.
QTLs and candidate genes associated with salt tolerance of rice at the germination stage
Linkage mapping using a bi-parental population benefits from high statistical power resulting from many individuals sharing the identical genotype at a given locus, but it has low resolution because of the limited number of recombination events. In contrast, association mapping has higher resolution because of the long recombination histories of natural populations, but has lower statistical power because most genotypes occur in only a few individuals [26]. The distance of LD is an important factor in determining the efficiency of GWAS [27]. In this study, more rapid LD decay was observed in the indica subgroup than in the non-indica subgroup comprising mainly japonica and aus (Additional file 5: Figure S3). This is consistent with the LD patterns reported for different subpopulations in rice [28, 29], suggesting that there has been a stronger bottleneck in japonica than in indica during domestication. The extent of LD decay detected in indica in this study (~50 kb) was smaller than that detected in another set of indica germplasm in another study (~100 kb) [29], partly because of the larger number of SNPs, which provides high resolution for GWAS.
To date, more than 70 QTLs related to salt tolerance in rice have been mapped using bi-parental linkage mapping populations [5], but this is the first study to use association mapping to detect potentially novel QTLs for salt tolerance at the seed germination stage. We identified 11 loci associated with salt tolerance at the germination stage in rice using a conservative threshold based on a Bonferroni-adjusted P value (0.05/total markers) to reduce false positives. We compared the loci identified in this study with previously reported QTLs related to salinity or other stresses in the QTL Annotation Rice Online (Q-TARO) database [30]. This comparison showed that seven loci on chromosome 1, 5, 6, 11, and 12 were co-located with six previously detected QTLs related to tolerance to salinity or other abiotic stresses in rice (Table 3). For example, Koyama et al. [31] reported an initial area, from markers RM1 to R886 on chromosome 1, which was associated with QTLs controlling the Na+: K+ ratio, total Na+ uptake, and total K+ concentration. Two regions significantly associated with the SSI of MGT on chromosome 1 were detected in this area in this study.
Our results demonstrate that the gene OsNRT2.2 (LOC_Os02g02190), encoding a nitrate transporter, is a candidate gene in the hot region for the SSI of VI (on rice chromosome 2) in different GWAS panels. In rice, OsNAR2.1 interacts with OsNRT2.2 and plays a key role in enabling plants to cope with a variable nitrate supply [32]. Salt stress affects the expression level of the OsNRT gene family, which might contribute to the accumulation of NO3 − in the leaves of salt-stressed rice [33]. From the haplotype analysis, we found that OsNRT2.2 (LOC_Os02g02190) was associated with subpopulation differentiation (Fig. 3). The minor/rare tolerant haplotype (Hap7) of OsNRT2.2 (LOC_Os02g02190) could be useful in further studies and in breeding for salt tolerance at the rice germination stage.
Conclusions
We conducted genome-wide association mapping for salt tolerance at the germination stage based on high-density SNPs using 478 rice accessions. We identified 11 loci using the SSIs of germination-related traits, which co-located with six previously detected rice QTLs related to tolerance to salinity or other abiotic stresses. The minor/rare salt-tolerant haplotype of OsNRT2.2 (LOC_Os02g02190) was detected in a haplotype analysis. This gene is a candidate for the associated hot region on rice chromosome 2. Our study provides new insights into the genetic basis of salt tolerance in rice. Identification of varieties with high salt tolerance at the germination stage, as well as knowledge of the associated SNPs and haplotype, could be useful for rice production and for improvement of direct-seeding varieties.
Methods
Rice germplasms and evaluation of seed GRs under salt stress
We selected 478 accessions from 46 countries and areas with an appropriate growth period and without distinct unfavorable traits in southern China (Additional file 2: Table S2) in the 3 K RGP [13] to evaluate salt tolerance at the seed germination stage. In total, there were 305 indica accessions, 85 japonica accessions (9 japonica, 34 temperate japonica, and 43 tropical japonica), 65 aus accessions, 16 basmati accessions, and 7 intermediate accessions. All seeds used in your study were from the International Rice Genebank Collection at the International Rice Research Institute.
A total of 120 yellow-ripe seeds of each accession were dried at 50 °C for 3 days to break seed dormancy. The seeds were surface-sterilized with 15% sodium hypochlorite solution for 20 min and then rinsed three times with sterile distilled water before the germination experiment. Two replications for each treatment, each consisting of 30 seeds from each accession, were placed in 9-cm-diameter Petri dishes on two layers of filter paper, to which 10 mL of NaCl solution was added to simulate salt-stress conditions. In the control, the filter paper was soaked with 10 mL water. The seeds were incubated in a growth chamber at 30 °C under a 12-h light/12-h dark photoperiod with 80% relative humidity for 10 days. To determine the most suitable salt concentration, we selected 35 accessions for treatment with NaCl at three different concentrations (60, 80, and 100 mM). More genetic variation was observed in the 60 mM NaCl treatment than in the other treatments. Therefore, the seed GRs were evaluated among all the accessions under a 60 mM NaCl treatment and control (water) conditions. The solution was replaced every day to maintain the NaCl concentration and the distilled water volume.
Dry seeds, and seeds incubated for 24 and 48 h, were weighed independently to calculate seed IR (mg/g) using the method of Wang et al. [6], as follows: IR = (W 2 – W 1)/W 1 × 1000, where W 1 (g) represents the dry seed weight, and W 2 (g) represents the total seed weight after imbibition for 24 h or 48 h. The seeds that germinated were observed each day to calculate the GR and GI. We considered a seed germinated when its shoot length was greater than half of the seed length and the root length was greater than the seed length. The GR was the germination percentage at a certain day (GR = N t/N 0 × 100%, where N t represents the number of germinated seeds at day t and N 0 represents the total number of experimental seeds). The GI was calculated by the method of Wang et al. [6] as follows: GI = ∑(G t/T t), where G t is the accumulated number of germinated seeds at day t and T t is the time corresponding to G t in days. The MGT was calculated for the GR using the following formula: MGT = ∑T i N i/∑N i, where N i is the number of newly germinated seeds at day t [34]. We chose 10 germinated seeds to measure the average shoot length after 10 days, and the seed VI was calculated using the formula [6]: VI = GI × average shoot length. The responses of the genotypes to salt stress were expressed according to the SSIs calculated using the methods of Fischer and Maurer [35] as follows: SSI = (1 − Ys/Yp)/D, where Ys = mean performance of a genotype under stress; Yp = mean performance of the same genotype without stress; D (stress intensity) = 1 − (mean Ys of all of the genotypes/mean Yp of all of the genotypes).
LD decay
The 3 K RG 6.5mio SNP dataset was downloaded from the Rice SNP-Seek Database (http://www.oryzasnp.org/) [14]. We randomly selected 20% of SNPs with MAF > 10% and missing rate < 20% for the LD decay analysis of each panel (whole panel: 478 accessions with 683,037 SNP, indica panel: 305 accessions with 473,146 SNPs, and non-indica panel: 173 accessions with 720,819 SNPs). We calculated r 2 as an estimation of LD using plink software [36] with the parameter ‘--r2 --ld-window-kb 1000 --ld-window 99999 --ld-window-r2 0’, and the parameter ‘--thin 0.2’ to randomly select 20% of the SNPs.
Genome-wide association mapping
A total of 6,361,920, 4,736,461, and 6,029,780 SNPs with a minor allele frequency > 5% and accession numbers with minor alleles ≥6 were filtered for the association analyses in the whole population, indica, and non-indica panels, respectively. The GWAS was performed with a linear mixed-effects model to determine the association between each SNP and the evaluated phenotypes and SSIs using an efficient mixed-model analysis with EMMA eXpedited (EMMAX) software [37]. An identical-by-state matrix based on genome-wide SNP data was used to create the kinship matrix that measured the genetic similarities between individuals. The effective number of independent markers (N) was calculated using GEC software [38], and significant thresholds (0.05/N) were calculated. The effective number of independent SNPs was 1,546,501, 1,336,375, and 1,255,892 in the whole, indica, and non-indica panels, respectively. Based on a significance level of 0.05, the genome-wide significance thresholds were P = 3.23 × 10−8, 3.74 × 10−8, and 3.98 × 10−8 for the whole, indica, and non-indica panels, respectively. To obtain independent association signals, multiple SNPs passing the threshold on the same chromosome were clustered as one association locus by r 2 of LD ≥ 0.2 in each panel, and the SNP with the minimum P value in a cluster was considered as the lead SNP. Candidate genes near hits were extracted from the literature and the Q-TARO database [30]. The phenotypic variation explained (R 2) by the multiple lead SNPs of association loci in the whole, indica, and non-indica panels was estimated by a linear regression using SAS PROC GLM [39] after coding one allele as 1 and the other as 0. Manhattan and Q–Q plots were created by the R package ‘qqman’ [40] using the GWAS results.
Significant signal analyses and annotations
Synonymous and nonsynonymous SNPs, and SNPs with large effect changes, were annotated based on gene models of the IRGSP 1.0 ‘Nipponbare’ annotated reference genome [41] using snpEff software version 4.0 [42]. All significant SNPs located in genes and the annotation information from the IRGSP 1.0 ‘Nipponbare’ annotated reference genome [41] are listed in Additional file 8 and Table S5.
Statistical analyses
Correlation analyses were conducted between pairs of tested traits using R software. Differences in phenotypic and SSI values of accessions among haplotypes or subgroups were evaluated by one-way ANOVA and Duncan’s multiple mean comparison test at the 5% level of significance using SAS [39].
Additional files
Phenotypic evaluation of seed germination for 35 randomly selected rice accessions under different NaCl concentrations and in the control. (DOCX 15 kb)
Rice accessions used in this study and related phenotypic data. (XLSX 141 kb)
Frequency distributions of germination index, vigor index, mean germination time, imbibition rate, and germination rate for 478 rice accessions under salt-stress and control conditions, and stress susceptibility indices of these traits. Under salt stress (a) and control (b) conditions. Stress susceptibility index (c). (TIFF 1093 kb)
Box plots for stress susceptibility indices of germination-related traits in 478 rice accessions. (TIFF 1970 kb)
Differences in linkage disequilibrium among panels. (TIFF 422 kb)
Filtered and effective numbers of SNPs across subpopulations and adjusted significant P value thresholds based on Bonferroni correction. (XLSX 9 kb)
List of SNPs with significant P values detected by genome-wide association analysis in this study. (XLSX 24 kb)
Haplotype analysis of LOC_Os02g02190 using 31 SNPs in 2-kb upstream promoter region, 5′ and 3′ untranslated regions, and coding region based on IRGSP 1.0 ‘Nipponbare’ annotated reference genome. (XLSX 15 kb)
Acknowledgements
Not applicable.
Funding
This research was supported by grants from the National High-tech Program of China (No.2014AA10A603) and the CAAS Innovative Team Award, and partly supported by the Bill & Melinda Gates Foundation (No. OPP51587) and the Shenzhen Peacock Plan (No.20130415095710361).
Availability of data and materials
The SNP dataset used during this study is available in the Rice SNP-Seek Database (http://www.oryzasnp.org/). Any other datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
Authors’ contributions
Conceived and designed the experiments: YZ, ZL. Performed the experiments: YS, LG, XZ, MW, CZ. Analyzed the data: FZ, ZW. Wrote the paper: FZ, YZ. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- 3 K RGP
3000 rice genomes project
- Chr
Chromosome
- GI
Germination index
- GR
Germination rate
- GR-10d
Germination rate at 10 d
- GR-5d
Germination rate at 5 d
- GWAS
Genome-wide association study
- Hap
Haplotype
- IR
Imbibition rate
- IR-24 h
Imbibition rate at 24 h
- IR-48 h
Imbibition rate at 48 h
- LD
Linkage disequilibrium
- MAF
Minor allele frequency
- MGT
Mean germination time
- Q-TARO
QTL Annotation Rice Online
- QTL
Quantitative trait loci
- SNP
Single nucleotide polymorphism
- SSI
Stress-susceptibility index
- VI
Vigor index
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-017-1044-0) contains supplementary material, which is available to authorized users.
Contributor Information
Yingyao Shi, Email: shiyy123@163.com.
Lingling Gao, Email: 1428605765@qq.com.
Zhichao Wu, Email: yeswzc@163.com.
Xiaojing Zhang, Email: 1009364219@qq.com.
Mingming Wang, Email: 1448267443@qq.com.
Congshun Zhang, Email: 530565355@qq.com.
Fan Zhang, Email: zhangfan03@caas.cn.
Yongli Zhou, Email: zhouyongli@caas.cn.
Zhikang Li, Email: lizhikang@caas.cn.
References
- 1.Farooq M, Siddique KHM, Rehman H, Aziz T, Lee D-J, Wahid A. Rice direct seeding: experiences, challenges and opportunities. Soil Tillage Res. 2011;111(2):87–98. doi: 10.1016/j.still.2010.10.008. [DOI] [Google Scholar]
- 2.Zhu JK. Plant salt tolerance. Trends Plant Sci. 2001;6(2):66–71. doi: 10.1016/S1360-1385(00)01838-0. [DOI] [PubMed] [Google Scholar]
- 3.Zeng L, Shannon MC, Lesch SM. Timing of salinity stress affects rice growth and yield components. Agric Water Manag. 2001;48(3):191–206. doi: 10.1016/S0378-3774(00)00146-3. [DOI] [Google Scholar]
- 4.Johnson DW, Smith SE, Dobrenz AK. Genetic and phenotypic relationships in response to NaCl at different developmental stages in alfalfa. Theor Appl Genet. 1992;83(6–7):833–838. doi: 10.1007/BF00226705. [DOI] [PubMed] [Google Scholar]
- 5.Hu S, Tao H, Qian Q, Guo L. Genetics and molecular breeding for salt-tolerance in Rice. Rice Genomics Genet. 2012;3(7):39–49. [Google Scholar]
- 6.Wang Z, Wang J, Bao Y, Wu Y, Su X, Zhang H. Inheritance of rice seed germination ability under salt stress. Rice Sci. 2010;17(2):105–110. doi: 10.1016/S1672-6308(08)60112-2. [DOI] [Google Scholar]
- 7.Wang Z, Wang J, Bao Y, Wu Y, Zhang H. Quantitative trait loci controlling rice seed germination under salt stress. Euphytica. 2011;178:297–307. doi: 10.1007/s10681-010-0287-8. [DOI] [Google Scholar]
- 8.Zang J, Sun Y, Wang Y, Yang J, Li F, Zhou Y, et al. Dissection of genetic overlap of salt tolerance QTLs at the seedling and tillering stages using backcross introgression lines in rice. Sci China Ser C Life Sci. 2008;51(7):583–591. doi: 10.1007/s11427-008-0081-1. [DOI] [PubMed] [Google Scholar]
- 9.Huang X, Zhao Y, Wei X, Li C, Wang A, Zhao Q, et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat Genet. 2012;44(1):32–39. doi: 10.1038/ng.1018. [DOI] [PubMed] [Google Scholar]
- 10.Han B, Huang X. Sequencing-based genome-wide association study in rice. Curr Opin Plant Biol. 2013;16(2):133–138. doi: 10.1016/j.pbi.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 11.Chen W, Gao Y, Xie W, Gong L, Lu K, Wang W, et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat Genet. 2014;46(7):714–721. doi: 10.1038/ng.3007. [DOI] [PubMed] [Google Scholar]
- 12.Yang W, Guo Z, Huang C, Duan L, Chen G, Jiang N, et al. Combining high-throughput phenotyping and genome-wide association studies to reveal natural genetic variation in rice. Nat Commun. 2014;5:5087. doi: 10.1038/ncomms6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.3K RGP The 3,000 rice genomes project. Gigascience. 2014;3:7. doi: 10.1186/2047-217X-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexandrov N, Tai S, Wang W, Mansueto L, Palis K, Fuentes RR, et al. SNP-Seek database of SNPs derived from 3000 rice genomes. Nucleic Acids Res. 2015;43(Database issue):D1023–D1027. doi: 10.1093/nar/gku1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell MT, Knecht AC, Berger B, Brien CJ, Wang D, Walia H. Integrating image-based phenomics and association analysis to dissect the genetic architecture of temporal salinity responses in rice. Plant Physiol. 2015. doi:10.1104/pp.15.00450. [DOI] [PMC free article] [PubMed]
- 16.Al-Tamimi N, Brien C, Oakey H, Berger B, Saade S, Ho YS, et al. Salinity tolerance loci revealed in rice using high-throughput non-invasive phenotyping. Nat Commun. 2016;7:13342. doi: 10.1038/ncomms13342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar V, Singh A, Mithra SV, Krishnamurthy SL, Parida SK, Jain S, et al. Genome-wide association mapping of salinity tolerance in rice (Oryza sativa) DNA Res. 2015;22(2):133–145. doi: 10.1093/dnares/dsu046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thiry AA, Chavez Dulanto PN, Reynolds MP, Davies WJ. How can we improve crop genotypes to increase stress resilience and productivity in a future climate? A new crop screening method based on productivity and resistance to abiotic stress. Journal of Experimental Botany. 2016;67(19):5593–603. doi:10.1093/jxb/erw330. [DOI] [PMC free article] [PubMed]
- 19.Tiwari S, Sl K, Kumar V, Singh B, Rao AR, Mithra Sv A, et al. Mapping QTLs for salt tolerance in Rice (Oryza sativa L.) by bulked Segregant analysis of recombinant inbred lines using 50K SNP Chip. PLoS One. 2016;11(4):e0153610. doi: 10.1371/journal.pone.0153610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mondal AB, Pramanik SC. In vitro studies of salt tolerance of three rice races in Bay Islands. J Indian Soc Coastal Agric Res. 1995;13(2):127–131. [Google Scholar]
- 21.Hossain MR, Pritchard J, Ford-Lloyd BV. Qualitative and quantitative variation in the mechanisms of salinity tolerance determined by multivariate assessment of diverse rice (Oryza sativa L.) genotypes. Plant Genet Resour. 2015;14(02):91–100. doi: 10.1017/S1479262115000118. [DOI] [Google Scholar]
- 22.Cheng L, Wang Y, Meng L, Hu X, Cui Y, Sun Y, et al. Identification of salt-tolerant QTLs with strong genetic background effect using two sets of reciprocal introgression lines in rice. Genome. 2012;55(1):45–55. doi: 10.1139/g11-075. [DOI] [PubMed] [Google Scholar]
- 23.Lee KS, Choi WY, Ko JC, Kim TS, Gregorio GB. Salinity tolerance of japonica and indica rice (Oryza sativa L.) at the seedling stage. Planta. 2003;216(6):1043–1046. doi: 10.1007/s00425-002-0958-3. [DOI] [PubMed] [Google Scholar]
- 24.De Leon TB, Linscombe S, Gregorio G, Subudhi PK. Genetic variation in southern USA rice genotypes for seedling salinity tolerance. Front Plant Sci. 2015;6:374. doi: 10.3389/fpls.2015.00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He D, Yang P. Proteomics of rice seed germination. Front Plant Sci. 2013;4:246. doi: 10.3389/fpls.2013.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang N, Gibon Y, Wallace JG, Lepak N, Li P, Dedow L, et al. Genome-wide association of carbon and nitrogen metabolism in the maize nested association mapping population. Plant Physiol. 2015;168(2):575–583. doi: 10.1104/pp.15.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipka AE, Kandianis CB, Hudson ME, Yu J, Drnevich J, Bradbury PJ, et al. From association to prediction: statistical methods for the dissection and selection of complex traits in plants. Curr Opin Plant Biol. 2015;24:110–118. doi: 10.1016/j.pbi.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 28.Xu X, Liu X, Ge S, Jensen JD, Hu F, Li X, et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotechnol. 2011;30(1):105–111. doi: 10.1038/nbt.2050. [DOI] [PubMed] [Google Scholar]
- 29.Zhao K, Tung CW, Eizenga GC, Wright MH, Ali ML, Price AH, et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun. 2011;2:467. doi: 10.1038/ncomms1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamamoto E, Yonemaru J, Yamamoto T, Yano M. OGRO: the overview of functionally characterized genes in Rice online database. Rice (N Y) 2012;5(1):26. doi: 10.1186/1939-8433-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koyama ML, Levesley A, Koebner RM, Flowers TJ, Yeo AR. Quantitative trait loci for component physiological traits determining salt tolerance in rice. Plant Physiol. 2001;125(1):406–422. doi: 10.1104/pp.125.1.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan M, Fan X, Feng H, Miller AJ, Shen Q, Xu G. Rice OsNAR2.1 interacts with OsNRT2.1, OsNRT2.2 and OsNRT2.3a nitrate transporters to provide uptake over high and low concentration ranges. Plant Cell Environ. 2011;34(8):1360–1372. doi: 10.1111/j.1365-3040.2011.02335.x. [DOI] [PubMed] [Google Scholar]
- 33.Wang H, Zhang M, Guo R, Shi D, Liu B, Lin X, et al. Effects of salt stress on ion balance and nitrogen metabolism of old and young leaves in rice (Oryza sativa L.) BMC Plant Biol. 2012;12:194. doi: 10.1186/1471-2229-12-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alvarado AD, Bradford KJ, Hewitt JD. Osmotic priming of tomato seed: effect on germination, field emergence, seedling growth and fruit yield. J Am Soc Hortic Sci. 1987;112:427–432. [Google Scholar]
- 35.Fischer RA, Maurer R. Drought resistance in spring wheat cultivars. I. Grain yield responses. Aust J Agric Res. 1978;29(5):897–912. doi: 10.1071/AR9780897. [DOI] [Google Scholar]
- 36.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang HM, Sul JH, Service SK. Zaitlen NA, Kong SY, Freimer NB, et al. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42(4):348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li MX, Yeung JM, Cherny SS, Sham PC. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet. 2012;131(5):747–756. doi: 10.1007/s00439-011-1118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.SAS Institute Inc . SAS/STAT 9.1 User’ s guide. Cary: SAS Institute Inc; 2004. [Google Scholar]
- 40.Turner SD. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. bioRxiv. 2014. doi:10.1101/005165.
- 41.Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice (N Y) 2013;6(1):4. doi: 10.1186/1939-8433-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.You J, Li Q, Yue B, Xue WY, Luo LJ, Xiong LZ. Identification of quantitative trait loci for ABA sensitivity at seed germination and seedling stages in rice. Yi Chuan Xue Bao. 2006;33(6):532–541. doi: 10.1016/S0379-4172(06)60082-6. [DOI] [PubMed] [Google Scholar]
- 44.Cui H, Peng B, Xing Z, Xu G, Yu B, Zhang Q. Molecular dissection of seedling-vigor and associated physiological traits in rice. Theor Appl Genet. 2002;105(5):745–753. doi: 10.1007/s00122-002-0908-2. [DOI] [PubMed] [Google Scholar]
- 45.Lin HX, Yanagihara S, Zhuang JY, Senboku T, Zheng KL, Yashima S. Identification of QTL for salt tolerance in rice via molecular markers. Chin J Rice Sci. 1998;12(2):72–78. doi: 10.1007/BF02984515. [DOI] [Google Scholar]
- 46.Toojinda T, Siangliw M, Tragoonrung S, Vanavichit A. Molecular genetics of submergence tolerance in rice: QTL analysis of key traits. Ann Bot. 2003;91 Spec No:243–253. doi: 10.1093/aob/mcf072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andaya VC, Tai TH. Fine mapping of the qCTS12 locus, a major QTL for seedling cold tolerance in rice. Theor Appl Genet. 2006;113(3):467–475. doi: 10.1007/s00122-006-0311-5. [DOI] [PubMed] [Google Scholar]
- 48.Qu Y, Mu P, Zhang H, Chen CY, Gao Y, Tian Y, et al. Mapping QTLs of root morphological traits at different growth stages in rice. Genetica. 2007;133(2):187–200. doi: 10.1007/s10709-007-9199-5. [DOI] [PubMed] [Google Scholar]
- 49.Bernier J, Kumar A, Venuprasad R, Spaner D, Verulkar S, Mandal NP, et al. Characterization of the effect of a QTL for drought resistance in rice, qtl12.1, over a range of environments in the Philippines and eastern India. Euphytica. 2008;166(2):207–217. doi: 10.1007/s10681-008-9826-y. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phenotypic evaluation of seed germination for 35 randomly selected rice accessions under different NaCl concentrations and in the control. (DOCX 15 kb)
Rice accessions used in this study and related phenotypic data. (XLSX 141 kb)
Frequency distributions of germination index, vigor index, mean germination time, imbibition rate, and germination rate for 478 rice accessions under salt-stress and control conditions, and stress susceptibility indices of these traits. Under salt stress (a) and control (b) conditions. Stress susceptibility index (c). (TIFF 1093 kb)
Box plots for stress susceptibility indices of germination-related traits in 478 rice accessions. (TIFF 1970 kb)
Differences in linkage disequilibrium among panels. (TIFF 422 kb)
Filtered and effective numbers of SNPs across subpopulations and adjusted significant P value thresholds based on Bonferroni correction. (XLSX 9 kb)
List of SNPs with significant P values detected by genome-wide association analysis in this study. (XLSX 24 kb)
Haplotype analysis of LOC_Os02g02190 using 31 SNPs in 2-kb upstream promoter region, 5′ and 3′ untranslated regions, and coding region based on IRGSP 1.0 ‘Nipponbare’ annotated reference genome. (XLSX 15 kb)
Data Availability Statement
The SNP dataset used during this study is available in the Rice SNP-Seek Database (http://www.oryzasnp.org/). Any other datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.