

Abstract
Human indoleamine 2,3-dioxygenase catalyzes the oxidative cleavage of tryptophan to N-formyl kynurenine, the initial and rate-limiting step in the kynurenine pathway. Additionally, this enzyme has been identified as a possible target for cancer therapy. A 20-amino acid protein segment (the JK loop), which connects the J and K helices, was not resolved in the reported hIDO crystal structure. Previous studies have shown that this loop undergoes structural rearrangement upon substrate binding. In this work, we apply a combination of replica exchange molecular dynamics simulations and site-directed mutagenesis experiments to characterize the structure and dynamics of this protein region. Our simulations show that the JK loop can be divided into two regions: the first region (JK loopC) displays specific and well-defined conformations and is within hydrogen bonding distance of the substrate, while the second region (JK loopN) is highly disordered and exposed to the solvent. The peculiar flexible nature of JK loopN suggests that it may function as a target for post-translational modifications and/or a mediator for protein–protein interactions. In contrast, hydrogen bonding interactions are observed between the substrate and Thr379 in the highly conserved “GTGG” motif of JK loopC, thereby anchoring JK loopC in a closed conformation, which secures the appropriate substrate binding mode for catalysis. Site-directed mutagenesis experiments confirm the key role of this residue, highlighting the importance of the JK loopC conformation in regulating the enzymatic activity. Furthermore, the existence of the partially and totally open conformations in the substrate-free form suggests a role of JK loopC in controlling substrate and product dynamics.
Graphical abstract
Human indoleamine 2,3-dioxygenase (hIDO) is a heme-containing enzyme, which catalyzes the oxidative cleavage of tryptophan (Trp) to N-formyl kynurenine (NFK), the initial and rate-limiting step in the kynurenine pathway,1–5 by adding both dioxygen atoms to the C2=C3 bond of the indole moiety of the Trp. hIDO is a monomeric enzyme, which is ubiquitously expressed in many tissues.6,7 Its expression is induced by interferon-γ, lipopolysaccharide, and tumor necrosis factor.1,3,4,6–11 In addition, hIDO suppresses the immune response by starving cytotoxic T-cells by consuming local Trp and by producing Trp metabolites, which activate regulatory T-cells. The immunosuppressive role of hIDO is important for regulating various physiological and pathophysiological conditions, feto-maternal tolerance, and cancer immune escape.6,7,10–12 It has been shown that hIDO inhibitors are able to facilitate regression of established solid tumors in mice,13–15 some of which are currently in clinical trials for human cancer therapy.16,17
IDO has been extensively characterized from a biochemical,6,18,19 spectroscopic,20–25 and structural perspective.26,27 Although the crystal structure of hIDO has been obtained in its substrate-free forms,27,28 the structure and function of the loop connecting helix J to helix K (residues 360–380), named the JK loop hereafter, remains unknown due to its highly flexible nature.
Human tryptophan 2,3-dioxygenase (hTDO) is another heme dioxygenase, which catalyzes the same reaction as hIDO.18,29,30 It has been shown that hTDO is more specific towards l-Trp,31,32 while hIDO can react with both l- and d-Trp, as well as different indole derivatives.7,18,33 The structures of hTDO,34 Drosophila melanogaster TDO,35 Ralstonia metallidurans TDO,36 and Xanthomonas campestris TDO (xcTDO)29 show that TDO is a homotetrameric enzyme. The structure of xcTDO is particularly important as it was crystallized in the substrate-free and substrate-bound forms.29,34,36
Additionally, the second enzyme in the pyrrolnitrin biosynthesis pathway from Pseudomonas fluorescens, called PrnB, which converts 7-Cl-tryptophan into monodechloroaminopyrrolnitrin, belongs to the dioxygenase superfamily.37 Its structure was resolved for the cyanide complex and two ternary complexes with both l-tryptophan or 7-Cl-l-tryptophan and cyanide.37,38
Comparison of the structures of the dioxygenases shows that the TDO monomer, PrnB, and hIDO display a high degree of structural similarity (see Figure S1). In addition, structure-based sequence alignment (see Figure S2) shows that the JK loop in hIDO is ∼20 and 15 residues longer than that in xcTDO and that in PrnB, respectively. In xcTDO, Trp binding induces the organization of the JK loop, which like that in hIDO is disordered in the substrate-free form, into an Ω-loop structure.29 The JK loop, hence, could play an important role in control substrate and product dynamics.
In a previous work,26 we have used a combination of docking and molecular dynamics simulation to characterize the active sites of both hIDO and xcTDO. We found that the active site of hIDO is larger and more flexible than that of xcTDO. This allows the coexistence of two different Trp conformations in hIDO (called Cf1 and Cf2). In contrast, only one conformation is observed in xcTDO (Figure 1). In Cf1, the Trp has a perpendicular orientation with respect to the heme plane and resembles the Trp binding mode in xcTDO (see Figure S3) and as such was attributed to the reactive structure. In Cf2, the Trp lies almost parallel to the heme and farther from the Fe–O2 moiety, which could account for a Trp molecule migrating from solvent into the active site.
Figure 1.
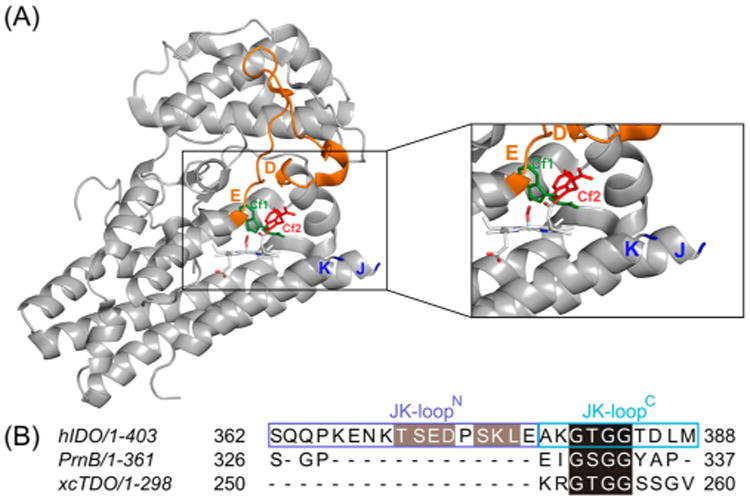
(A) Three-dimensional structure of hIDO and (B) structure-based sequence alignment of the JK loops of hIDO, PrnB, and xcTDO. The JK loop connecting the helix J and helix K is disordered, and hence missing in the structure. The DE loop is colored orange. The Trp residues labeled in green sticks and red sticks represent the Cf1 and Cf2 conformations, respectively. The sequence motifs labeled in green and red in panel B highlight in black the highly conserved “GT(S)GG” motif and the potential phosphorylation sites in gray.
In a separate work,39 we have characterized the functional role of a specific residue in the JK loop of hTDO, T342, in the highly conserved “GTGG” motif of the JK loop in hTDO (Uniprot entry P48775). We found that the single T342A mutation results in a total loss of substrate stereoselectivity.39 By means of classical molecular dynamics simulations, we determined that this mutation affects not only the specific interaction of this residue with the Trp but also the position and dynamics of the JK loop because of the perturbation of the interaction between the JK loop and the DE loop [the loop connecting helix D and helix E (residues 120–125) (Figure S3)]. As a result, the position of the Trp backbone is affected and the enzymatic reaction is significantly inhibited.39
Although classical molecular dynamics simulations are widely used to describe conformational dynamics in protein systems, on occasions when extensive sampling is required, advanced sampling techniques, such as temperature replica exchange molecular dynamics (T-REMD), can be applied to properly explore the free energy landscape and avoid being trapped in local energy minima. T-REMD is a commonly used technique for improving the sampling of the configurational space in biomolecules.40–42 In T-REMD simulations, several replica of the system are run at different temperatures in parallel. After a user-defined amount of MD steps, temperatures in two different contiguous replicas are allowed to exchange according to a Monte Carlo probabilistic scheme. In this way, the conformational sampling is significantly improved with respect to constant-temperature MD simulations, which are typically run at lower temperature values. The number of replicas and the temperature values are chosen to guarantee an acceptable interchange ratio of ∼10–20%.43 If the interchange value were within this range, it would mean that the temperatures were well-distributed and that the number of replicas was sufficient, indicative of proper performance of the replica exchange simulation.
Here, we used a combination of T-REMD and classical molecular dynamics simulations (MD) to sample the conformations of the JK loop in hIDO and to analyze its role in Trp binding and catalysis. Our results indicate that the JK loop conformation stabilizes the reactive conformation by specific H-bond interactions. They allow us to hypothesize a possible role of the conformational flexibility of the JK loop in modulating the activity of hIDO.
Materials and Methods
Computational Methods
Starting Structures
Initial structures for full length hIDO were built from the hIDO available structure [Protein Data Bank (PDB) entry 2D0T], which corresponds to the complex with Phenyl-Imidazol (PI) and not the l-Trp substrate. As in previous works,26 we removed the PI ligand and built the oxy in silico. Trp-bound structures (in both conformations) were built on the oxy complexes derived from our previous work.45 In this structure, the JK loop (residues 360–380) is absent. Sequence structure-based alignment of hIDO with xcTDO (PDB entry 2NW8) and PrnB (PDB entry 2×67) [which show a high degree of overall structural similarity (see Figures S1 and S2)] indicates that they share a common motif consisting of four residues in the C-terminal segment of the loop (residues 377–383 in hIDO, which we will term JK loopC hereafter): GTGG in xcTDO and hIDO and GSGG in PrnB. Because of the similarity in this portion of the sequence, we used both xcTDO and PrnB as templates for the JK loopC region. Overall, 30 different JK loop models were built using the Modeller program (version 9.14),44 which differ mostly in the N-terminal region, for which no template is available (see results for further details).
T-REMD Calculations
Starting from the aforementioned structures, we performed temperature replica exchange molecular dynamics (T-REMD) simulations. Simulations were performed in the oxy complexes of hIDO in the SF state and in the substrate-bound states with Trp bound in either binding site (termed Cf1 and Cf2). For each case, 30 replicas (corresponding to the different starting JK loop structures) were run spanning a temperature range of 290–472 K. Each replica was run for 50 ns. This represents a total of 1.5 μs per system (total simulation time of 4.5 μs). All simulations were performed using the GPU (CUDA) version of the PMEMD module of the Amber14 package,46–49 using the Amber99SB force field. Details about additional parameters (including heme parametrization) can be found in our previous publications.21,26,30,33 To properly explore the conformational space of the JK loop and to avoid unfolding events, the residues in the JK loop region were completely free to move, whereas a position restraint was applied on the rest of the protein Cα atoms, using a force constant of 50 kcal mol−1 Å−2.
After the simulations, the three lowest-temperature trajectories (at 290, 295, and 300 K) were analyzed. Clustering analysis was performed on these three trajectories using Cpptraj, a program within the Amber14 package.50 A cutoff of 2.5 Å was used [i.e., structures where the loop structures with a root-mean-square deviation (rmsd) of <2.5 Å are included in the same cluster]. We considered the three most populated clusters as representative structures.
Equilibrium Molecular Dynamics Simulations of the hIDO Complexes
Starting from the central structures of the three most populated clusters (populations of >10%), we performed classical molecular dynamics (MD) in explicit solvent. Structures were immersed in an octahedral box of TIP3P waters,51 using a 10 Å minimal distance from the protein surface to the box. All simulations were performed with the PMEMD module of the AMBER14 package49 using standard simulation parameters, under periodic boundary conditions. We used Ewald sums to treat long-range electrostatic interactions,52,53 the SHAKE algorithm to keep bonds including hydrogen atoms at their equilibrium length, a 2 fs time step for the integration of Newton's equations,54 and the Langevin thermostat55 to control the system temperature. A 12 Å cutoff for explicit electrostatic interactions was used in all cases. Equilibration consisted of an energy minimization of the initial structures, followed by a slow heating to 300 K at a constant volume. MD runs consisted of 45 ns trajectories and were run in the NPT ensemble.
Pocket Shape and Volume Characterization
To estimate the pocket shape and volume characteristics of each representative structure of the three conformations, we use the pocket volume measurer program (POVME 2.0)43,56 using the trajectories calculated for each case.
Solvent-Accessible Surface Area Calculations
For each representative structure of the Trp-bound hIDO, the solvent-accessible surface area (SASA) was calculated using the “rolling ball” algorithm57 where the SASA corresponds to the surface swept out by a probe sphere with a radius of 1.4 Å, which approximates the radius of a water molecule.
Binding Free Energy Calculations
The binding free energy was computed from the molecular dynamics trajectories, using the MM-GBSA (molecular mechanics, general-Born surface area) approach58 implemented in AMBER14.49 The binding free energy, , was calculated by
where corresponds to the substrate binding energy in vacuum and is the corresponding free energy for the solvation of each species. We performed an energy decomposition for the residues involved in the main interactions with the Trp.
Experimental Methods
Chemicals and Protein Expression and Purification
Hemin chloride and Tris base (triple crystallized, ≥99.9% pure) were obtained from Porphyrin Producs, Inc. (Logan, UT) and Fisher Scientific (Fair Lawn, NJ), respectively. All other chemicals were of the highest available purity (>99%) from Sigma-Aldrich. Wild-type hIDO1 was prepared as described elsewhere.59 The mutants were constructed by using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The proteins were expressed and purified with the same protocol as the wild-type enzyme.59 The purified proteins were stored in pH 7.4 Tris buffer (50 mM) at −80 °C until they were used.
Optical Absorption Studies
All the samples were prepared in a pH 7.4, 100 mM Tris buffer in the presence or absence of 30 mM l-Trp. All the optical absorption spectra were recorded in a 1 cm path length or cylindrical Raman quartz cuvette with a UV2100 spectrophotometer (Shimadzu Scientific Instruments, Inc.) with a spectral slit width of 1 nm.
Steady-State Activity Measurements
The steady-state activities of the proteins were measured with a standard protocol as described elsewhere.31,33 All the reactions were controlled at 25 °C by a Neslab RTE-111 water bath circulator unless specified otherwise. The initial linear velocities of the reactions, V, were obtained by monitoring the formation of the product, NFK, at 321 nm (ε = 3750 M−1 cm−1) as a function of time with a UV2100 spectrophotometer with a spectral slit width of 2 nm. V was calibrated with the concentration of the enzyme and plotted as a function of substrate concentration, [S]. All the data were analyzed with Origin 6.1 (Microcal Software, Inc.).
Results
JK Loop Structural Modeling
As stated above, all the hIDO structures available in the Protein Data Bank correspond to a substrate-free (SF) state, where the JK loop (residues 360–380) was not resolved because of the lack of a substrate and its highly flexible nature.27,28,60 In contrast, the crystal structures of xcTDO (PDB entries 2NW7 and 2NW8)29 and PrnB (PDB entries 2×66 and 2×67)37 were determined in both substrate-free and Trp-bound forms. In the Trp-bound structures, either a complete JK loop (xcTDO) or a fragment of it (PrnB) was ordered in these structures.29,37 These two enzymes display a high degree of structural similarity with hIDO (Figure S1); in addition, the JK loop anchoring points with the bound Trp are well-conserved. Furthermore, sequence comparison (Figure S2) indicates the presence of a four-residue motif, “GT(S)GG”, in the JK loopC region. The conserved motif results in many JK loopC structures harboring similar characteristic glycine turn conformations.61 On the other hand, the N-terminal segment of the loop (residues 359–376 in hIDO, named JK loopN hereafter) shows higher sequence diversity and a larger set of possible conformations. Taking into account the sequence identity in the JK loopC region, we constructed 30 different initial conformations as the starting points for the T-REMD simulations (vide supra).
JK Loop Structure and Dynamics
To gain insight into the JK loop conformation and dynamics, we performed three independent T-REMD simulations, each consisting of 30 replicas, corresponding to SF and two Trp-bound complexes of hIDO (termed Cf1 and Cf2 as shown in Figure 1).26 Overall, the simulations show that the JK loop explores an enormous range of conformational space (the rmsd of the loop against the averaged conformation is >16 Å).
To gain a first glimpse of the possible conformations of the JK loop, we performed a clustering analysis of the three different T-REMD simulations, corresponding to the SF, Cf1, and Cf2 states. This joint analysis indicates that the JK loopC region adopts specific and well-defined conformations (vide infra). On the other hand, JK loopN is far more flexible (rmsd of >10 Å). It adopts transient conformations with secondary structure elements, but there is no prevalent conformation.
The clustering analysis of the JK loopC region of the three different states (SF, Cf1, and Cf2 states) shows that the observed conformations can be classified into three groups that we named closed, intermediate, or open-extended conformations (Figure 2). Additionally, the closed conformation displays a quite rigid loop, especially in the Trp-bound states (the rmsd in Cf1, for example, is 1.6 Å). Visual comparison of the closed and intermediate conformations indicates that the closed conformation is closer to the distal pocket, as indicated by the Gly380 NH–heme propionate 7 distance (dashed line in Figure 2), which ranges from 2 to 5 Å in the closed conformation and from 5 to 10 Å in the intermediate conformation. Indeed, in the Cf1 structure, the GTGG motif of JK loopC interacts with the Trp backbone, isolating the active site from the solvent (vide infra). The T-REMD simulations show that the three conformations are interconvertible, manifesting the high flexibility of the loop.
Figure 2.
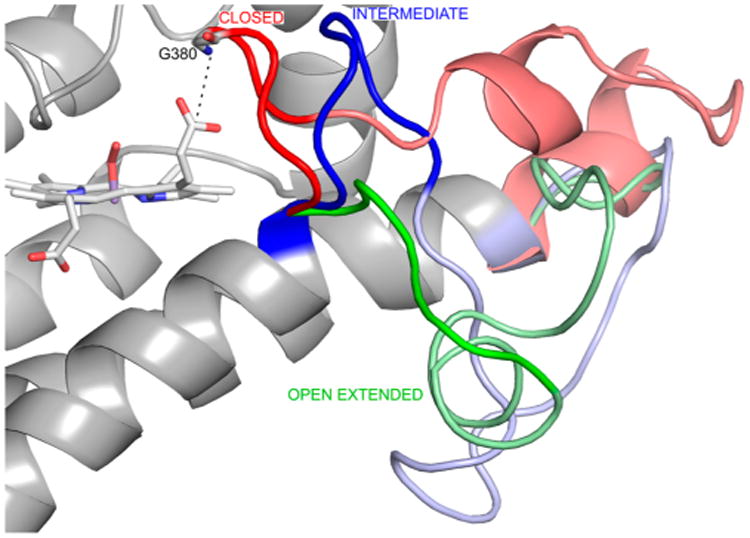
Three JK loop conformations resulting from clustering analysis of JK loopC in the SF, Cf1, and Cf2 states. The closed, intermediate, and open-extended conformations are colored red, blue, and green, respectively. In each conformation, the JK loopC and JK loopN regions are highlighted by the bright and pale colors, respectively. The dashed line indicates the distance between the backbone N atom of Gly380 and the carbon atom of the heme propionate 7 group.
Analysis of the JK Loop Conformational State and Its Role in Trp Binding
With the relevant JK loop conformational states described above, we analyzed the relevant conformations of both JK loop regions in each state, Cf1, Cf2, and SF. Figure 3 shows the predominant structures of each state derived from cluster analysis of the complete loop. For Cf1, the predominant conformations show a high variability in the JK loopN region (with a rmsd of 12.4 Å) while the JK loopC region stays tightly in a similar position (with a rmsd of 1.6 Å) (Figure 3A). For Cf2, there is higher variability in both the JK loopN and JK loopC regions (Figure 3B). The closed conformation of the JK loopC region is scarcely populated (for population analysis of JK loopC, see Table S1). In addition, the Trp in the Cf2 is more mobile than that in Cf1 and can adopt multiple conformations, in contrast to the Trp in Cf1. Finally, for SF, although the closed conformation is extensively visited, several intermediate or open-extended conformations are observed. Interestingly, in the SF state (Figure 3C), the closed conformations show a variability much greater than that in the Cf1, as evidenced by the higher RMSD of the JK loopC region (3.9 Å in the SF state vs 1.6 Å in Cf1). This increased flexibility is consistent with the loss of specific interactions between the substrate and the GTGG motif in JK loopC (vide infra).
Figure 3.
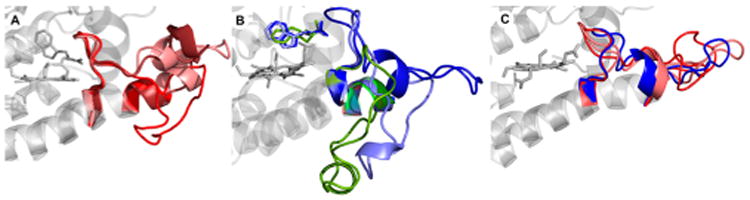
Three most populated clusters for (A) Cf1, (B) Cf2, and (C) SF resulting from cluster analysis of the whole JK loop. The loops colored red, blue, and green represent the closed, intermediate, and open-extended conformations, respectively.
Detailed Structural and Functional Characterization of JK Loop Conformational States
Having determined the main conformational states of the JK loop and their relation to Trp binding, we then analyzed interactions responsible for defining these states and their impact on the functional properties of hIDO. We performed additional 45 ns molecular dynamics simulations of the most representative conformations of each hIDO complex. Analysis of the interactions between Trp and the JK loopC region in the Cf1 “closed” state shows the existence of strong H-bonds of the NH and OH groups of Thr379 in the “GTGG” motif with the Trp backbone (see Figure 4A). These H-bonds cannot be established in the “open-extended” state because of the large distance between these residues and the substrate. Additionally, Thr382 OH and Gly380 NH also interact with a heme propionate, which holds JK loopC in the closed state. These interactions are lost when the loop opens, reaching the open-extended conformation, allowing Trp to move farther into the solvent, like that found in the Cf2 state. In this open-extended JK loop conformation, the “GTGG” motif is completely exposed to solvent.
Figure 4.

(A) Main interactions among the Trp, the heme group, and the residues of JK loopC in Cf1. (B–D) Free volume in the Trp binding pocket: in the closed state, with Arg231 and Thr379 represented as the doorknob (B), in the open-extended conformation (C), or in the closed conformation of Cf1 in the T379A mutant (D).
To further address the role of Thr379 in relation to the JK loop conformation, we performed a 70 ns MD simulation for the T379A mutant starting from the closed conformation of Cf1. The results show that although during the time scale of our simulation the main interactions between the residues of the active site and the Trp are retained, they are significantly weakened with respect to those found in the wild-type (wt) enzyme (see Table S2 for details). In the T379A mutant, the interaction between the Thr379 OH group and the Trp is lost; in addition, the extent of the observed interaction between Trp and Ala379 is reduced to 48% (with respect to 91% of the comparable interactions in the wt enzyme) (Table S2). This results in a larger variation in the JK loop position compared to that of the Cf1 conformation of the wt enzyme. As expected, Ala379 cannot strongly interact with the Trp backbone, promoting the opening of the JK loop.
Visual inspection of different loop states (Figure 2) shows that when JK loopC is in the closed conformation it isolates the active site from the bulk solvent, which only opens in the open-extended state. The data suggest that JK loopC controls Trp and NFK dynamics.
To characterize the accessibility, we determined the volume and shape of the Trp binding pocket in each JK loop conformation. The results show that in the closed Cf1 state the average pocket volume is 1778 Å3 (the volume of Trp is 227 Å3). Solvent accessibility between the active site and bulk is blocked, mainly because of the presence of Thr379 and Arg231, both of which interact strongly with the substrate. In the open-extended Cf2 state, the pocket volume increases to 5079 Å3 and bulk solvent can clearly access the active site (Figure 4C). Panels B and C of Figure 4 illustrate the differences between the closed and open conformations and highlight the gating function of JK loopC. Figure 4D shows the results of the same analysis for T379A mutant. Interestingly, in spite of conservation of the Cf1 closed conformation, the pocket volume of the closed state increases to 4399 Å3, similar to the volume of the open conformation of the wt enzyme. Because of the weaker A379–Trp interaction, the pocket also extends toward the protein surface (see Figure 4D). Additionally, in this closed state, the T379A binding site and solvent are connected in contrast to the isolation of the active site from the bulk solvent observed in the wt enzyme for Cf1.
To further monitor the Trp accessibility to the active site in the different conformational states, we computed the Trp solvent-accessible surface area (SASA) for both the closed Cf1 and open-extended Cf2 states. The SASA indicates how deeply buried the substrate is inside the hIDO structure. The results show Trp-SASA is very small (1.8 Å2 on average) in the Cf1 closed state, while that in the Cf2 open-extended states is 89.2 Å2 on average. These data suggest that in the Cf2 open-extended state, Trp is probably dissociating from the protein. It is important to highlight that in the Cf1 T379A mutant the Trp-SASA is 4.2 Å2 on average, showing that the Trp is more exposed to the solvent. Figure 5 depicts the protein surface in the closed Cf1 state and open Cf2 state, clearly showing that in Cf1 the Trp is buried in the protein matrix while in Cf2 it is much more exposed to the solvent because of the opening of JK loopC.
Figure 5.
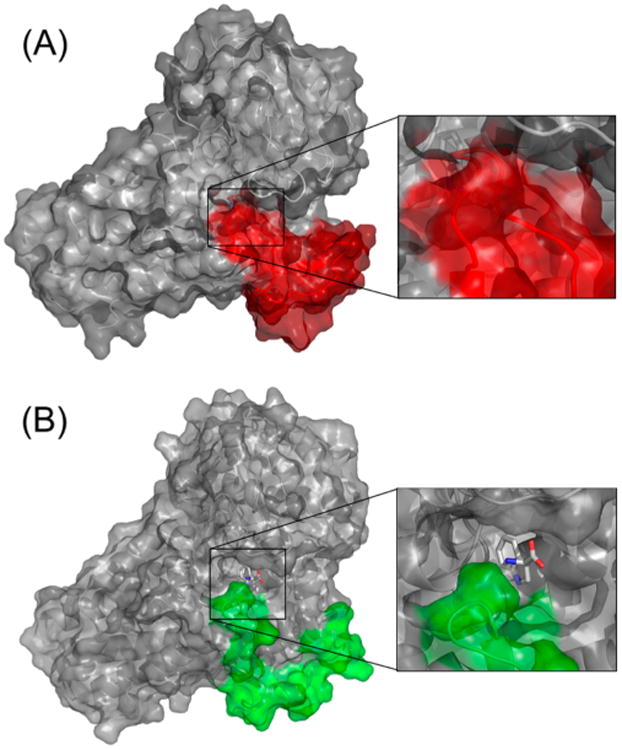
Protein surface view of hIDO in the (A) Cf1 and (B) Cf2 states. The surfaces corresponding to the loop region are colored (A) red or (B) green.
To obtain insight into the Trp binding affinity in the different JK loop conformations, the binding free energy and the contribution from both Thr379 and Arg231 were computed using an MM-GBSA scheme. This kind of approach typically yields overestimated binding free energy values, but they are very useful for extracting comparative trends.62 The results, presented in Table 1, show that the Trp binding energy is doubled in the closed loop conformation (most populated state in Cf1) compared to the open-extended loop conformation observed in Cf2. Interestingly, in the open-extended loop conformation, the interaction with Thr379 is weakend and the interaction with Arg231 is almost lost. Additionally, when comparing the results for wt-Cf1 and T379A-Cf1, we observe a significant decrease in Trp binding energy for the mutant protein, confirming the role of Thr379 in Cf1 stabilization. Similar to the previous observation, the mutant weakens not only the interaction between the backbone CO of this residue and the Trp but also that between Arg231 and the Trp (see Table S2).
Table 1. MM-GBSA Binding Free Energies (kilocalories per mole) for Trp Calculated for the Most Populated Cluster of Each hIDO-Bound Conformation of the wt Enzyme and the T379A Mutant.
| wt Cf1 | wt Cf2 | T379A Cf1 | |
|---|---|---|---|
| total | −58 ± 3 | −24 ± 3 | −51 ± 3 |
| Thr/Ala379 | −4.9 ± 0.8 | −3.5 ± 0.6 | −2.8 ± 0.8 |
| Arg231 | −15 ± 1 | −0.8 ± 0.7 | −13 ± 1 |
Activity Studies of the T379A and R231L Mutants
To confirm role of Thr379 and Arg231 in controlling substrate–protein interactions, we prepared and studied the T379A and R231L mutants. As shown in Figure 6A, in the SF state, the ferric T379A mutant exhibits a Soret band at 403 nm and visible bands at 500 and 533 nm, as well as a charge-transfer band at 632 nm (Figure 6A). They are consistent with a water-bound ferric heme with a mixed six-coordinate high-spin (6CHS) and low-spin (6CLS) electronic configuration.25 Binding of l-Trp to the ferric T379A mutant causes the shift of the Soret band to 407 nm, as well as the diminishment of the intensity of the 500 nm band and increases in the intensities of the 533 and 562 nm bands. It is also associated with the disappearance of the charge-transfer band at 632 nm. The spectral change indicates the conversion of the water-bound species to a 6CLS species, which has been attributed to hydroxide-bound ferric protein in wt hIDO1.63 The spectral features of T379A are similar to those of the comparative derivatives of wt hIDO.19 On the other hand, the R231L mutant (Figure 6B) shows spectral features similar to those of T379A and wt hIDO1, except that the presence of 30 mM l-Trp only slightly perturbs the spectrum, presumably because of incomplete binding of l-Trp.
Figure 6.
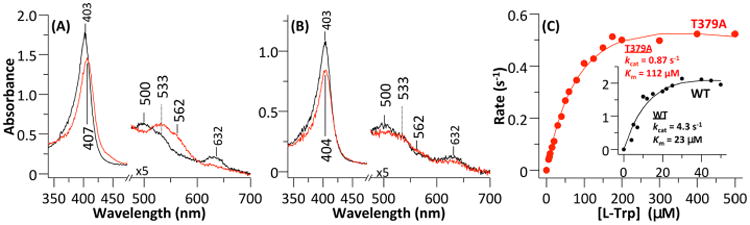
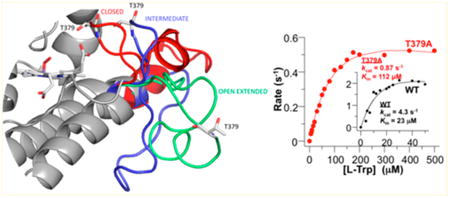
Absorption spectra of (A) T379A and (B) R231L mutants of hIDO and (C) Michaelis–Menten plots of the T379A mutant and wild-type hIDO as a function of l-Trp concentration. Panels A and B show the optical absorption spectra of ferric T379A in the absence (black line) and presence of ∼30 mM l-Trp (red line). The visible bands (475–700 nm) are expanded by a factor of 5. Spectra were generated in a sealed Raman quartz cuvette in the presence of ∼50 μM enzyme in pH 7.4, 100 mM Tris buffer. The Michaelis–Menten plots were generated using 100 nM enzyme. The reaction was initiated by addition of ascorbate to the reaction mixture in pH 7.4 Tris buffer at 25 °C.
The dioxygenase reaction catalyzed by hIDO1 does not consume any electrons.18 However, an electron source is required to sustain the multiple turnovers of the reaction, as the active ternary complex is prone to autoxidation, via the release of superoxide. Accordingly, we used a widely used methylene blue/ascorbate system64 as the reductant to measure the steady-state activity of the enzyme. Kinetic studies show that the T379A mutation results in a 25-fold reduction in the activity toward l-Trp, as evidenced by the ∼5-fold reduction in kcat (4.3 and 0.87 s−1 for wt hIDO and T379A, respectively) and a 5-fold increase in KM (23 and 112 μM for wt hIDO and T379A, respectively). In contrast, the R231L mutation abolishes all the activity toward l-Trp.
Discussion
In this work, we used a combination of T-REMD simulations and site-directed mutagenesis experiments to shed light on the structure and dynamics of the flexible active site JK loop that is absent in the crystal structure of hIDO. This loop is significantly longer than that of the other proteins of the same superfamily. Additionally, there is a high degree of sequence similarity in the JK loopC region characterized by the “GTGG” motif. Our simulation results showed that the JK loopC segment is able to adopt different conformations characterized as closed, intermediate, and open-extended conformations. The JK loopN region is far more flexible, and although it can adopt transient conformations with secondary structure elements, there are no dominant conformations. The JK loopC conformation is linked with substrate binding. When Trp binds in the reactive conformation (Cf1), JK loopC adopts the predominantly closed conformation. In the absence of substrate, or when Trp is in the alternative conformation (Cf2), JK loopC is more flexible and more likely to be in the open conformation. In this context, it is interesting to note that this kind of loop opening–closing mechanism that regulates substrate entry has also been proposed for other heme-based oxygenases, as shown for the widely studied P450Cam.65–67 Although their fold is different, the common mechanism highlights the relevance and plasticity of the heme oxygenase active site beyond the IDO/TDO family.
Our results suggest that the dynamic nature of JK loopC allows it to directly modulate substrate and product dynamics. In the absence of substrate, the high flexibility of the loop, dominated by the open conformations, allows Trp to enter the protein. This entry is followed by loop closure, resulting in tight binding of Trp, which is held in place in an active conformation by Thr379 and Arg231, allowing the enzymatic reaction to occur. After catalysis, these interactions are possibly weakened, leading to the opening of JK loopC to allow product release. The role of the loop, and in particular of Thr379, in binding and positioning of the substrate is consistent with the activity studies of the T379A mutant that show a 5-fold reduction in kcat and a 5-fold increase in Km toward Trp, which, although representing moderate variations on the kinetic parameters, indicate that Thr379, and hence the JK loop, participate in Trp binding and catalysis. The increase in Km is possibly related to a higher loop flexibility and a larger population of the open-extended conformation, which weakens substrate protein interactions. Previous studies of TDO45,68 have shown that the reaction barrier is tightly related to the proper positioning of the Trp, which is perturbed in the T379A mutant.
The role of JK loopN is far more difficult to address. Our results show that it displays high flexibility without secondary structures, and more importantly, it extends out from the protein core domain into the solvent. This behavior is not uncommon in protein regions that interact with other proteins and/or become chemically modified for regulatory purposes. With this in mind, we performed a database search in Prosite,69 which resulted in the prediction of two phosphorylation sites: 367TSED and 372SKL (the latter is also reported in http://www.phosphosite.org/). The JK loopN post-translational modification, through phosphorylation, for example, or its binding to another protein, could prevent JK loopC from opening, which in turn would inhibit substrate binding, rendering hIDO inactive. Interestingly, the nearby Tyr353 in helix J has been shown to become nitrated upon exposure to peroxynitrite, leading to IDO inhibition.70 This regulation role is consistent with the fact that JK loopN is present only in hIDO, where it has to regulate the immune system, and not hTDO or its bacterial counterparts, which are involved in maintaining Trp homeostasis. However, further studies are needed to confirm this idea.
Taking our results into account, we conclude the JK loop in hIDO is a highly dynamic loop, which presents large structural fluctuations, in agreement with the fact that this loop was disordered in the X-ray crystallographic structure. The T-REMD simulations show that JK loopC is able to adopt closed structures and open-extended conformations, which are in dynamic equilibrium. The closed structure, which is predominant in the Trp-bound enzyme, helps to bind and position the substrate properly for catalysis, thereby modulating hIDO activity. The lack of structure in the JK loopN and its dynamic nature suggest that it could be the target of post-translational modifications and or protein–protein interactions, thereby providing an additional layer of control over hIDO activity
Supplementary Material
Structural alignment, sequence comparison, and additional MD results (PDF)
Acknowledgments
The authors thank the High-Performance Computing Center at the University of Florida for providing further computational resources.
Funding: This work was supported in part by grants from Universidad de Buenos Aires (UBA-CYT Project 20020120300025BA), from Agencia Nacional de Producción Cientifica y Tecnológica (Project PICT 2012-2571), from CONICET (PIP 112 201101 0085), from the Ministerio de Ciencia, Tecnología e Innovación Productiva (RAICES-Siembra), and from the National Institutes of Health (Grant GM086482). Additionally, this research is part of the Blue Waters Sustained-Petascale Computing Project, which is supported by the National Science Foundation (Award OCI 07-25070) and the state of Illinois. Blue Waters is a joint effort of the University of Illinois at Urbana-Champaign and its National Center for Supercomputing Applications. Research funding comes from the National Science Foundation (OCI-1440031).
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b00077.
Notes: The authors declare no competing financial interest.
References
- 1.Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 1991;5:2516–2522. [PubMed] [Google Scholar]
- 2.Thomas SR, Stocker R. Redox reactions related to indoleamine 2,3-dioxygenase and tryptophan metabolism along the kynurenine pathway. Redox Rep. 1999;4:199–220. doi: 10.1179/135100099101534927. [DOI] [PubMed] [Google Scholar]
- 3.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 4.King NJC, Thomas SR. Molecules in focus: Indoleamine 2,3-dioxygenase. Int J Biochem Cell Biol. 2007;39:2167–2172. doi: 10.1016/j.biocel.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Takikawa O. Biochemical and medical aspects of the indoleamine 2,3-dioxygenase- initiated L-tryptophan metabolism. Biochem Biophys Res Commun. 2005;338:12–19. doi: 10.1016/j.bbrc.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 6.Hayaishi O. Properties and function of indoleamine 2,3-dioxygenase. J Biochem. 1976;79:13P–21P. doi: 10.1093/oxfordjournals.jbchem.a131115. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto S, Hayaishi O. Tryptophan Pyrrolase of Rabbit Intestine: d- and l-tryptophan-cleaving enzyme or enzymes. J Biol Chem. 1967;242:5260–5266. [PubMed] [Google Scholar]
- 8.Werner-Felmayer G, Werner ER, Fuchs D, Hausen A, Reibnegger G, Wachter H. Neopterin formation and tryptophan degradation by a human myelomonocytic cell line (THP-1) upon cytokine treatment. Cancer Res. 1990;50:2863–2867. [PubMed] [Google Scholar]
- 9.Löb S, Königsrainer A, Zieker D, Brücher BDM, Rammensee HG, Opelz G, Terness P. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol Immun-other. 2009;58:153–157. doi: 10.1007/s00262-008-0513-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Croitoru-Lamoury J, Lamoury FMJ, Caristo M, Suzuki K, Walker D, Takikawa O, Taylor R, Brew BJ. Interferon-γ Regulates the Proliferation and Differentiation of Mesenchymal Stem Cells via Activation of Indoleamine 2,3 Dioxygenase (IDO) PLoS One. 2011;6:e14698. doi: 10.1371/journal.pone.0014698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller AJ, Prendergast GC. Indoleamine 2,3-dioxygenase in immune suppression and cancer. Curr Cancer Drug Targets. 2007;7:31–40. doi: 10.2174/156800907780006896. [DOI] [PubMed] [Google Scholar]
- 12.Godin-Ethier J, Hanafi LA, Piccirillo Ca, Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Cancer Res. 2011;17:6985–6991. doi: 10.1158/1078-0432.CCR-11-1331. [DOI] [PubMed] [Google Scholar]
- 13.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11:312–319. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 14.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 15.Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, Mellor AL, Prendergast GC, Munn DH. Inhibition of Indoleamine 2,3-Dioxygenase in Dendritic Cells by Stereoisomers of 1-Methyl-Tryptophan Correlates with Antitumor Responses. Cancer Res. 2007;67:792–801. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- 16.Vacchelli E, Aranda F, Eggermont A, Sautès-Fridman C, Tartour E, Kennedy EP, Platten M, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology. 2014;3:e957994. doi: 10.4161/21624011.2014.957994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Platten M, von Knebel Doeberitz N, Oezen I, Wick W, Ochs K. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Front Immunol. 2015;5:673. doi: 10.3389/fimmu.2014.00673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-Containing Oxygenases. Chem Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 19.Chauhan N, Basran J, Efimov I, Svistunenko DA, Seward HE, Moody PCE, Raven EL. The role of serine 167 in human indoleamine 2,3-dioxygenase: A comparison with tryptophan 2,3-dioxygenase. Biochemistry. 2008;47:4761–4769. doi: 10.1021/bi702405a. [DOI] [PubMed] [Google Scholar]
- 20.Lu C, Yeh SR. Ferryl Derivatives of Human Indoleamine 2,3-Dioxygenase. J Biol Chem. 2011;286:21220–21230. doi: 10.1074/jbc.M111.221507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis-Ballester A, Batabyal D, Egawa T, Lu C, Lin Y, Marti Ma, Capece L, Estrin Da, Yeh SR. Evidence for a ferryl intermediate in a heme-based dioxygenase. Proc Natl Acad Sci U S A. 2009;106:17371–17376. doi: 10.1073/pnas.0906655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davydov RM, Chauhan N, Thackray SJ, Anderson JLR, Papadopoulou ND, Mowat CG, Chapman SK, Raven EL, Hoffman BM. Probing the ternary complexes of indoleamine and tryptophan 2,3-dioxygenases by cryoreduction epr and endor spectroscopy. J Am Chem Soc. 2010;132:5494–5500. doi: 10.1021/ja100518z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yanagisawa S, Yotsuya K, Hashiwaki Y, Horitani M, Sugimoto H, Shiro Y, Appelman EH, Ogura T. Identification of the Fe–O2 and the Fe=O Heme Species for Indoleamine 2,3-Dioxygenase during Catalytic Turnover. Chem Lett. 2010;39:36–37. [Google Scholar]
- 24.Lu C, Lin Y, Yeh SR. Spectroscopic studies of ligand and substrate binding to human indoleamine 2,3-dioxygenase. Biochemistry. 2010;49:5028–5034. doi: 10.1021/bi1005078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papadopoulou ND, Mewies M, McLean KJ, Seward HE, Svistunenko DA, Munro AW, Raven EL. Redox and Spectroscopic Properties of Human Indoleamine 2,3-Dioxygenase and A His303Ala Variant: Implications for Catalysis†. Biochemistry. 2005;44:14318–14328. doi: 10.1021/bi0513958. [DOI] [PubMed] [Google Scholar]
- 26.Capece L, Arrar M, Roitberg AE, Yeh SR, Marti Ma, Estrin Da. Substrate stereo-specificity in tryptophan dioxygenase and indoleamine 2,3-dioxygenase. Proteins: Struct, Funct, Genet. 2010;78:2961–2972. doi: 10.1002/prot.22819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugimoto H, Oda S, Otsuki T, Hino T, Yoshida T, Shiro Y. Crystal structure of human indoleamine 2,3-dioxygenase: catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc Natl Acad Sci U S A. 2006;103:2611–2616. doi: 10.1073/pnas.0508996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tojo S, Kohno T, Tanaka T, Kamioka S, Ota Y, Ishii T, Kamimoto K, Asano S, Isobe Y. Crystal Structures and Structure–Activity Relationships of Imidazothiazole Derivatives as IDO1 Inhibitors. ACS Med Chem Lett. 2014;5:1119–1123. doi: 10.1021/ml500247w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forouhar F, Anderson JLR, Mowat CG, Vorobiev SM, Hussain A, Abashidze M, Bruckmann C, Thackray SJ, Seetharaman J, Tucker T, Xiao R, Ma LC, Zhao L, Acton TB, Montelione GT, Chapman SK, Tong L. Molecular insights into substrate recognition and catalysis by tryptophan 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2007;104:473–478. doi: 10.1073/pnas.0610007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greengard O, Feigelson P. The purification and properties of liver tryptophan pyrrolase. J Biol Chem. 1962;237:1903–1907. [PubMed] [Google Scholar]
- 31.Batabyal D, Yeh SR. Human tryptophan dioxygenase: A comparison to indoleamine 2,3-dioxygenase. J Am Chem Soc. 2007;129:15690–15701. doi: 10.1021/ja076186k. [DOI] [PubMed] [Google Scholar]
- 32.Rafice Sa, Chauhan N, Efimov I, Basran J, Raven EL. Oxidation of L-tryptophan in biology: a comparison between tryptophan 2,3-dioxygenase and indoleamine 2,3-dioxygenase. Biochem Soc Trans. 2009;37:408–412. doi: 10.1042/BST0370408. [DOI] [PubMed] [Google Scholar]
- 33.Shimizu T, Nomiyama S, Hirata F, Hayaishi O. Indoleamine 2,3-dioxygenase Purification and some properties. J Biol Chem. 1978;253:4700–4706. [PubMed] [Google Scholar]
- 34.Meng B, Wu D, Gu J, Ouyang S, Ding W, Liu ZJ. Structural and functional analyses of human tryptophan 2,3-dioxygenase. Proteins: Struct, Funct, Genet. 2014;82:3210–3216. doi: 10.1002/prot.24653. [DOI] [PubMed] [Google Scholar]
- 35.Searles LL, Ruth RS, Pret AM, Fridell RA, Ali AJ. Structure and transcription of the Drosophila melanogaster vermilion gene and several mutant alleles. Mol Cell Biol. 1990;10:1423–1431. doi: 10.1128/mcb.10.4.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Kang SA, Mukherjee T, Bale S, Crane BR, Begley TP, Ealick SE. Crystal structure and mechanism of tryptophan 2,3-dioxygenase, a heme enzyme involved in tryptophan catabolism and in quinolinate biosynthesis. Biochemistry. 2007;46:145–155. doi: 10.1021/bi0620095. [DOI] [PubMed] [Google Scholar]
- 37.Zhu X, van Pèe KH, Naismith JH. The Ternary Complex of PrnB (the Second Enzyme in the Pyrrolnitrin Biosynthesis Pathway), Tryptophan, and Cyanide Yields New Mechanistic Insights into the Indolamine Dioxygenase Superfamily. J Biol Chem. 2010;285:21126–21133. doi: 10.1074/jbc.M110.120485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Laurentis W, Khim L, Anderson JLR, Adam A, Phillips RS, Chapman SK, van Pee KH, Naismith JH. The Second Enzyme in Pyrrolnitrin Biosynthetic Pathway Is Related to the Heme-Dependent Dioxygenase Superfamily†. Biochemistry. 2007;46:12393–12404. doi: 10.1021/bi7012189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Capece L, Lewis-Ballester A, Marti Ma, Estrin Da, Yeh SR. Molecular basis for the substrate stereoselectivity in tryptophan dioxygenase. Biochemistry. 2011;50:10910–10918. doi: 10.1021/bi201439m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 41.Rhee YM, Pande VS. Multiplexed-Replica Exchange Molecular Dynamics Method for Protein Folding Simulation. Biophys J. 2003;84:775–786. doi: 10.1016/S0006-3495(03)74897-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou R. Trp-cage: Folding free energy landscape in explicit water. Proc Natl Acad Sci U S A. 2003;100:13280–13285. doi: 10.1073/pnas.2233312100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Durrant JD, De Oliveira CAF, McCammon JA. POVME: An algorithm for measuring binding-pocket volumes. J Mol Graphics Modell. 2011;29:773–776. doi: 10.1016/j.jmgm.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fiser A, Šali A. Macromolecular Crystallography, Part D. Academic Press; San Diego: 2003. Modeller: Generation and Refinement of Homology-Based Protein Structure Models; pp. 461–491. [DOI] [PubMed] [Google Scholar]
- 45.Capece L, Lewis-Ballester A, Batabyal D, Di Russo N, Yeh SR, Estrin Da, Marti Ma. The first step of the dioxygenation reaction carried out by tryptophan dioxygenase and indoleamine 2,3-dioxygenase as revealed by quantum mechanical/molecular mechanical studies. JBIC, J Biol Inorg Chem. 2010;15:811–823. doi: 10.1007/s00775-010-0646-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le Grand S, Götz AW, Walker RC. SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput Phys Commun. 2013;184:374–380. [Google Scholar]
- 47.Götz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J Chem Theory Comput. 2012;8:1542–1555. doi: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, III, DeBolt S, Ferguson D, Seibel G, Kollman P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun. 1995;91:1–41. [Google Scholar]
- 49.Case DA, Berryman JT, Betz RM, Cerruti DS, Cheatham TE, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Luchko T, Luo R, Madej B, Merz KM, Monard G, Needham P, Nguyen H, Nguyen HT, Omelayan I, Onufriev A, Roe DR, Roitberg A, Salomon-Ferrer R, Simmerling CL, Smith W, Swails JM, Walker RC, Wang J, Wolf RM, Wu X, York DM, Kollman PA. Amber 2015. University of California; San Francisco: 2015. [Google Scholar]
- 50.Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 51.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926. [Google Scholar]
- 52.Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J Chem Theory Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 53.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577. [Google Scholar]
- 54.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 55.Wu X, Brooks BR. Self-guided Langevin dynamics simulation method. Chem Phys Lett. 2003;381:512–518. [Google Scholar]
- 56.Durrant JD, Votapka L, Sørensen J, Amaro RE. POVME 2.0: An Enhanced Tool for Determining Pocket Shape and Volume Characteristics. J Chem Theory Comput. 2014;10:5047–5056. doi: 10.1021/ct500381c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shrake A, Rupley JA. Environment and exposure to solvent of protein atoms Lysozyme and insulin. J Mol Biol. 1973;79:351–371. doi: 10.1016/0022-2836(73)90011-9. [DOI] [PubMed] [Google Scholar]
- 58.Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: An efficient program for end-state free energy calculations. J Chem Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 59.Samelson-Jones BJ, Yeh SR. Interactions between nitric oxide and indoleamine 2,3-dioxygenase. Biochemistry. 2006;45:8527–8538. doi: 10.1021/bi060143j. [DOI] [PubMed] [Google Scholar]
- 60.Peng YH, Ueng SH, Tseng CT, Hung MS, Song JS, Wu JS, Liao FY, Fan YS, Wu MH, Hsiao WC, Hsueh CC, Lin SY, Cheng CY, Tu CH, Lee LC, Cheng MF, Shia KS, Shih C, Wu SY. Important Hydrogen Bond Networks in Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Design Revealed by Crystal Structures of Imidazoleisoindole Derivatives with IDO1. J Med Chem. 2016;59:282–293. doi: 10.1021/acs.jmedchem.5b01390. [DOI] [PubMed] [Google Scholar]
- 61.Trevino SR, Schaefer S, Scholtz JM, Pace CN. Increasing Protein Conformational Stability by Optimizing β-Turn Sequence. J Mol Biol. 2007;373:211–218. doi: 10.1016/j.jmb.2007.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oehme DP, Brownlee RTC, Wilson DJD. Effect of atomic charge, solvation, entropy, and ligand protonation state on MM-PB(GB)SA binding energies of HIV protease. J Comput Chem. 2012;33:2566–2580. doi: 10.1002/jcc.23095. [DOI] [PubMed] [Google Scholar]
- 63.Terentis aC. The Heme Environment of Recombinant Human Indoleamine 2,3-Dioxygenase Structural properties and substrate-ligand interactions. J Biol Chem. 2002;277:15788–15794. doi: 10.1074/jbc.M200457200. [DOI] [PubMed] [Google Scholar]
- 64.Lu C, Lin Y, Yeh SR. Inhibitory substrate binding site of human indoleamine 2,3-dioxygenase. J Am Chem Soc. 2009;131:12866–12867. doi: 10.1021/ja9029768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee YT, Wilson RF, Rupniewski I, Goodin DB. P450cam visits an open conformation in the absence of substrate. Biochemistry. 2010;49:3412–3419. doi: 10.1021/bi100183g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Skinner SP, Liu WM, Hiruma Y, Timmer M, Blok A, Hass MAS, Ubbink M. Delicate conformational balance of the redox enzyme cytochrome P450cam. Proc Natl Acad Sci U S A. 2015;112:9022–9027. doi: 10.1073/pnas.1502351112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stoll S, Lee YT, Zhang M, Wilson RF, Britt RD, Goodin DB. Double electron–electron resonance shows cytochrome P450cam undergoes a conformational change in solution upon binding substrate. Proc Natl Acad Sci U S A. 2012;109:12888–12893. doi: 10.1073/pnas.1207123109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Capece L, Lewis-Ballester A, Yeh SR, Estrin Da, Marti Ma. Complete reaction mechanism of indoleamine 2,3-dioxygenase as revealed by QM/MM simulations. J Phys Chem B. 2012;116:1401–1413. doi: 10.1021/jp2082825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Castro E, Sigrist CJA, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006;34:W362–W365. doi: 10.1093/nar/gkl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fujigaki H, Seishima M, Saito K. Posttranslational modification of indoleamine 2,3-dioxygenase. Anal Bioanal Chem. 2012;403:1777–1782. doi: 10.1007/s00216-012-5946-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Structural alignment, sequence comparison, and additional MD results (PDF)