

Abstract
Introduction
Multidrug resistance-associated protein 3 (MRP3), an efflux transporter on the hepatic basolateral membrane, may function as a compensatory mechanism to prevent the accumulation of anionic substrates (e.g., bile acids) in hepatocytes. Inhibition of MRP3 may disrupt bile acid homeostasis and is one hypothesized risk factor for the development of drug-induced liver injury (DILI). Therefore, identifying potential MRP3 inhibitors could help mitigate the occurrence of DILI.
Methods
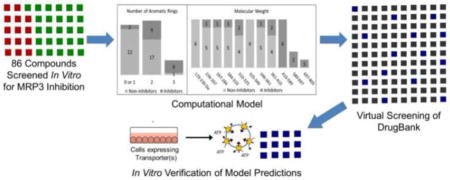
Bayesian models were developed using MRP3 transporter inhibition data for 86 structurally diverse drugs. The compounds were split into training and test sets of 57 and 29 compounds, respectively, and six models were generated based on distinct inhibition thresholds and molecular fingerprint methods. The six Bayesian models were validated against the test set and the model with the highest accuracy was utilized for a virtual screen of 1,470 FDA-approved drugs from DrugBank. Compounds that were predicted to be inhibitors were selected for in vitro validation. The ability of these compounds to inhibit MRP3 transport at a concentration of 100 μM was measured in membrane vesicles derived from stably transfected MRP3-over-expressing HEK-293 cells with [3H]-estradiol-17β-D-glucuronide (E217G; 10 μM; 5 min uptake) as the probe substrate.
Results
A predictive Bayesian model was developed with a sensitivity of 73% and specificity of 71% against the test set used to evaluate the six models. The area under the Receiver Operating Characteristic (ROC) curve was 0.710 against the test set. The final selected model was based on compounds that inhibited substrate transport by at least 50% compared to the negative control, and functional-class fingerprints (FCFP) with a circular diameter of six atoms, in addition to one-dimensional physicochemical properties. The in vitro screening of predicted inhibitors and non-inhibitors resulted in similar model performance with a sensitivity of 64% and specificity of 70%. The strongest inhibitors of MRP3-mediated E217G transport were fidaxomicin, suramin, and dronedarone. Kinetic assessment revealed that fidaxomicin was the most potent of these inhibitors (IC50 = 1.83±0.46 μM). Suramin and dronedarone exhibited IC50 values of 3.33±0.41 and 47.44±4.41 μM, respectively.
Conclusion
Bayesian models are a useful screening approach to identify potential inhibitors of transport proteins. Novel MRP3 inhibitors were identified by virtual screening using the selected Bayesian model, and MRP3 inhibition was confirmed by an in vitro transporter inhibition assay. Information generated using this modeling approach may be valuable in predicting the potential for DILI and/or MRP3-mediated drug-drug interactions.
Keywords: transporters, Bayesian models, multidrug resistance-associated protein 3, cholestasis
Graphical Abstract
1. Introduction
Drug-induced liver injury (DILI) is a major safety concern in drug development and can lead to drug withdrawal from the market (Kaplowitz, 2001). DILI is also of great clinical interest because it is the underlying cause of liver transplantation in approximately 15% of cases (Russo et al., 2004). Patients with DILI can present with either a hepatocellular, cholestatic, or mixed pattern of injury. Cholestatic DILI is caused by impaired bile acid secretion and bile flow (Trauner et al., 1998; Wagner et al., 2009). Bile acids are key signaling molecules involved in lipid and glucose metabolism, however, they can be cytotoxic due to their hydrophobic, detergent-like properties. Therefore, bile acid synthesis, cellular accumulation, and bile flow is tightly regulated in vivo (Attili et al., 1986; Staels and Fonseca, 2009).
Under normal conditions, bile acids are excreted from the hepatocyte into bile, undergo intestinal reabsorption via the enterocyte, and cycle back to the hepatocyte via the portal circulation. Conjugated bile acids also can be excreted from the hepatocyte into plasma across the hepatic basolateral membrane. Inhibition of bile acid transporters at the canalicular and/or basolateral membranes can disrupt bile acid homeostasis, alter the normal routes of bile acid excretion, and may even lead to intracellular accumulation of bile acids. The bile salt export pump (BSEP) is the primary transporter that excretes bile acids from hepatocytes into bile, and inhibition of BSEP is a known risk factor for the development of cholestatic DILI (Dawson et al., 2012; Morgan et al., 2010). However, BSEP inhibition alone is a poor predictor of a compound’s potential to cause DILI (Pedersen et al., 2013). Factoring in the inhibition of other compensatory bile acid efflux transporters, including multidrug resistance associated protein (MRP) 2, MRP3, and MRP4, can improve DILI predictions (Köck et al., 2014; Morgan et al., 2013).
MRP3 (ABCC3) is an ATP binding cassette (ABC) transporter localized on the hepatic basolateral membrane that primarily transports glucuronide conjugated anionic compounds, including bilirubin glucuronides, bile acids, organic anions, acyl glucuronides, and some anticancer drugs (Hirohashi et al., 2000; König et al., 1999; Kool et al., 1999; Lee et al., 2004). Under normal physiological conditions, MRP3 is expressed at low levels in human hepatocytes. However, MRP3 is upregulated in certain liver diseases and cholestatic conditions, including nonalcoholic steatohepatitis (NASH), Dubin-Johnson syndrome, and primary biliary cirrhosis (PBC) (Canet et al., 2015; Corpechot et al., 2006; Donner and Keppler, 2001; Zollner et al., 2003). In these situations, MRP3 serves as an important compensatory pathway for the clearance of conjugated bile acids and other substrates from the hepatocyte due to decreased expression and/or function of MRP2. Inhibition of MRP3 can alter the disposition of endogenous substrates, such as conjugated bilirubin and bile acids, which may have toxicological implications including hyperbilirubinemia and cholestatic DILI (Keppler, 2014). Inhibition of MRP3 also may contribute to toxicity mediated directly by drugs and/or drug metabolites. For example, the acyl glucuronide metabolite of diclofenac, a non-steroidal anti-inflammatory drug (NSAID), is excreted into bile via MRP2. This reactive metabolite can cause ulcerations and tissue damage in the small intestine (Seitz and Boelsterli, 1998). MRP3 can minimize this toxicity by redirecting the route of hepatic excretion of this acyl glucuronide across the basolateral membrane into plasma, thereby reducing biliary excretion and subsequent intestinal exposure to the reactive metabolite (Scialis et al., 2015).
Currently, there are no published computational models or broad-based studies of MRP3 inhibitors that have focused on identifying the physicochemical and structural features that mediate MRP3 inhibition. It has been demonstrated with other membrane transporters that a comparative analysis of molecular features among inhibitors and non-inhibitors enables the development of predictive models and an enhanced understanding of the underlying mechanisms that govern inhibition and non-inhibition (Ekins et al., 2007). Among ligand-based modeling techniques, Bayesian modeling is well-suited for membrane transporters with broad specificity versus pharmacophore modeling. The difficulty with using substrate transport inhibition data for ligand-based modeling is that there is an uncertainty of where each ligand binds to mediate inhibition. Furthermore, there is evidence that MRP3, as well as other MRPs, have multiple binding sites (Russel et al., 2008; Zehnpfennig et al., 2009). Pharmacophore modeling relies on a method that generates several low-energy conformations of molecules and then develops a pharmacophore by overlaying the features of known inhibitors; the software attempts to maximize the number of overlaid features. If the inhibition dataset contains compounds binding in different locations, then this approach may not be successful. Bayesian modeling, however, is a probabilistic strategy that does not rely on a rigid pharmacophore and instead determines molecular features and properties associated with inhibition. A probabilistic approach is also better able to handle the noise in data. Based on the molecular features and properties associated with inhibitors, insight can be gained regarding the features underlying inhibition of specific transport proteins.
The primary objective of this study was to develop and apply a computational model to identify novel MRP3 inhibitors. An inverted membrane vesicle assay was used to measure the degree of MRP3 substrate inhibition in order to validate the model predictions.
2. Methods
2.1 Datasets
The MRP3 inhibition dataset used for model development and refinement was derived from uptake inhibition studies (tested at a concentration of 100 μM) previously published by our laboratory (Köck et al., 2014). This dataset comprises of 86 structurally diverse compounds across various therapeutic areas. Molecular structures were retrieved from PubChem and standardized to remove counterions, add charges to common groups, and generate the molecular ionization state at pH 7.4 (Kim et al., 2016). The dataset of 86 compounds was split at a 2:1 ratio into a training set of 57 compounds and a test set of 29 compounds using the “generate training and test data” protocol contained in Biovia Discovery Studio (v4.5.0.15071, Dassault Systèmes, 2016.) with the split method parameter set to diverse molecules. The diverse molecules algorithm selects a maximally diverse subset of molecules for the training set by clustering the molecules based on AlogP, molecular weight, number of hydrogen bond donors/acceptors, number of rotatable bonds, number of rings, number of aromatic rings, and fractional polar surface area, as well as selecting molecules from different clusters. The clustering method is a relocation clustering algorithm based on maximal dissimilarity by calculating the Euclidean distance of the molecular descriptors. The training and test set molecules and descriptors are contained in Supplementary Table 1.
The external dataset used to predict novel MRP3 inhibitors was composed of a structure database containing 1,470 FDA-approved drugs from DrugBank (Wishart et al., 2006). Approved drugs were chosen for screening because these compounds are commercially available and in clinical use, with the potential for high clinical impact.
2.2 QSAR Bayesian Inhibition Modeling
Biovia Discovery Studio (v4.5.0.15071, Dassault Systèmes, 2016.) was employed to build Laplacian-corrected Naïve Bayesian models that predict the likelihood that a compound is an MRP3 inhibitor based on molecular features such as molecular properties and structure (Glick et al., 2006; Rogers et al., 2005; Xia et al., 2004). A score was calculated for each feature based on how strongly that feature was associated with inhibition or non-inhibition, and that score was summed with the scores of other features to generate a combined Bayesian score for a molecule. The Laplacian correction adjusts the weight of the inhibition probability estimate based on the frequency of features in the training set to correct for potential bias from undersampled features. This summed score was then used to calculate the probability that a compound was an inhibitor.
During model development, the threshold of MRP3 inhibition required for a compound to be labeled as an inhibitor was varied to optimize the model’s predictive ability by ensuring the number of compounds classified as inhibitors in the training set was sufficient to develop a robust model. Inhibition of MRP3 substrate transport by 50% was selected for the final model resulting in the training set containing 17 inhibitors and 40 non-inhibitors. The structural features used to generate models were Functional-class Fingerprint of 6 Bond Diameters (FCFP) and Extended-connectivity Fingerprint of 6 Bond Diameters (ECFP). ECFP generates models at a defined atom level (6 bond lengths, in this case) while FCFP treats functionally similar atoms as the same such that, for example, halogen atoms are treated equivalently. For each molecular property, the molecules were binned with a maximum of ten bins per molecular property. Molecular properties with continuous values were subdivided to bins such that the bins had approximately an equal number of molecules per bin.
2.3 Materials
[3H]-Estradiol-17β-glucuronide (E217G) and Microscint-O liquid scintillation fluid were purchased from Perkin Elmer Life and Analytical Sciences (Waltham, MA). Cell culture supplies, including Dulbeco’s modified essential medium (DMEM), fetal bovine serum (FBS), trypsin, and geneticin were obtained from Gibco (Life Technologies, Grand Island, NY). Unlabeled E217G and the compounds tested for inhibition were purchased from either Sigma-Aldrich (St. Louis, MO) or Cayman Chemicals (Ann Arbor, Michigan).
2.4 Cell Culture and Membrane Vesicle Preparation
Stably transfected MRP3 over-expressing HEK293 cells and mock transfected control HEK293 cells were generously provided by Dr. Elaine Leslie at the University of Alberta (Banerjee et al., 2014). Cells were cultured in DMEM supplemented with 10% FBS and 1 mg/ml geneticin in a humidified incubator (37 ºC, 5% CO2) (Banerjee et al., 2014). Membrane expression of MRP3 was characterized using immunocytochemistry staining and goat derived polyclonal MRP3 antibody (sc-5774) from Santa Cruz Biotechnology (Dallas, Texas).
To prepare membrane vesicles, cells were collected, centrifuged, washed twice in 10 ml of Tris-sucrose buffer (TSB), snap frozen in liquid nitrogen, and stored at −80 ºC. Frozen cell pellets were thawed, resuspended in TSB and exploded by N2 cavitation. EDTA was added and the suspension was centrifuged using density-gradient ultracentrifugation as described previously (Ghibellini et al., 2008). Briefly, the supernatants were pooled and were layered over 35% (w/w) sucrose/50 mM Tris in high speed ultra-centrifuge tubes and TSB was added. The samples were centrifuged at 100,000 g for 1 hour at 4 ºC, the opaque interphase was collected and added to a clean high speed centrifuge tube and centrifuged again at 100,000 g for 30 min at 4 ºC. The supernatant was discarded and the pellet was resuspended in TSB and centrifuged at 100,000 g for 10 min. The resultant pellet was resuspended in TSB and the suspension was homogenized with a 27-gauge needle to aid in vesicle formation and the membrane vesicle suspension was aliquoted and stored at −80 ºC. The protein concentration of the membrane vesicle suspension was measured using the bicinchoninic acid (BCA) assay (Thermo Scientific, Rockford, Illinois).
2.5 Membrane Vesicle Assay
Membrane vesicles were incubated with [3H]-E217G (10 μM) in reaction buffer in a final volume of 50 μL in the presence and absence of ATP. Both the single tube format and the 96-well format were used for this assay. In the single tube format, the reaction was stopped by adding 800 μl of ice-cold stopping buffer at the end of the incubation period. Samples were immediately filtered through glass fiber filters presoaked overnight in stopping buffer containing 3 mM reduced glutathione/10 mM dithiothreitol. Filters were washed three times with ice-cold stopping buffer, air dried, and dissolved in 10 mL scintillation cocktail for scintillation counting. The high-throughput assay has been described previously (Köck et al., 2014); membrane vesicles were incubated with [3H]-E217G (10 μM) in reaction buffer in a 96-well plate. After incubation, the reaction was stopped by addition of 150 μL of ice-cold stopping buffer. The samples were immediately filtered through GF/B unifilter plates (Perkin Elmer) presoaked overnight in stopping buffer containing 3 mM reduced glutathione/10 mM dithiothreitol. Filters were washed three times with ice-cold stopping buffer, air dried, and dissolved in 75 μL scintillation cocktail for scintillation counting.
The initial characterization of MRP3 vesicles was conducted to determine steady-state kinetics of E217G uptake in order to establish the appropriate incubation time for subsequent experiments. Time-dependent activity of membrane vesicles was characterized over 3, 5, and 10 min. The activity of vesicles derived from MRP3 over-expressing cells was compared to activity of vesicles from mock transfected HEK293 cells. Subsequently, experiments were conducted to determine the effect of model-predicted inhibitors and non-inhibitors on E217G uptake. Membrane vesicles were incubated at 37 °C with [3H]-E217G (10 μM) and the compounds of interest (100 μM), positive control MK-571 (50 μM), or vehicle control DMSO in reaction buffer in a final volume of 50 μL, in the presence and absence of ATP. Each screening assay included vehicle control (DMSO) and positive control (MK-571); all samples were measured in triplicate. Compounds that inhibited MRP3-mediated E217G transport > 75% of control in the screening assay were selected for further evaluation to determine IC50 values. In separate studies, IC50 values were measured using five different inhibitor concentrations (0.1 to 100 μM) in triplicate.
2.6 Data Analysis and Statistics
Experiments were conducted in triplicate in three independent experiments (unless otherwise specified), and data are expressed as mean ± SEM. Inhibition of MRP3-mediated uptake was determined by normalizing values to vehicle control (DMSO). Differences between groups were evaluated using ANOVA with post-hoc corrections for multiple comparisons using Dunnett’s test. Activity in the vehicle control group was used as the comparator for the statistical analysis of the model-predicted non-inhibitors. The comparator group for the model-predicted inhibitors was based on the inhibition threshold that was used for the generation of the model (50% inhibition). This inhibition threshold, rather than a statistically significant difference from vehicle control, was used to classify compounds as inhibitors to ensure consistency with the model. IC50 values were calculated by non-linear regression using GraphPad Prism 6 (San Diego, California).
3. Results
3.1 Bayesian Model Selection
Six Bayesian models were developed through a combination of three MRP3 inhibition thresholds and two fingerprinting methods. The three inhibition threshold levels tested, which represent the extent of MRP3 inhibition required for classification as an inhibitor, were 90%, 75%, and 50% inhibition. The number of molecules classified as inhibitors for each threshold were 17, 24, and 32, respectively, and the molecules classified as non-inhibitors were 69, 62, and 54, respectively. The number of compounds in each classification is important because there is a trade-off between developing models that predict strong inhibitors versus the number of classified inhibitors available to generate a predictive model. With a higher threshold of inhibition, there are fewer experimental inhibitors to generate the model resulting in a less robust model.
Of the six Bayesian models, the model with the highest predictive accuracy against the test set used a 50% inhibition threshold and the FCFP6 method for structure fingerprinting. The performance of this model and the other models is shown in Table 1. The selected model also demonstrated near equivalent sensitivity and specificity against the test set indicating no bias towards positive or negative predictive values.
Table 1.
Summary statistics for the Bayesian models.
| MRP3 Inhibition Threshold | Number of Inhibitors / Non-Inhibitors in Training Set | Fingerprint Method | Sensitivity | Specificity | Accuracy | Area Under the ROC Curve |
|---|---|---|---|---|---|---|
| 50% | 17 /40 | FCFP | 73% | 71% | 72% | 0.710 |
| 50% | ECFP | 86% | 28% | 58% | 0.667 | |
| 75% | 13 / 44 | FCFP | 42% | 68% | 62% | 0.558 |
| 75% | ECFP | 28% | 68% | 58% | 0.539 | |
| 90% | 9 / 48 | FCFP | 75% | 47% | 55% | 0.696 |
| 90% | ECFP | 75% | 61% | 65% | 0.661 |
The Bayesian model selected for subsequent predictions is indicated in bold.
ROC: Receiver Operating Characteristic
FCFP: Functional-class Fingerprints
ECFP: Extended-connectivity Fingerprints
3.2 Molecular Properties Associated with MRP3 Inhibition and Non-inhibition
Correlating the in vitro transport inhibition of a compound with molecular properties and structure enables the development of predictive models through an indirect understanding of the mechanism that governs molecular recognition. Of the eight molecular features we studied, five displayed a significant difference between inhibitors and non-inhibitors (Fig 1).
Fig 1. Statistical significance of differences in molecular properties between inhibitors and non-inhibitors.

The calculated p-value is plotted for the differences in molecular properties between the compounds classified as inhibitors and non-inhibitors. The following molecular properties were compared: octanol-water partition coefficient (AlogP), molecular weight, number of aromatic rings, number of rings, number of rotatable bonds, number of hydrogen bond acceptors, molecular fractional polar surface area, and number of hydrogen bond donors. The dotted line represents p=0.01; statistical significance was determined by two-way ANOVA and Bonferroni’s post hoc test.
The molecular properties with the strongest correlation to MRP3 inhibition were 1) high molecular weight and 2) multiple aromatic rings, whereas the properties that negatively correlated with MRP3 inhibition were 1) lack of rotatable bonds and aromatic rings and 2) low molecular weight and logP (Fig. 2A). Common structural features among inhibitors included a combination of both deprotonated oxygens and aromatic rings, and the features identified among non-inhibitors were nitrogen-containing structures (Fig. 2B). The prediction accuracy of the model was 72% and correctly identified 73% of inhibitors and 71% of non-inhibitors of the 29 compounds in the test set.
Fig 2. Molecular properties associated with MRP3 inhibitors and non-inhibitors.

The number of inhibitors and non-inhibitors from the training set is plotted with each corresponding molecular feature. (A) The number of inhibitors and non-inhibitors binned according to the number of aromatic rings contained with the molecule. The number of inhibitors and non-inhibitors binned according to their respective molecular weight. (B) Structural features predictive of MRP3 inhibition based on the training set. The number of “good” features indicate the number of compounds classified as inhibitors that have that structure within the training set. The Bayesian score is used to calculate the probability that a compound is an inhibitor.
3.3 Virtual Screen of DrugBank Database
This model was subsequently used to predict novel inhibitors of MRP3 transport through a virtual screen of the DrugBank database of FDA-approved drugs. The applicability of using the MRP3 inhibition dataset to screen the DrugBank database was assessed by performing principal component analysis to compare the chemical space, defined by the calculated molecular and structural features, of the two datasets (Fig. 3). Principal component analysis reduces all the molecular properties generated to a 2-dimensional plot while maximizing variance between properties. Of the 1,470 drugs screened in the DrugBank database, 554 were predicted to be inhibitors of MRP3 and based on the model’s performance against the test set, theoretically 393 of those drugs are MRP3 inhibitors. Eleven of the predicted inhibitors with high likelihood of MRP3 inhibition were selected to validate the model predictions in vitro. The selection of the 11 drugs was based on the probability of inhibition (high calculated Bayesian score), clinical significance, and commercial availability. In addition, 10 drugs with the lowest probability of MRP3 inhibition were selected to validate the negative predictive value of the model. Molecular descriptors of the compounds selected for in vitro validation and additional virtual screening predictions from the Bayesian model are shown in Supplementary Table 2 and 3 respectively.
Fig 3. Principal component analysis of datasets.

The principal component analysis of the calculated molecular features for the MRP3 inhibitor dataset used to develop the Bayesian model and the FDA-approved dataset that was virtually screened. The plot demonstrates that the MRP3 inhibition training and test sets occupy a similar chemical space to a majority of the virtually screened dataset.
3.4 Characterization of MRP3 Over-Expressing Cells and Membrane Vesicles
Stably transfected MRP3 over-expressing HEK293 cells had high levels of membrane expression and localization of MRP3 based on immunocytochemistry staining (Fig. 4). ATP-dependent transport of E217G was measured over 3–10 min and appeared to be linear at 3 and 5 min, with some saturation of transport at the 10 min time point with mean uptake of 70.5, 69.2, and 49.2 pmol/mg protein/min at 3, 5, and 10 min, respectively (Fig. 4). The 5-min time point was chosen for subsequent experiments. The activity of mock transfected vesicles was similar to transport in the absence of ATP in the MRP3 vesicles.
Fig 4. Characterization of MRP3 membrane vesicles.

Membrane localization of MRP3 over-expressing cells stained using polyclonal antibody (inset). MRP3 vesicles were co-incubated with [3H]-E217G (10 μM) for 3, 5, or 10 min at 37 °C to evaluate time-dependent MRP3-mediated E217G transport (mean ± SD; n=1 in triplicate). Activity of MRP3 vesicles and mock vesicles was tested at 5 min (mean ± SD; n=1 in triplicate).
3.5 In Vitro Validation of Model Predictions
Eleven predicted MRP3 inhibitors and 10 predicted MRP3 non-inhibitors were screened for inhibition of MRP3-mediated E217G transport. The tested compounds at 100 μM had varying degrees of inhibition (Fig. 5). Of the predicted inhibitors, amprenavir, bortezomib, darunavir, and pravastatin did not inhibit MRP3. The mean E217G uptake in the presence of these compounds was 119.4, 119.3, 98.3 and 100.5%, respectively, expressed as a percentage of control. The model classified a compound as an inhibitor if it inhibited MRP3-mediated uptake of E217G by at least 50%; therefore, a representative model inhibitor with 50% inhibition was used as a comparator group for statistical analysis. Amiodarone, atazanavir, dronedarone, fidaxomicin, lovastatin, regorafenib, and suramin were not statistically different from this model threshold. Lovastatin, regorafenib, amiodarone, and atazanavir were weak inhibitors and the mean E217G uptake in the presence of these compounds was 63.5, 69.5, 58.4 and 55.0%, respectively, expressed as a percentage of control. At a concentration of 100 μM, fidaxomicin, suramin, and dronedarone were the strongest inhibitors; mean E217G uptake in the presence of these compounds was only 18.1, 10.1 and 14.2%, respectively, compared to control. Fidaxomicin was the most potent inhibitor with an IC50 (mean ± SEM) of 1.83 ± 0.46 μM. Suramin was also a strong inhibitor with an IC50 of 3.33 ± 0.41 μM. Dronedarone was a moderately potent inhibitor with an IC50 of 47.44 ± 4.41 μM (Fig. 6). The predicted non-inhibitors acetylsalicylic acid, amantadine, atenolol, clonidine, isoniazid, 6-mercaputopurine, and methimazole were not statistically different from vehicle control and did not inhibit MRP3 transport. Chlorothiazide, felodipine, and dipyridamole had some degree of MRP3 inhibition; E217G uptake in the presence of these compounds was 37.4, 45.8, and 52.1 %, respectively, compared to control (Fig. 7).
Fig 5. Inhibitory effects of model-predicted inhibitors on MRP3-mediated E217G transport.
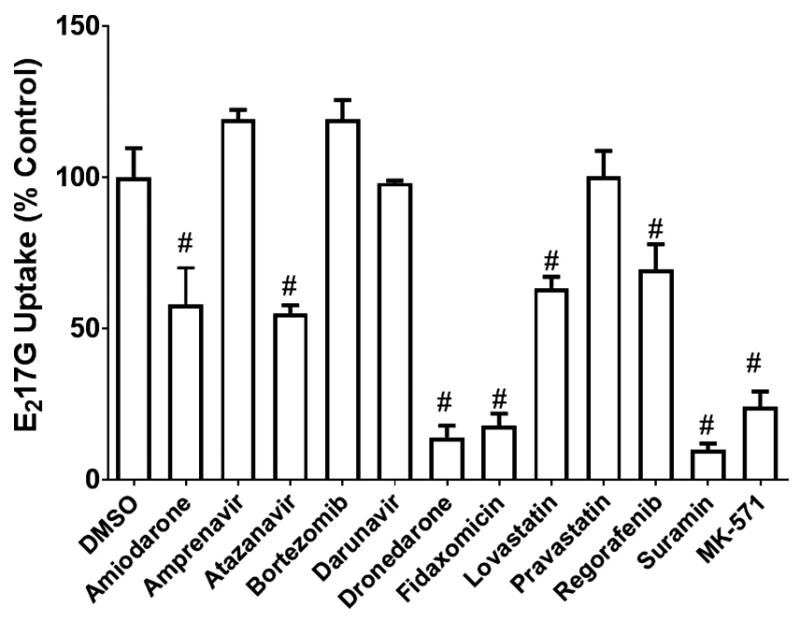
MRP3 vesicles were co-incubated with model-predicted inhibitors (100 μM) or MK-571 (50 μM) and [3H]-E217G (10 μM) for 5 min at 37 °C. Values are expressed as percentage of vehicle control (DMSO) (mean ± SEM; n=3 in triplicate). Compounds that caused a decrease in [3H]-E217G uptake that was not statistically different from the model threshold of 50% inhibition are indicated by #, p < 0.05.
Fig 6. Inhibitory potency of dronedarone, fidaxomicin, and suramin on MRP3-mediated E217G transport.

IC50values were determined at concentrations of 0.1 to 100 μM for dronedarone (A), fidaxomicin (B), and suramin (C) on MRP3-mediated [3H]-E217Gtransport (10 μM; 5-min incubation). Values are expressed as percentage of vehicle control; each value represents the mean ± SD. IC50 values (mean ± SEM; n=3 in triplicate) were calculated using non-linear least square regression for dronedarone (47.44±4.41 μM), fidaxomicin (1.83±0.46 μM), and suramin (3.33±0.41 μM).
Fig 7. Inhibitory effects of model-predicted non-inhibitors on MRP3-mediated E217G transport.
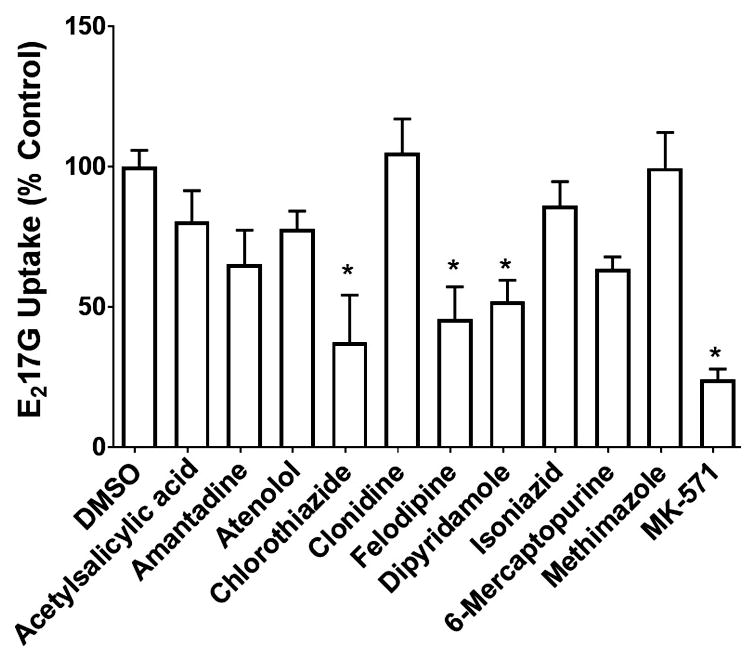
MRP3 vesicles were co-incubated with non-inhibitors (100 μM) and [3H]-E217G (10 μM) for 5 min at 37 °C. Values are expressed as percentage of vehicle control (DMSO) (mean ± SEM; n=3 in triplicate). Compounds that caused a decrease in [3H]-E217G uptake that was statistically different from vehicle control are indicated by *p < 0.05.
4. Discussion
In this study, a computational model was developed to predict MRP3 inhibitors based on compounds previously identified in our laboratory as MRP3 inhibitors (Köck et al., 2014). Results from a virtual screening performed using this computational model were tested in vitro to evaluate the model. Compounds that were strong inhibitors of MRP3 transport at a concentration of 100 μM were characterized further using an IC50 assay to quantify the potency of inhibition.
A predictive computational model of MRP3 inhibition was developed from the structurally diverse MRP3 inhibition dataset using Bayesian modeling; molecular structures and properties that correlated with inhibition and non-inhibition were identified. The features identified as predictive of MRP3 inhibition were molecular weight and multiple aromatic rings. There was a distinct decline in the number of compounds with a molecular weight less than 410 Da that were classified as MRP3 inhibitors (Fig. 2). As our group and others have demonstrated previously, molecular weight was also significantly associated with inhibition of the hepatic bile acid transporters BSEP, MRP2 and MRP4. These findings indicate that large molecular weight compounds may inhibit multiple transporters, which increases the likelihood that a drug may inhibit bile acid transport and induce cholestasis (Pedersen et al., 2008; Pedersen et al., 2013; Welch et al., 2015). Interestingly, the association between inhibition of MRP3-mediated transport and multiple aromatic rings appears to be more correlated with MRP3 inhibition compared to models we and others previously developed for BSEP and MRP4 inhibition. For example, Pedersen et al. noted that MRP2 inhibitors had significantly more aromaticity compared to non-inhibitors as measured by unsaturated non-polar surface area, based on the MRP2 model they developed. Across the four transporters, there is a correlation between transport inhibition and hydrophobicity, but the influence of aromaticity is notable for MRP3 and MRP2 compared to BSEP and MRP4 (Matsson et al., 2007; Pedersen et al., 2008; Warner et al., 2012). Physicochemical features that correlated strongly with non-inhibition were absence of rotatable bonds, low logP values, low molecular weight, and lack of aromatic rings. Fewer rotatable bonds suggests a more rigid molecule that may not be able to conform in order to interact with the transporter; low molecular weight molecules may not have requisite features to bind to MRP3. Low logP values and lack of aromatic rings likely indicates that it is more energetically favorable for a non-inhibitor molecule to remain solvated than to bind to the transporter. These features associated with non-inhibition are common across ABC transporters (Matsson et al., 2007; Pedersen et al., 2008; Warner et al., 2012)..
Among the fragment-like fingerprints that were correlated with inhibition, the most predictive contained deprotonated oxygen atoms, which is expected since MRP3 primarily transports anions; additionally, aromatic rings were featured, which reiterates the observation that multiple aromatic rings are predictive of inhibition. The fingerprints associated with non-inhibition contained nitrogen atoms, most of which were ionizable tertiary amines that would result in a positive charge.
The next step in model validation was to test predicted MRP3 inhibitors in a stably transfected MRP3 over-expressing HEK-293 cell line in vitro. Experiments were conducted using both the single tube and the high-throughput assay format to increase the robustness of the data. Drugs that were predicted to be MRP3 inhibitors in the virtual screening were tested in vitro to evaluate model sensitivity. Seven out of the 11 (64%) predicted inhibitors that were tested in vitro inhibited MRP3-mediated E217G transport. The drugs that were found to be false positives were evaluated retrospectively to determine where the model underperformed. For example, bortezomib had 9 rotatable bonds and a dipeptide core structure similar to the macrocyclic MRP3 inhibitors cyclosporine A and valinomycin, and these combined features increased its Bayesian score. Pravastatin was another false positive that shares structural similarities to fluvastatin, an MRP3 inhibitor, and it contains a high number of hydrogen bond acceptors and rotatable bonds that increase this drug’s calculated score. Interestingly, dronedarone and amiodarone, which are structurally similar compounds, were both model-predicted MRP3 inhibitors but exhibited different degrees of inhibition in the in vitro assay. Although speculative, the difference in activity between the two compounds is likely driven by the increase in the number of rotatable bonds: amiodarone possesses 11 whereas dronedarone has 17 rotatable bonds due to two elongated alkyl sidechains and the addition of a methanesulfonamide functional group. Two other false positive drugs assayed in the in vitro screen were amprenavir and darunavir, which are both protease inhibitors. Retrospectively, this is likely due to an inadvertently introduced bias in the 86 compound dataset that contained three protease inhibitors (lopinavir, ritonavir, and indinavir) all of which inhibit MRP3 by greater than 50%.
The specificity of the model was tested by in vitro evaluation of the model predicted non-inhibitors. Seven of the 10 (70%) predicted non-inhibitors that were tested did not inhibit MRP3 activity. These results were in agreement with the prediction accuracy of the model, which was able to correctly identify 71% of inhibitors and 72% of non-inhibitors.
The novel findings that dronedarone, fidaxomicin, and suramin are strong MRP3 inhibitors may have important implications for patient care. For example, dronedarone, which is an antiarrhythmic that is indicated for the treatment of atrial fibrillation (“Dronedarone Package Insert,” 2009), has been associated with case reports of hepatotoxicity (Jahn et al., 2013). Although the exact mechanisms of dronedarone-associated hepatotoxicity are unknown, mitochondrial toxicity along with other factors are thought to play a role (Felser et al., 2013). Inhibition of MRP3 may contribute to dronedarone-associated hepatotoxicity by altering disposition of bile acids and other MRP3 substrates.
In the present study, a computational model was developed by leveraging existing data to predict MRP3 inhibitors; model predictions were validated in vitro and potent MRP3 inhibitors were identified. Large-scale in vitro screening studies to identify transporter inhibitors are time-consuming and resource-intensive. The present predictive computational model could be used to identify drug candidates that are likely MRP3 inhibitors, thereby increasing the efficiency of accurately identifying, a priori, potential compounds that might increase the risk of DILI. In addition, potent MRP3 inhibitors could increase susceptibility to cholestasis and liver injury in some patients who may rely more extensively on MRP3 for hepatic excretion of organic anions (e.g., patients with NASH, Dubin-Johnson’s syndrome or primary biliary cirrhosis). An integrated approach utilizing both computational modeling and in vitro screening, as demonstrated in this study, can be a valuable tool to predict transporter inhibition, and identify novel and potent transporter inhibitors.
Supplementary Material
Acknowledgments
The assistance of Dr. Melina Malinen with microscopy and immunocytochemistry, and the technical assistance of Yi Zeng with the conduct of experiments is gratefully acknowledged. We also acknowledge Dr. Alex MacKerell for providing access to Biovia Discovery Studio 4.5.
Funding: This work was supported by the National Institutes of Health [R01 GM041935] and M-CERSI Scholars program FDA grant 1U01FD005946.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Attili AF, Angelico M, Cantafora A, Alvaro D, Capocaccia L. Bile acid-induced liver toxicity: relation to the hydrophobic-hydrophilic balance of bile acids. Med Hypotheses. 1986;19:57–69. doi: 10.1016/0306-9877(86)90137-4. [DOI] [PubMed] [Google Scholar]
- Banerjee M, Carew MW, Roggenbeck BA, Whitlock BD, Naranmandura H, Le XC, Leslie EM. A novel pathway for arsenic elimination: human multidrug resistance protein 4 (MRP4/ABCC4) mediates cellular export of dimethylarsinic acid (DMAV) and the diglutathione conjugate of monomethylarsonous acid (MMAIII) Mol Pharmacol. 2014;86:168–79. doi: 10.1124/mol.113.091314. [DOI] [PubMed] [Google Scholar]
- Canet MJ, Merrell MD, Hardwick RN, Bataille AM, Campion SN, Ferreira DW, Xanthakos SA, Manautou JE, A-Kader HH, Erickson RP, Cherrington NJ. Altered regulation of hepatic efflux transporters disrupts acetaminophen disposition in pediatric nonalcoholic steatohepatitis. Drug Metab Dispos. 2015;43:829–35. doi: 10.1124/dmd.114.062703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpechot C, Ping C, Wendum D, Matsuda F, Barbu V, Poupon R. Identification of a novel 974C-->G nonsense mutation of the MRP2/ABCC2 gene in a patient with Dubin-Johnson syndrome and analysis of the effects of rifampicin and ursodeoxycholic acid on serum bilirubin and bile acids. Am J Gastroenterol. 2006;101:2427–32. doi: 10.1111/j.1572-0241.2006.00695.x. [DOI] [PubMed] [Google Scholar]
- Dawson S, Stahl S, Paul N, Barber J, Kenna JG. In vitro inhibition of the bile salt export pump correlates with risk of cholestatic drug-induced liver injury in humans. Drug Metab Dispos. 2012;40:130–138. doi: 10.1124/dmd.111.040758. [DOI] [PubMed] [Google Scholar]
- Donner MG, Keppler D. Up-regulation of basolateral multidrug resistance protein 3 (Mrp3) in cholestatic rat liver. Hepatology. 2001;34:351–9. doi: 10.1053/jhep.2001.26213. [DOI] [PubMed] [Google Scholar]
- [accessed 11.30.16];Dronedarone Package Insert [WWW Document] 2009 http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/022425lbl.pdf.
- Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br J Pharmacol. 2007;152:9–20. doi: 10.1038/sj.bjp.0707305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felser A, Blum K, Lindinger PW, Bouitbir J, Krähenbühl S. Mechanisms of hepatocellular toxicity associated with dronedarone--a comparison to amiodarone. Toxicol Sci. 2013;131:480–90. doi: 10.1093/toxsci/kfs298. [DOI] [PubMed] [Google Scholar]
- Ghibellini G, Leslie EM, Pollack GM, Brouwer KLR. Use of Tc-99m Mebrofenin as a Clinical Probe to Assess Altered Hepatobiliary Transport: Integration of In Vitro, Pharmacokinetic Modeling, and Simulation Studies. Pharm Res. 2008;25:1851–1860. doi: 10.1007/s11095-008-9597-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick M, Jenkins JL, Nettles JH, Hitchings H, Davies JW. Enrichment of High-Throughput Screening Data with Increasing Levels of Noise Using Support Vector Machines, Recursive Partitioning, and Laplacian-Modified Naive Bayesian Classifiers. J Chem Inf Model. 2006;46:193–200. doi: 10.1021/ci050374h. [DOI] [PubMed] [Google Scholar]
- Hirohashi T, Suzuki H, Takikawa H, Sugiyama Y. ATP-dependent Transport of Bile Salts by Rat Multidrug Resistance-associated Protein 3 (Mrp3) J Biol Chem. 2000;275:2905–2910. doi: 10.1074/jbc.275.4.2905. [DOI] [PubMed] [Google Scholar]
- Jahn S, Zollner G, Lackner C, Stauber RE. Severe toxic hepatitis associated with dronedarone. Curr Drug Saf. 2013;8:201–2. doi: 10.2174/15748863113089990031. [DOI] [PubMed] [Google Scholar]
- Kaplowitz N. Drug-induced liver disorders: implications for drug development and regulation. Drug Saf. 2001;24:483–90. doi: 10.2165/00002018-200124070-00001. [DOI] [PubMed] [Google Scholar]
- Keppler D. Special Section on Transporters in Toxicity and Disease—Minireview The Roles of MRP2, MRP3, OATP1B1, and OATP1B3 in Conjugated Hyperbilirubinemia. DRUG Metab Dispos Drug Metab Dispos. 2014;42:561–565. doi: 10.1124/dmd.113.055772. [DOI] [PubMed] [Google Scholar]
- Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Han L, He J, He S, Shoemaker BA, Wang J, Yu B, Zhang J, Bryant SH. PubChem Substance and Compound databases. Nucleic Acids Res. 2016;44:D1202–D1213. doi: 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köck K, Ferslew BC, Netterberg I, Yang K, Urban TJ, Swaan PW, Stewart PW, Brouwer KLR. Risk Factors for Development of Cholestatic Drug-Induced Liver Injury: Inhibition of Hepatic Basolateral Bile Acid Transporters Multidrug Resistance-Associated Proteins 3 and 4. Drug Metab Dispos. 2014;42:665–674. doi: 10.1124/dmd.113.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König J, Rost D, Cui Y, Keppler D. Characterization of the human multidrug resistance protein isoform MRP3 localized to the basolateral hepatocyte membrane. Hepatology. 1999;29:1156–63. doi: 10.1002/hep.510290404. [DOI] [PubMed] [Google Scholar]
- Kool M, van der Linden M, de Haas M, Scheffer GL, de Vree JM, Smith AJ, Jansen G, Peters GJ, Ponne N, Scheper RJ, Elferink RP, Baas F, Borst P. MRP3, an organic anion transporter able to transport anti-cancer drugs. Proc Natl Acad Sci U S A. 1999;96:6914–9. doi: 10.1073/pnas.96.12.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YMA, Cui Y, König J, Risch A, Jäger B, Drings P, Bartsch H, Keppler D, Nies AT. Identification and functional characterization of the natural variant MRP3-Arg1297His of human multidrug resistance protein 3 (MRP3/ABCC3) Pharmacogenetics. 2004;14:213–23. doi: 10.1097/00008571-200404000-00001. [DOI] [PubMed] [Google Scholar]
- Matsson P, Englund G, Ahlin G, Bergström CAS, Norinder U, Artursson P. A Global Drug Inhibition Pattern for the Human ATP-Binding Cassette Transporter Breast Cancer Resistance Protein (ABCG2) J Pharmacol Exp Ther. 2007:323. doi: 10.1124/jpet.107.124768. [DOI] [PubMed] [Google Scholar]
- Morgan RE, Trauner M, van Staden CJ, Lee PH, Ramachandran B, Eschenberg M, Afshari CA, Qualls CW, Lightfoot-Dunn R, Hamadeh HK. Interference with bile salt export pump function is a susceptibility factor for human liver injury in drug development. Toxicol Sci. 2010;118:485–500. doi: 10.1093/toxsci/kfq269. [DOI] [PubMed] [Google Scholar]
- Morgan RE, van Staden CJ, Chen Y, Kalyanaraman N, Kalanzi J, Dunn RT, Afshari CA, Hamadeh HK. A multifactorial approach to hepatobiliary transporter assessment enables improved therapeutic compound development. Toxicol Sci. 2013;136:216–41. doi: 10.1093/toxsci/kft176. [DOI] [PubMed] [Google Scholar]
- Pedersen JM, Matsson P, Bergström CAS, Hoogstraate J, Norén A, LeCluyse EL, Artursson P. Early identification of clinically relevant drug interactions with the human bile salt export pump (BSEP/ABCB11) Toxicol Sci. 2013;136:328–43. doi: 10.1093/toxsci/kft197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen JM, Matsson P, Bergström CAS, Norinder U, Hoogstraate J, Artursson P. Prediction and Identification of Drug Interactions with the Human ATP-Binding Cassette Transporter Multidrug-Resistance Associated Protein 2 (MRP2; ABCC2) J Med Chem. 2008;51:3275–3287. doi: 10.1021/jm7015683. [DOI] [PubMed] [Google Scholar]
- Rogers D, Brown RD, Hahn M. Using Extended-Connectivity Fingerprints with Laplacian-Modified Bayesian Analysis in High-Throughput Screening Follow-Up. J Biomol Screen. 2005;10:682–686. doi: 10.1177/1087057105281365. [DOI] [PubMed] [Google Scholar]
- Russel F, Koenderink J, Masereeuw R. Multidrug resistance protein 4 (MRP4/ABCC4): a versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol Sci. 2008;29:200–207. doi: 10.1016/j.tips.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Russo MW, Galanko JA, Shrestha R, Fried MW, Watkins P. Liver transplantation for acute liver failure from drug induced liver injury in the United States. Liver Transplant. 2004;10:1018–1023. doi: 10.1002/lt.20204. [DOI] [PubMed] [Google Scholar]
- Scialis RJ, Csanaky IL, Goedken MJ, Manautou JE. Multidrug Resistance-Associated Protein 3 Plays an Important Role in Protection against Acute Toxicity of Diclofenac. Drug Metab Dispos. 2015;43:944–950. doi: 10.1124/dmd.114.061705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz S, Boelsterli UA. Diclofenac acyl glucuronide, a major biliary metabolite, is directly involved in small intestinal injury in rats. Gastroenterology. 1998;115:1476–1482. doi: 10.1016/S0016-5085(98)70026-5. [DOI] [PubMed] [Google Scholar]
- Staels B, Fonseca VA. Bile acids and metabolic regulation: mechanisms and clinical responses to bile acid sequestration. Diabetes Care. 2009;32(Suppl 2):S237–45. doi: 10.2337/dc09-S355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauner M, Meier PJ, Boyer JL. Molecular pathogenesis of cholestasis. N Engl J Med. 1998;339:1217–27. doi: 10.1056/NEJM199810223391707. [DOI] [PubMed] [Google Scholar]
- Wagner M, Zollner G, Trauner M. New molecular insights into the mechanisms of cholestasis. J Hepatol. 2009;51:565–580. doi: 10.1016/j.jhep.2009.05.012. [DOI] [PubMed] [Google Scholar]
- Warner DJ, Chen H, Cantin LD, Kenna JG, Stahl S, Walker CL, Noeske T. Mitigating the Inhibition of Human Bile Salt Export Pump by Drugs: Opportunities Provided by Physicochemical Property Modulation, In Silico Modeling, and Structural Modification. Drug Metab Dispos. 2012;40:2332–2341. doi: 10.1124/dmd.112.047068. [DOI] [PubMed] [Google Scholar]
- Welch MA, Köck K, Urban TJ, Brouwer KLR, Swaan PW. Toward Predicting Drug-Induced Liver Injury: Parallel Computational Approaches to Identify Multidrug Resistance Protein 4 and Bile Salt Export Pump Inhibitors. Drug Metab Dispos. 2015;43:725–734. doi: 10.1124/dmd.114.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–72. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Malisk EG, Gallant P, Rogers D. Classification of Kinase Inhibitors Using a Bayesian Model. 2004 doi: 10.1021/JM0303195. [DOI] [PubMed] [Google Scholar]
- Zehnpfennig B, Urbatsch IL, Galla HJ. Functional Reconstitution of Human ABCC3 into Proteoliposomes Reveals a Transport Mechanism with Positive Cooperativity. Biochemistry. 2009;48:4423–4430. doi: 10.1021/bi9001908. [DOI] [PubMed] [Google Scholar]
- Zollner G, Fickert P, Silbert D, Fuchsbichler A, Marschall HU, Zatloukal K, Denk H, Trauner M. Adaptive changes in hepatobiliary transporter expression in primary biliary cirrhosis. J Hepatol. 2003;38:717–27. doi: 10.1016/s0168-8278(03)00096-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.