

ABSTRACT
A bimolecular fluorescence complementation (BiFC) approach was used to study the molecular interactions between different components of the postsynaptic protein complex at the neuromuscular junction of living mice. We show that rapsyn forms complex with both α-dystrobrevin and α-syntrophin at the crests of junctional folds. The linkage of rapsyn to α-syntrophin and/or α-dystrobrevin is mediated by utrophin, a protein localized at acetylcholine receptor (AChR)-rich domains. In mice deficient in α-syntrophin, in which utrophin is no longer present at the synapse, rapsyn interaction with α-dystrobrevin was completely abolished. This interaction was completely restored when either utrophin or α-syntrophin was introduced into muscles deficient in α-syntrophin. However, in neuromuscular junctions deficient in α-dystrobrevin, in which utrophin is retained, complex formation between rapsyn and α-syntrophin was unaffected. Using fluorescence recovery after photobleaching, we found that α-syntrophin turnover is 5-7 times faster than that of AChRs, and loss of α-dystrobrevin has no effect on rapsyn and α-syntrophin half-life, whereas the half-life of AChR was significantly altered. Altogether, these results provide new insights into the spatial distribution of dystrophin glycoprotein components and their dynamics in living mice.
KEY WORDS: AChR, Dystrophin glycoprotein complex, DGC, Neuromuscular junction, NMJ
Summary: Utrophin is a key component bridging rapsyn and AChRs to α-dystrobrevin and α-syntrophin in the AChR-rich domain, and we show that α-syntrophin and rapsyn turn over more rapidly than AChRs at the NMJ.
INTRODUCTION
In the nervous system, the stability of the postsynaptic scaffold protein complex at both central and peripheral synapses is crucial for the effectiveness of synaptic transmission. At the neuromuscular junction (NMJ) – the synapse between the motor neuron and muscle fiber – the dystrophin glycoprotein complex (DGC; comprises α-syntrophin, α-dystrobrevin, dystrophin, utrophin and dystroglycans), which connects the extracellular basal lamina to the intracellular cytoskeleton, plays an important role in preserving the structural integrity of the synapse and of the skeletal muscle fiber (Adams et al., 2000; Blake et al., 2002; Cohn and Campbell, 2000; Deconinck et al., 1997; Enigk and Maimone, 2001; Ervasti, 2007; Ervasti and Campbell, 1993; Straub and Campbell, 1997; Martinez-Pena y Valenzuela et al., 2011). Mutations in genes encoding one or more of the DGC components cause muscular dystrophy in both humans and animal models, and impair the structural integrity of the synapse (Durbeej and Campbell, 2002).
Rapsyn, a 43 kDa scaffold protein that is specifically localized to NMJs, is also required for the clustering of acetylcholine receptors (AChRs) at contacts between the nerve and muscles, and for the stability of other postsynaptic proteins at the synapse. In mice deficient in rapsyn, postsynaptic apparatus fails to form, and mice die immediately after birth (Gautam et al., 1995). In humans, mutations in the rapsyn gene cause the disassembly of the postsynaptic apparatus, which leads to neuromuscular diseases (Lochmuller et al., 2006; Muller et al., 2006). Likewise, in mice deficient in α-syntrophin or α-dystrobrevin, NMJs are structurally aberrant and exhibit a low level of AChRs. At the NMJ, the molecular interaction between different components of the synaptic apparatus has been extensively studied at the biochemical level (Murphy and Ohlendieck, 2016; Schmidt et al., 2011; Yoon et al., 2012). It has been shown that both α-syntrophin and α-dystrobrevin form complexes with utrophin (Yoon et al., 2012), which is confined only in the crests of the post-junctional folds that colocalize with the AChRs (Kramarcy and Sealock, 2000) and with dystrophin, which is concentrated only in the depths of the post-junctional folds (Bewick et al., 1996). However, the molecular interactions within and between DGC proteins and other scaffold proteins in their native environment in vivo remain largely unknown. In this work, we sought to address the dynamics of the molecular interaction between α-syntrophin, α-dystrobrevin and rapsyn, and whether these synaptic proteins are trafficked and inserted into the postsynaptic apparatus as individual proteins or as a pre-assembled complex.
In normal innervated muscle fibers, the AChRs are highly concentrated at the crests of the junctional folds, where they are held in clusters to ensure synaptic transmission. This stability of AChRs is established through an equilibrium between rates of removal and insertion of AChRs (Bruneau et al., 2005; Bruneau and Akaaboune, 2006; Martinez-Pena y Valenzuela et al., 2011). At functioning NMJs, receptor lifetime in the junctional membrane is quite long; however, the lifetime of DGC proteins at NMJs remains unknown.
Using bimolecular fluorescence complementation (BiFC) assays and fluorescence recovery after photobleaching (FRAP), we show that DGC components are distributed in a spatially ordered fashion, and that α-syntrophin and rapsyn are highly dynamic within a stable structure at the postsynaptic apparatus of the synapse.
RESULTS
Spatial distribution of rapsyn, AChRs and DGC components at the NMJ of a living mouse
To visualize the molecular interactions between components of the dystrophin glycoprotein complex – particularly those of α-syntrophin and α-dystrobrevin within the complex – and rapsyn in their native environment at the NMJ, we used a BiFC assay (Hu and Kerppola, 2003). A series of BiFC constructs were generated containing α-syntrophin, α-dystrobrevin and rapsyn fused to either N-terminal (VN173) or C-terminal (VC155) fragments of Venus fluorescent protein (see Materials and Methods; Fig. 1A). For each DGC component, a pair of constructs containing the N- (VN) and C- (VC) terminal fragments of Venus protein were then electroporated into the sternomastoid muscle of a wild-type animal to test for self-interaction as a positive BiFC signal (Ramarao et al., 2001). At 7 days after electroporation, the sternomastoid muscle was exposed, and postsynaptic nicotinic AChRs (marker of the synapse) were labeled with α-bungarotoxin conjugated to Alexa-Fluor-594 (BTX–Alexa594, red). Muscles were then fixed with paraformaldehyde (PFA), and synapses expressing BiFC signals were imaged with a confocal microscope. Rapsyn–VN and rapsyn–VC exhibited a strong BiFC signal, which was restricted to the crest of junctional folds where it precisely colocalizes with AChRs, consistent with the localization of rapsyn as the BiFC signal showed the same pattern as that of rapsyn–GFP signal (Fig. 1B,C). However, when constructs containing the N- (VN) and C- (VC) terminal fragments of Venus protein alone were electroporated into the sternomastoid muscle, no fluorescence was observed at the synapses. α-syntrophin–VN and α-syntrophin–VC BiFC signals were present on both AChR-rich crests and at the bottom of the postsynaptic folds (shown by the lack of overlap between green with red signals), consistent with the localization of α-syntrophin, as shown by α-syntrophin–GFP signal (Fig. 1D,E). These results indicate that these BiFC constructs behave like their respective GFP fusions and that they are expressed at their proper locations.
Fig. 1.

In vivo BiFC shows self-interaction between rapsyn–VN and rapsyn–VC, and between α-syntrophin–VN and α-syntrophin–VC at the NMJ. (A) Schematic representation of BiFC assay. Association of two non-fluorescent protein fragments that are fused to proteins; when they come close to each other, the native three-dimensional structure is formed and emits a fluorescence signal. (B–E) Sternomastoid muscles were electroporated with constructs expressing either rapsyn–VN and rapsyn–VC, α-syntrophin–VC (Synt–VC) and α-syntrophin–VN (Synt–VN), rapsyn–GFP, or α-syntrophin–GFP, and NMJs were identified by labeling AChRs with BTX–Alexa594 (red). (B) A representative image showing BiFC signal of rapsyn–VC and rapsyn–VN. (C) A representative image of an NMJ expressing rapsyn–GFP fusion protein. Note that BiFC and rapsyn–GFP signals localized perfectly with AChRs labeled with BTX–Alexa594 at synaptic sites (see arrowheads). (D) A representative image showing the BiFC signal of α-syntrophin–VC with α-syntrophin–VN. (E) A representative image of an NMJ expressing α-syntrophin–GFP. Note that BiFC and GFP signals are present in crests and at the bottom of the junctional folds (see arrows). Six mice were used in each experiment. Scale bars: 5 µm.
Next we examined whether rapsyn, a protein that has a direct binding site for AChRs, interacts with either or both of α-syntrophin and α-dystrobrevin. To do this, rapsyn–VC and syntrophin–VN, or rapsyn–VC and α-dystrobrevin–VN fusion constructs were co-electroporated into sternomastoid muscle. After 7 days, muscles were bathed with BTX–Alexa594, fixed with paraformaldehyde (PFA), and NMJs expressing BiFC signals were imaged with a confocal microscope. A strong BiFC signal was observed specifically at AChR-rich domains (crests of the junctional folds only) between rapsyn and α-syntrophin (Fig. 2A) or between rapsyn and α-dystrobrevin (Fig. 2B) but not in the bottom of the folds as no green signal was observed outside AChR boundaries. This result indicates that rapsyn specifically interacts with both α-syntrophin and α-dystrobrevin at the crests of the junctional folds. Similarly, specific BiFC signals were observed between a VN-tagged muscle-specific CaMKIIβm isoform (Martinez-Pena y Valenzuela et al., 2010) and rapsyn–VC at the crests of the junctional folds (Fig. 2D). These results indicate the specificity of BiFC signals of electroporated constructs.
Fig. 2.
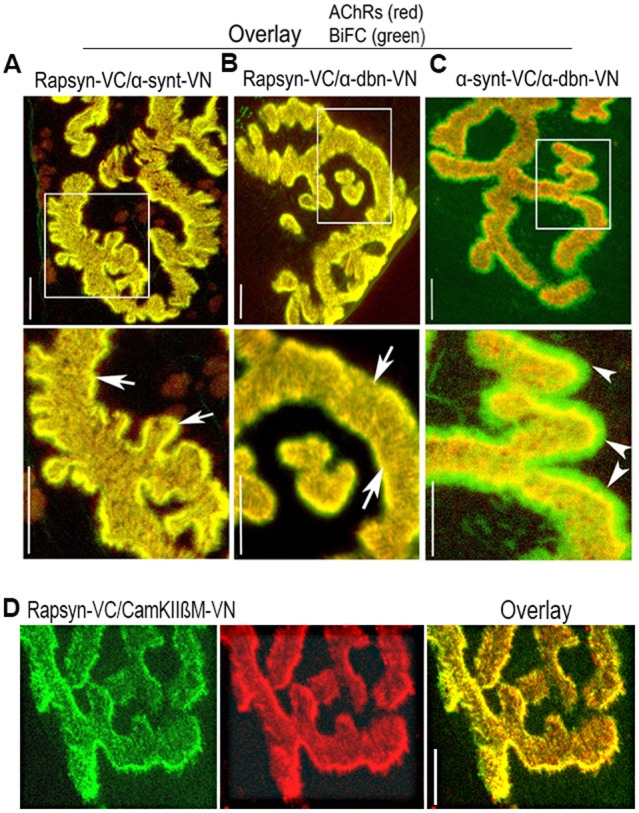
In vivo BiFC shows interaction between rapsyn, α-dystrobrevin, α-syntrophin and CamKIIβM at the NMJ. Sternomastoid muscles were co-electroporated either with rapsyn–VC and α-syntrophin–VN (α-synt–VN), or rapsyn–VC and α-dystrobrevin–VN (α-dbn–VN), or α-dystrobrevin–VN and α-syntrophin–VC (α-synt–VC); 7 days later, muscles were bathed with BTX–Alexa594 to label AChRs, and synapses expressing BiFC signals were imaged. (A) A representative synapse showing that the fluorescence complementation between rapsyn–VC and α-syntrophin–VN is restricted to the crests of junctional folds where they precisely colocalized with AChRs (see arrows). (B) A representative synapse image showing fluorescence complementation between rapsyn–VC and α-dystrobrevin–VN (α-dbn–VN) only at the crests of junctional folds that were identified by AChR labeling (see arrows). (C) A representative synapse image showing the fluorescence complementation between α-syntrophin and α-dystrobrevin at both the crest and troughs of junctional folds (see arrowheads). Boxed areas (above) are enlarged in the images below. (D) A representative image showing the BiFC signal between rapsyn–VC and CamKIIβM–VN. Scale bars: 5 µm. Eight mice were used in each experiment.
Next we examined the spatial distribution of α-dystrobrevin and α-syntrophin in the postsynaptic apparatus. α-Dystrobrevin–VN and α-syntrophin–VC fusion constructs were co-electroporated into the sternomastoid muscle, and the BiFC signal at the NMJ was imaged. As shown in Fig. 2C, a strong BiFC signal was observed in both the troughs and the crests of the folds (as green signal was observed outside AChR boundaries), indicating a broad distribution of the α-dystrobrevin–α-syntrophin complex, as opposed to a specific concentration of these proteins at the crest of the folds when complexed with rapsyn.
Having found that the fluorescence complementation between rapsyn and α-syntrophin or α-dystrobrevin at the crest of the folds, we wanted to know whether the association of these components with rapsyn was direct or via a third protein. If α-syntrophin and α-dystrobrevin interact with rapsyn at the same site, one should expect to see a decrease of α-dystrobrevin–rapsyn BiFC signal when a competing high concentration of α-syntrophin–VN construct is electroporated and vice versa. To examine this, we performed an in vivo competition assay using a multicolor BiFC approach. The sternomastoid muscle was co-electroporated with a fixed amount of rapsyn tagged with cyan (rapsyn–CC) (5 µg), and either equal amounts of α-syntrophin–VN (5 µg) and cerulean-tagged α-syntrophin (5 μg)–rapsyn (5 μg)–α-dystrobrevin (5 µg) (S5-R5-D5) or α-syntrophin (5 µg)–rapsyn (5 μg)–α-dystrobrevin (10 µg) (S5-R5-D10), or α-syntrophin (10 µg)–rapsyn (5 μg)–α-dystrobrevin (5 µg) (S10-R5-D5). Complementation between α-Syn–VN and rapsyn–CC generate a green BiFC signal, whereas α-dystrobrevin–CrN and rapsyn–CC generate a cyan BiFC signal. As shown in Fig. 3A,B, we found that BiFC signals were unaffected by changing the concentrations of electroporated constructs (α-syn–VN and α-dystrobrevin–CrN; one-way ANOVA, P=0.89). Quantification of the green and cyan intensity ratio values at synapses expressing BiFC signals at different concentrations showed that the ratio remained roughly the same (Fig. 3B). These results strongly suggest that α-syntrophin and α-dystrobrevin interact with rapsyn, either at different sites or via a third protein (see below).
Fig. 3.
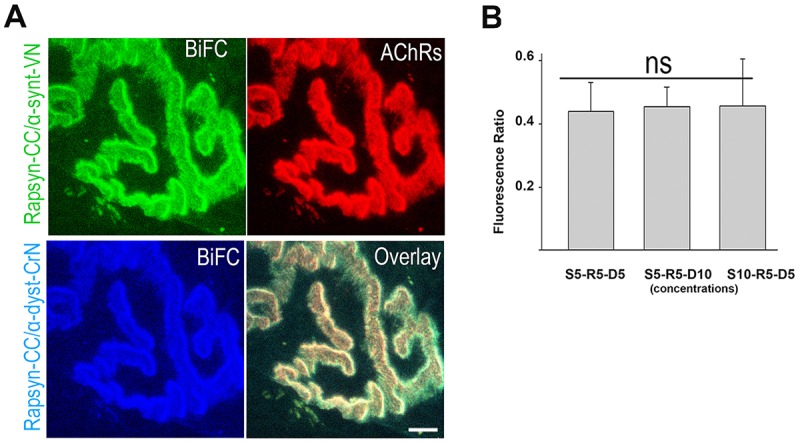
Rapsyn interacts with both α-syntrophin and α-dystrobrevin. The sternomastoid muscle was co-electroporated with a fixed amount of rapsyn–CC with one of the following: (i) the same amount of α-syntrophin–VN and α-dystrobrevin–CrN (α-dyst–CrN) (S5-R5-D5); (ii) a low concentration of α-syntrophin–VN (α-synt–VN) and high concentration of α-dystrobrevin–CrN (S5-R5-D10); (iii) or a high concentration of α-syntrophin–VN and low concentration of α-dystrobrevin–CrN (S10-R5-D5). After 7 days, the muscle was bathed with BTX–Alexa594 to label AChRs, fixed and imaged. (A) A representative image of a synapse showing the complementation of rapsyn–CC with α-syntrophin–VN, and rapsyn–CC with α-dystrobrevin–CrN (α-dbn-CrN). (B) Graph showing quantification of BiFC signals between rapsyn–CC and α-syntrophin–VN and rapsyn–CC and α-dystrobrevin–CrN at different concentrations. Note that the fluorescence ratio value between green and cyan remains the same between different constructs concentrations. Each bar represents the mean±s.d. of 40 junctions (six mice). Scale bar: 5 µm. One-way ANOVA test, P=0.89; ns, not significant. R, rapsyn; S, α-syntrophin; D, α-dystrobrevin; numbers indicate the amount of plasmid (µg).
Utrophin is required for the formation of a rapsyn and α-dystrobrevin complex at the synapse
Given the fact that α-dystrobrevin forms a complex with α-syntrophin and that rapsyn forms complexes with α-syntrophin and α-dystrobrevin, we wanted to know whether the BiFC complementation of rapsyn with these two DGC components requires the presence of either these components or others in their native environment. To address this, we used α-syntrophin- and α-dystrobrevin-knockout mice. When α-syntrophin–VN and rapsyn–VC constructs were electroporated into sternomastoid muscles of mice deficient in α-dystrobrevin, a strong BiFC signal was generated at the synapse, similar to the one observed at synapses in wild-type mice, indicating that α-dystrobrevin is not required for the interaction between rapsyn and α-syntrophin (Fig. 4A). In contrast, when α-dystrobrevin–VN and rapsyn–VC constructs were electroporated into sternomastoid muscle of mice deficient in α-syntrophin, BiFC signals were no longer visible at synapses (Fig. 4B). The fluorescence complementation of rapsyn with α-dystrobrevin was completely restored at NMJs deficient in α-syntrophin when sternomastoid muscles of these mutant mice were co-electroporated with α-dystrobrevin–CrN, rapsyn–CC and wild-type α-syntrophin–GFP (Fig. 4C). This result led us to investigate whether there is an intermediate protein that is controlled by α-syntrophin and that is required for the BiFC complementation between rapsyn and α-dystrobrevin, and/or between rapsyn and α-syntrophin. Previous studies have shown that in the adult synapses, both α-syntrophin and α-dystrobrevin bind to utrophin, a major component of the dystrophin glycoprotein complex that is highly concentrated at the crest of AChR-rich postsynaptic membranes (Bewick et al., 1996) and to dystrophin in the troughs of the junctional folds with roughly equal levels. In mice lacking α-syntrophin, utrophin is completely lost from the postsynaptic membrane, whereas it remains concentrated at crests of AChR-rich folds in mice deficient in α-dystrobrevin (Grady et al., 2000). These observations prompted us to examine whether utrophin, which is present at crests of synaptic folds, is the protein that links rapsyn to α-dystrobrevin. To do this, the sternomastoid muscle of mice deficient in α-syntrophin was electroporated with α-dystrobrevin–CrN, rapsyn–CC and utrophin constructs, and 7 days later muscles were fixed with PFA and immunostained with antibody against utrophin, and the BiFC signal between α-dystrobrevin and rapsyn was evaluated at electroporated synapses. Fig. 5A showed strong BiFC signals only at synapses where utrophin was expressed, but these signals were not present in neighboring synapses that did not express utrophin (Fig. 5B). However, when α-dystrophin (instead of a utrophin construct) was electroporated into sternomastoid muscle deficient in α-syntrophin, no BiFC signal between rapsyn–VC and α-dystrobrevin–VN was observed (data not shown). Taken together, these results indicate that utrophin is required to establish the molecular interaction between α-dystrobrevin and rapsyn.
Fig. 4.
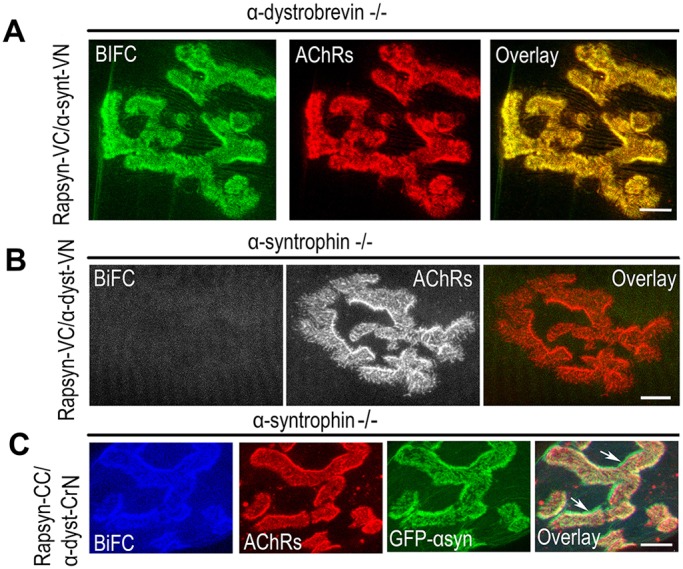
In vivo BiFC complementation between rapsyn and α-dystrobrevin is abolished at NMJs of mice deficient in α-syntrophin but not in synapses deficient in α-dystrobrevin. (A) The sternomastoid muscle of mice deficient in α-dystrobrevin was electroporated with rapsyn–VC and α-syntrophin–VN constructs (α-synt–VN); 7 days later, AChRs on muscles were labeled with BTX–Alexa594, fixed and imaged. A representative synapse expressing the BiFC signal is shown. Note that the BiFC signal at the NMJ is not impaired by the absence of α-dystrobrevin. (B) A representative synapse from the sternomastoid muscle of mice deficient in α-syntrophin that had been co-electroporated with rapsyn–VC and α-dystrobrevin–VN (α-dyst–VN) constructs were bathed with BTX–Alexa594 to label AChRs. Note the complete absence of BiFC signal at the NMJ. (C) A representative synapse from the sternomastoid muscle of mice deficient in α-syntrophin that had been electroporated with rapsyn–CC, α-dystrobrevin–CrN (α-dyst–CrN) and α-syntrophin–GFP (GFP–αsyn). Note that the BiFC signal at the synapses was rescued at NMJs expressing α-syntrophin–GFP. AChRs were labeled with BTX–Alexa594. Scale bars: 5 µm. Seven mice were used in each experiment.
Fig. 5.
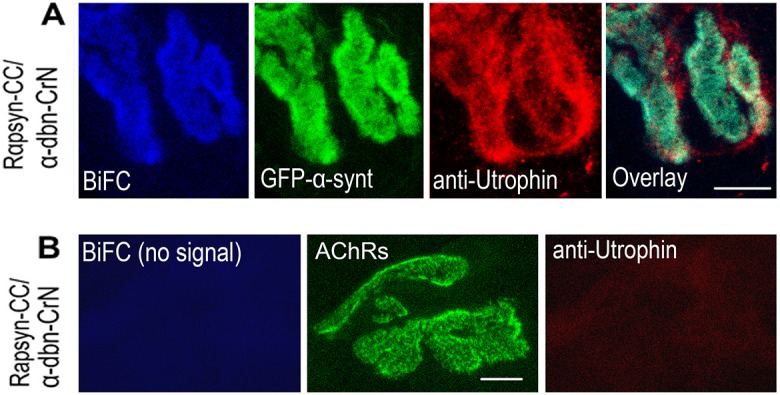
Utrophin expression is required for the fluorescence complementation between rapsyn and α-syntrophin and α-dystrobrevin. The sternomastoid muscle of mice deficient in α-syntrophin was co-electroporated with rapsyn–CC, α-dystrobrevin–CrN (α-dbn–CrN), α-syntrophin–GFP (GFP–α-synt) and utrophin. (A) Examples of high-resolution confocal images of an NMJ expressing α-syntrophin–GFP and BiFC signal between rapsyn–CC and α-dystrobrevin–CrN are shown. (B) Examples of an NMJ imaged from the sternomastoid muscle of mice deficient in α-syntrophin that had been co-electroporated with rapsyn–CC and α-dystrobrevin–CrN, and 7 days after electroporation, muscles were fixed and stained with antibody against utrophin and BTX–Alexa488. Note that there is no fluorescence complementation between rapsyn and α-dystrobrevin and no utrophin expression in the absence of α-syntrophin. Scale bars: 5 µm. Eight mice were used in each experiment.
Finally, we asked whether components of the DGC are inserted into the postsynaptic sites as pre-assembled complexes or as individual proteins, notably α-syntrophin and α-dystrobrevin and rapsyn. As above, the sternomastoid muscle was electroporated with a combination of BiFC DGC and rapsyn constructs, and 7 days later, the sternomastoid muscle was removed, fixed and imaged. If α-syntrophin or α-dystrobrevin is inserted as a complex with rapsyn, one should expect the presence of BiFC signals both on AChR-rich crests and at the bottom of the postsynaptic folds (since α-syntrophin and α-dystrobrevin are localized at both the crest and bottom of the NMJ). However, Fig. 6 shows that when rapsyn and α-syntrophin were co-electroporated, BiFC signals were restricted to the crests of the folds, indicating that these proteins are targeted to synaptic sites individually rather than as pre-assembled complexes.
Fig. 6.

Postsynaptic rapsyn, α-syntrophin and α-dystrobrevin are targeted to their specific locations as individual proteins. The sternomastoid muscle was electroporated with either rapsyn–VC and α-syntrophin–VN (Rapsyn–VC/α-synt-VN) or with α-syntrophin–VC and α-dystrobrevin–VN (α–synt–VC/α–dyst-VN). The muscle was bathed with BTX–Alexa594 to label AChRs, then fixed and imaged. (A) A representative image of a synapse showing fluorescence complementation between rapsyn and α-syntrophin at the crest of synaptic folds only, as indicated by the perfect colocalization of BiFC signal with AChR at the crest of synaptic folds of the synapses. (B) Example of a high-resolution confocal image of an NMJ expressing the BiFC signal of α-dystrobrevin and α-syntrophin at the crests and bottom of the junctional folds (see arrows). Scale bars: 5 µm. Six mice were used in each experiment.
The dynamics of α-syntrophin and rapsyn at wild-type and α-dystrobrevin-deficient NMJs
The molecular dynamics of DGC components at NMJs of living mice are unknown. Here, we wanted to determine the half-life of α-syntrophin and compared it to that of rapsyn and AChR at functioning NMJs. The sternomastoid muscle of adult wild-type mice was electroporated with either α-syntrophin–GFP or rapsyn–GFP. Two weeks after electroporation, synapses expressing fluorescent signals were imaged, and discrete areas of synapses were carefully photo-bleached with an argon laser. The recovery of fluorescence at bleached areas was measured and normalized to unbleached areas of the same synapses, as described in our previous work (Bruneau and Akaaboune, 2010). In mice that had been electroporated with α-syntrophin–GFP, we found that the recovery of fluorescence at bleached regions after 24 h was 49.8±6% (mean±s.d.) of the original fluorescence (after normalization; corresponding to a half-life of 1.02±0.21 days, n=21 NMJs, five mice; mean±s.d.). Similar to α-syntrophin, the recovery of fluorescence of rapsyn–GFP at bleached areas after 24 h was 47±9% of the original fluorescence (half-life of 1.16±0.36 days, n=14 NMJs, three mice; P=0.37) (Fig. 7A,B). We also found that the turnover rate of AChR was very slow (half-life of about 9–12 days), regardless of expression levels of α-syntrophin–GFP or rapsyn–GFP (low or high) at the synapse. This indicates that the dynamics of AChR are unaffected by exogenous expression of rapsyn or α-syntrophin (data not shown), consistent with previous studies (Bruneau and Akaaboune, 2010).
Fig. 7.

The DGC α-syntrophin half-life at wild-type and α-dystrobrevin mutant NMJs. Sternomastoid muscles of live wild-type and α-dystrobrevin mutant mice that had been electroporated with either rapsyn–GFP (rap–GFP) or α-syntrophin–GFP (α-syn–GFP); 7 days later, the fluorescence from discrete regions of NMJs expressing GFP was removed with a laser and re-imaged. The recovery of fluorescence was monitored over 24 h. Notice that only images of sternomastoid muscles electroporated with α-syntrophin–GFP are shown. (A) Example of a wild-type NMJ that was electroporated with α-syntrophin–GFP, imaged at time 0 and immediately bleached and re-imaged at 24 h (eight mice). (B) Graph summarizing data of the half-time of α-syntrophin, rapsyn and AChRs obtained from 47 synapses. (C) Example of NMJ deficient in α-dystrobrevin expressing α-syntrophin–GFP that was imaged at time 0 and then immediately bleached and re-imaged at 24 h. (D) Graph summarizing data of half-time of α-syntrophin, rapsyn and AChRs obtained from 42 mutant synapses (12 mice). All data represent mean±s.d. Scale bars: 10 µm.
In our previous studies, we have shown that the half-life of AChR is significantly reduced in synapses deficient in α-dystrobrevin (Akaaboune et al., 2002). Here, we asked whether the half-lives of α-syntrophin and rapsyn are altered by the loss from the postsynaptic apparatus of the DGC member α-dystrobrevin. The sternomastoid muscle of mice deficient in α-dystrobrevin was electroporated with either α-syntrophin–GFP or rapsyn–GFP, and 24 h after photobleaching, the recovery of GFP fluorescence into bleached synaptic areas was measured (the recovery of fluorescence for rapsyn–GFP was 47.6±9%, n=6 mice; the recovery for α-syntrophin–GFP was 43.00±10%, n=6 mice). It is clear that the half-lives of rapsyn (t1/2∼1.3 days) and α-syntrophin (t1/2∼1.25 days) were not significantly different from those of wild-type synapses (P=0.12). However, as expected, the half-life of AChRs at synapses of α-dystrobrevin was significantly reduced (t1/2∼1–2 days) compared to 9–12 days in wild-type animals (P<0.0001) (Fig. 7A-D) (Akaaboune et al., 2002). These results indicate that the loss of α-dystrobrevin has no effect on the dynamics of intracellular postsynaptic proteins (at least the ones investigated here) but that it does significantly alter the dynamics of AChRs.
DISCUSSION
In this study, we took advantage of the BiFC approach to examine the spatial distribution of key components of the dystrophin glycoprotein complex and their interaction with the scaffold protein rapsyn in their native environment. The major findings are that (1) α-dystrobrevin and α-syntrophin form complexes with rapsyn in the AChR-rich domain but are also present in the bottom of junctional folds (Na+channel-rich domain); (2) utrophin is a key component linking rapsyn to α-dystrobrevin and α-syntrophin; (3) rapsyn, α-dystrobrevin and α-syntrophin are inserted into the postsynaptic density as individual proteins rather than as a pre-assembled complex; (4) α-syntrophin is highly dynamic with a turnover rate much more rapid than AChRs, but similar to that of scaffold protein rapsyn; (5) the loss of α-dystrobrevin has no effect on the turnover rate of rapsyn and α-syntrophin but significantly increases the AChR turnover rate. Taken together, these results provide new insights into the spatial distribution of key components of the dystrophin glycoprotein complex and their dynamics in their native environment at the NMJ.
Immunocytochemical studies are capable of demonstrating whether two molecules are near each other, but they do not test whether they actually interact. For this reason, most previous knowledge of postsynaptic molecular interactions at the NMJ is based on biochemical studies (pull down or immunoprecipitation). Here, we exploited the properties of BiFC to address the molecular interactions between proteins in the postsynaptic apparatus of the NMJ. The interactions we demonstrated with BiFC are consistent with those identified by immunocytochemistry labeling (Ahn et al., 1996; Kramarcy and Sealock, 2000). The fact that BiFC signals between rapsyn–α-syntrophin and rapsyn–α-dystrobrevin were obtained only in the crest of junctional folds where rapsyn is confined, but not in the bottom of synaptic folds, suggests that DGC components, along with rapsyn, are inserted as individual components rather than as pre-assembled complexes, as evidenced by the confinement of BiFC signals at AChR-rich areas (Fig. 6).
Previous studies have shown that utrophin, a major component of the DGC, is strictly restricted to the crest of the folds and that α-syntrophin is important for utrophin stabilization and trafficking to the NMJ (Adams et al., 2010). Interestingly, synapses of mice deficient in utrophin are smaller and tend to be fragmented, and the number of AChRs was slightly reduced; the number of folds was also reduced by ∼50% compared to in utrophin wild-type synapses (Deconinck et al., 1997; Grady et al., 1997). The current work shows that utrophin is required for the BiFC complementation of rapsyn with α-dystrobrevin. This conclusion is based on the following findings: (i) in mice deficient in α-syntrophin, in which utrophin is lost from synapses, the rapsyn–α-dystrobrevin BiFC signal was completely abolished; (ii) in α-syntrophin-deficient muscle cells, electroporation of either utrophin or α-syntrophin–GFP completely restored the complementation signals between rapsyn and α-dystrobrevin at NMJs of only electroporated muscles, supporting previous results showing that even only a small amount of exogenous wild-type α-syntrophin in α-syntrophin−/− NMJs is sufficient to re-establish utrophin at the NMJ (Adams et al., 2010); (iii) in mice deficient in α-dystrobrevin, in which utrophin is retained at synapses, the rapsyn–α-syntrophin BiFC signal remained unaffected. Collectively, these observations indicate that utrophin is a core component of a structural building block that correctly localizes α-syntrophin and α-dystrobrevin in their native environments at the postsynaptic domain of NMJs, and that utrophin in partnership with rapsyn may serve as a backbone for consolidating postsynaptic domains of NMJs. Thus, it is reasonable to suggest that utrophin is a key component in linking AChR–rapsyn complexes at junctional folds to lateral DGCs, and to α-syntrophin and α-dystrophin along the sides of the folds, and this orderly spatial distribution of scaffold protein is critical not only for the anchorage and stability of AChRs, but possibly for connecting the endplate region with sub-synaptic myonuclei and other organelles through intermediate filaments and microtubules.
Taking advantage of GFP fusion constructs and FRAP, the current experiments show that, in sharp contrast to the slow turnover rate of AChRs (Akaaboune et al., 1999), α-syntrophin in intracellular DGCs is highly dynamic with a half-life of about a day, similar to the turnover rate of the AChR-associated scaffold protein rapsyn (Bruneau and Akaaboune, 2010). It is unlikely that the GFP fusion protein has an effect on the stability of α-syntrophin and rapsyn as these proteins functioned like the endogenous proteins, specifically, they clustered and colocalized with receptors at NMJs, and their expression did not alter the clustering and stability of AChR. Similar to peripheral neuromuscular synapses, intracellular scaffold proteins that anchor postsynaptic receptors at excitatory and inhibitory synapses in the central nervous system have a high turnover rate, as determined by FRAP (Gray et al., 2006; Hanus et al., 2006; Okabe et al., 2001; Rasse et al., 2005). From these studies and others, it is clear that the dystrophin glycoprotein complex proteins α-syntrophin and rapsyn are highly dynamic despite the overall stability of the NMJ, and such dynamism could serve as the basis for synaptic plasticity.
An interesting finding of this work is that the half-life of AChR is significantly impaired by the loss of α-dystrobrevin, but the half-lives of rapsyn and α-syntrophin do not change, even though they are in the same postsynaptic membrane. It is well documented that in mice deficient in either α-dystrobrevin or α-syntrophin, the turnover rate of AChR is significantly increased (Grady et al., 2003; Martinez-Pena y Valenzuela et al., 2011). Thus, it is possible that the stability of membrane receptors at synaptic sites depends on their tethering by postsynaptic proteins, while the stability of intracellular synaptic proteins within the postsynaptic apparatus is insensitive to changes in the makeup of postsynaptic components but regulated, instead, by other factors.
MATERIALS AND METHODS
Generation of BiFC constructs
To generate our BiFC constructs, we used the following vectors from Addgene: pBiFC-VN173, pBiFC-VC155, pBiFC-CrN173 and pBiFC-CC155. The Venus or cerulean cyan fragments (VN or CrN: amino acids 1–172 or VC or CC: amino acids 155-238) were fused to the C-terminal end of our protein of interest (rapsyn, α-syntrophin or α-dystrobrevin). As templates for PCR, we used rapsyn–GFP, α-syntrophin–GFP and α-dystrobrevin–GFP. We first cloned our protein-of-interest coding sequences by PCR into pBiFC-VN173 or pBiFC-VC155 using the primers indicated below, and then subcloned these sequences from pBiFC-VN173 to pBiFCCrN173 or from pBiFC-VC155 to pBiFC-CC155 for multicolor BiFC. pEDT-FLAG-Utrophin and pEDT-FLAG-Dystrophin plasmids were gifts from Dr Froenher (University of Washington, Seattle).
The primers for VN fusions were: Rapsyn: forward, 5′-TAAGCTTATGGGGCAGGAACCAGACCAAGCAGCAGATC; reverse, 5′-TATCTAGATACAAAGCCAGGCTTCATGGATGAGCGG (restriction sites HindII and Xbal); α-syntrophin: forward, 5′-TAAGCTTATGGCATCAGGCAGGCGCACTCCGCGCACC; reverse, 5′-AATTCTAGAGGCCAAGAGCCCCAAGCGGGTGACCTTGGC (restriction sites HindII and Xbal); α-dystrobrevin: forward, 5′-TATGATATCGATGATTGAAGATAGTGGAAAAAGAGGAAACACC; reverse, 5′-TTAGTCGACTCTAGAACCTTGCAAGCTGACCTGGTAGGCC (restriction sites EcoRV and SaII).
The primers for VC fusions were: Rapsyn: forward, 5′-TATGAATTCCGATGGGGCAGGACCAGACCAAGCAGCAGATC; reverse, 5-TTAGTCGACCATACAAAGCCAGGCTTCATGGATGAGCGG (restriction sites EcoRI and SaII); α-syntrophin: forward, 5-TATAAGCTTATGGCATCAGGCAGGCGCACTCCGCGCACC; reverse, 5-AATTCTAGAGGCCAAGAGCCCCAAGCGGGTGACCTTGGC (restriction sites EcoRV and SaII).
Electroporation of constructs into the sternomastoid muscle and confocal microscopy
Adult female non-Swiss albino mice (3 months old) and age-matched female mice deficient in α-dystrobrevin were anesthetized by injecting intraperitoneally a mixture of 80 mg/kg of body weight of ketamine and 20 mg/kg of body weight xylazine, and the sternomastoid muscle was surgically exposed. The solution containing the plasmid (10 μg) encoding the construct of interest, was placed over the sternomastoid muscle, as described in our previous work (Bruneau and Akaaboune, 2010; Martinez-Pena y Valenzuela et al., 2015). Gold electrodes were set parallel to the muscle fibers on either side of the muscle, and then eight monopolar square-wave pulses were applied perpendicularly to the long axis of the muscle. The animal was sutured and allowed to recover in a warm chamber. All animal usage methods were approved by the University of Michigan Institutional Animal Care and Use Committee.
Immunofluorescence and confocal microscopy
Sternomastoid muscle of mice that had been electroporated with the different constructs was bathed with a saturating dose of BTX–Alexa594 (5 μg/ml for 1 h) and then perfused with 2% PFA. The sternomastoid muscles were removed, and NMJs were imaged with a confocal scanning laser microscope (Leica SP5) using an HCX Plan Apochromat 100× objective (NA 1.46) and a resolution of 1024×1024 pixels. The z-stacks were then collapsed, processed with Leica confocal software, and the contrast was adjusted with Photoshop.
For immunocytochemistry, the sternomastoid muscle of α-syntrophin mutant mice was co-electroporated with rapsyn–CC and α-dystrobrevin–CrN, and either α-syntrophin–GFP or utrophin constructs. Seven days later, muscles were fixed with PFA, teased fiber bundles were permeabilized and then incubated overnight with primary monoclonal antibody against utrophin [5 µg/ml; MANCHO13(2G12); Developmental Studies Hybridoma Bank, University of Iowa) (Morris et al., 1998)]. Muscle cells were then bathed with Alexa-Fluor-594-conjugated anti-mouse secondary antibody (catalog no. A-11005, Molecular Probes; 10 µg/ml) for 1 h, mounted on a coverslip and imaged with a Leica SP5 confocal microscope.
FRAP experiments and quantitative fluorescence imaging
To determine the half-life of α-syntrophin and rapsyn, sternomastoid muscles were electroporated with the above GFP constructs, and NMJs expressing GFP were imaged; then discrete regions of individual synapses were bleached with an argon laser and re-imaged immediately. After 24 h, the recovery of green fluorescence was quantified both at the bleached region and the unbleached region. GFP recovery after bleaching was normalized to the non-bleached sections of the same synapse.
To determine whether the loss of α-dystrobrevin has an effect on the half-lives of rapsyn and α-syntrophin, the sternomastoid muscle of mice deficient in α-dystrobrevin was electroporated with rapsyn–GFP or α-syntrophin–GFP, and NMJs expressing GFP were bleached as described above. The half-life of each GFP-tagged construct was compared to the half-life of AChR.
The half-life of AChR was determined as previously described in our published work (Akaaboune et al., 1999; Martinez-Pena y Valenzuela et al., 2015). Briefly, the sternomastoid muscle was bathed with a non-saturating (2 μg/ml, 2 min) dose of BTX–Alexa594, and the fluorescence intensity of labeled receptors was assayed using a quantitative fluorescence imaging technique, as described in previous work (Akaaboune et al., 1999; Turney et al., 1996).
Statistical analysis
Data are shown as means±s.d. Quantitative comparisons of numerical datasets were tested for statistical significance by using t-test or one-way ANOVA test.
Acknowledgements
We thank Drs Richard Hume, Ryan Insolera (University of Michigan) for critical comments on the manuscript. We also thank Akaaboune laboratory members (Po-Ju Chen, Lyna Azzouz and Bavica Gummadi) for technical assistance and comments on the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: M. Aittaleb, M. Akaaboune; Methodology: M. Aittaleb, I.M.; Validation: M. Aittaleb, I.M.; Formal analysis: M. Aittaleb, I.M.; Writing - original draft: M. Aittaleb; Writing - review & editing: I.M., M. Akaaboune; Visualization: I.M.; Supervision: M. Akaaboune; Project administration: M. Akaaboune; Funding acquisition: M. Akaaboune.
Funding
This work was supported by the National Institutes of Health [grant number NS-047332 and NS-082615], and MCube (University of Michigan) to M. Akaaboune. Deposited in PMC for release after 12 months.
References
- Adams M. E., Kramarcy N., Krall S. P., Rossi S. G., Rotundo R. L., Sealock R. and Froehner S. C. (2000). Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J. Cell Biol. 150, 1385-1398. 10.1083/jcb.150.6.1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams M. E., Anderson K. N. E. and Froehner S. C. (2010). The alpha-syntrophin PH and PDZ domains scaffold acetylcholine receptors, utrophin, and neuronal nitric oxide synthase at the neuromuscular junction. J. Neurosci. 30, 11004-11010. 10.1523/JNEUROSCI.1930-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn A. H., Freener C. A., Gussoni E., Yoshida M., Ozawa E. and Kunkel L. M. (1996). The three human syntrophin genes are expressed in diverse tissues, have distinct chromosomal locations, and each bind to dystrophin and its relatives. J. Biol. Chem. 271, 2724-2730. 10.1074/jbc.271.5.2724 [DOI] [PubMed] [Google Scholar]
- Akaaboune M., Culican S. M., Turney S. G. and Lichtman J. W. (1999). Rapid and reversible effects of activity on acetylcholine receptor density at the neuromuscular junction in vivo. Science 286, 503-507. 10.1126/science.286.5439.503 [DOI] [PubMed] [Google Scholar]
- Akaaboune M., Grady R. M., Turney S., Sanes J. R. and Lichtman J. W. (2002). Neurotransmitter receptor dynamics studied in vivo by reversible photo-unbinding of fluorescent ligands. Neuron 34, 865-876. 10.1016/S0896-6273(02)00739-0 [DOI] [PubMed] [Google Scholar]
- Bewick G. S., Young C. and Slater C. R. (1996). Spatial relationships of utrophin, dystrophin, beta-dystroglycan and beta-spectrin to acetylcholine receptor clusters during postnatal maturation of the rat neuromuscular junction. J. Neurocytol. 25, 367-379. [PubMed] [Google Scholar]
- Blake D. J., Weir A., Newey S. E. and Davies K. E. (2002). Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 82, 291-329. 10.1152/physrev.00028.2001 [DOI] [PubMed] [Google Scholar]
- Bruneau E. G. and Akaaboune M. (2006). The dynamics of recycled acetylcholine receptors at the neuromuscular junction in vivo. Development 133, 4485-4493. 10.1242/dev.02619 [DOI] [PubMed] [Google Scholar]
- Bruneau E. G. and Akaaboune M. (2010). Dynamics of the rapsyn scaffolding protein at the neuromuscular junction of live mice. J. Neurosci. 30, 614-619. 10.1523/JNEUROSCI.4595-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau E., Sutter D., Hume R. I. and Akaaboune M. (2005). Identification of nicotinic acetylcholine receptor recycling and its role in maintaining receptor density at the neuromuscular junction in vivo. J. Neurosci. 25, 9949-9959. 10.1523/JNEUROSCI.3169-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn R. D. and Campbell K. P. (2000). Molecular basis of muscular dystrophies. Muscle Nerve 23, 1456-1471. 10.1002/1097-4598(200010)23:10<1456::AID-MUS2>3.0.CO;2-T [DOI] [PubMed] [Google Scholar]
- Deconinck A. E., Potter A. C., Tinsley J. M., Wood S. J., Vater R., Young C., Metzinger L., Vincent A., Slater C. R. Davies K. E. (1997). Postsynaptic abnormalities at the neuromuscular junctions of utrophin-deficient mice. J. Cell Biol. 136, 883-894. 10.1083/jcb.136.4.883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej M. and Campbell K. P. (2002). Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr. Opin. Genet. Dev. 12, 349-361. 10.1016/S0959-437X(02)00309-X [DOI] [PubMed] [Google Scholar]
- Enigk R. E. and Maimone M. M. (2001). Cellular and molecular properties of alpha-dystrobrevin in skeletal muscle. Front. Biosci. 6, D53-D64. 10.2741/A593 [DOI] [PubMed] [Google Scholar]
- Ervasti J. M. (2007). Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim. Biophys. Acta 1772, 108-117. 10.1016/j.bbadis.2006.05.010 [DOI] [PubMed] [Google Scholar]
- Ervasti J. M. and Campbell K. P. (1993). A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 122, 809-823. 10.1083/jcb.122.4.809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M., Noakes P. G., Mudd J., Nichol M., Chu G. C., Sanes J. R. and Merlie J. P. (1995). Failure of postsynaptic specialization to develop at neuromuscular-junctions of rapsyn-deficient mice. Nature 377, 232-236. 10.1038/377232a0 [DOI] [PubMed] [Google Scholar]
- Grady R. M., Merlie J. P. and Sanes J. R. (1997). Subtle neuromuscular defects in utrophin-deficient mice. J. Cell Biol. 136, 871-882. 10.1083/jcb.136.4.871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady R. M., Zhou H., Cunningham J. M., Henry M. D., Campbell K. P. and Sanes J. R. (2000). Maturation and maintenance of the neuromuscular synapse: genetic evidence for roles of the dystrophin–glycoprotein complex. Neuron 25, 279-293. 10.1016/S0896-6273(00)80894-6 [DOI] [PubMed] [Google Scholar]
- Grady R. M., Akaaboune M., Cohen A. L., Maimone M. M., Lichtman J. W. and Sanes J. R. (2003). Tyrosine-phosphorylated and nonphosphorylated isoforms of alpha-dystrobrevin: roles in skeletal muscle and its neuromuscular and myotendinous junctions. J. Cell Biol. 160, 741-752. 10.1083/jcb.200209045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray N. W., Weimer R. M., Bureau I. and Svoboda K. (2006). Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol. 4, 2065-2075. 10.1371/journal.pbio.0040370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanus C., Ehrensperger M. V. and Triller A. (2006). Activity-dependent movements of postsynaptic scaffolds at inhibitory synapses. J. Neurosci. 26, 4586-4595. 10.1523/JNEUROSCI.5123-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C.-D. and Kerppola T. K. (2003). Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 21, 539-545. 10.1038/nbt816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramarcy N. R. and Sealock R. (2000). Syntrophin isoforms at the neuromuscular junction: developmental time course and differential localization. Mol. Cell. Neurosci. 15, 262-274. 10.1006/mcne.1999.0823 [DOI] [PubMed] [Google Scholar]
- Lochmuller H., Mueller J. S., Baumeister S. K., Schara U., Huebner A. and Abicht A. (2006). A novel point mutation in the CHRND gene causes a congenital myasthenic syndrome by impairing co-clustering of the acetylcholine receptor with rapsyn. Neurology 66, A58-A59. 10.1212/WNL.66.10_suppl_4.S58 [DOI] [PubMed] [Google Scholar]
- Martinez-Pena y Valenzuela I., Mouslim C. and Akaaboune M. (2010). Calcium/calmodulin kinase II-dependent acetylcholine receptor cycling at the mammalian neuromuscular junction in vivo. J. Neurosci. 30, 12455-12465. 10.1523/JNEUROSCI.3309-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Pena y Valenzuela I., Mouslim C., Pires-Oliveira M., Adams M. E., Froehner S. C. and Akaaboune M. (2011). Nicotinic acetylcholine receptor stability at the NMJ deficient in alpha-syntrophin in vivo. J. Neurosci. 31, 15586-15596. 10.1523/JNEUROSCI.4038-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Pena y Valenzuela, Aittaleb M., Chen P.-J. and Akaaboune M. (2015). The knockdown of alphakap alters the postsynaptic apparatus of neuromuscular junctions in living mice. J. Neurosci. 35, 5118-5127. 10.1523/JNEUROSCI.3951-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. E., Sedgwick S. G., Ellis J. M., Pereboev A., Chamberlain J. S. and Nguyen thi M. (1998). An epitope structure for the C-terminal domain of dystrophin and utrophin. Biochemistry 37, 11117-11127. 10.1021/bi9805137 [DOI] [PubMed] [Google Scholar]
- Muller J. S., Baumeister S. K., Schara U., Cossins J., Krause S., von der Hagen M., Huebner A., Webster R., Beeson D., Lochmüller H. et al. (2006). CHRND mutation causes a congenital myasthenic syndrome by impairing co-clustering of the acetylcholine receptor with rapsyn. Brain 129, 2784-2793. 10.1093/brain/awl188 [DOI] [PubMed] [Google Scholar]
- Murphy S. and Ohlendieck K. (2016). The biochemical and mass spectrometric profiling of the dystrophin complexome from skeletal muscle. Comput. Struct. Biotechnol. J. 14, 20-27. 10.1016/j.csbj.2015.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S., Urushido T., Konno D., Okado H. and Sobue K. (2001). Rapid redistribution of the postsynaptic density protein PSD-Zip45 (Homer 1c) and its differential regulation by NMDA receptors and calcium channels. J. Neurosci. 21, 9561-9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramarao M. K., Bianchetta M. J., Lanken J. and Cohen J. B. (2001). Role of rapsyn tetratricopeptide repeat and coiled-coil domains in self-association and nicotinic acetylcholine receptor clustering. J. Biol. Chem. 276, 7475-7483. 10.1074/jbc.M009888200 [DOI] [PubMed] [Google Scholar]
- Rasse T. M., Fouquet W., Schmid A., Kittel R. J., Mertel S., Sigrist C. B., Schmidt M., Guzman A., Merino C., Qin G. et al. (2005). Glutamate receptor dynamics organizing synapse formation in vivo. Nat. Neurosci. 8, 898-905. 10.1038/nn1484 [DOI] [PubMed] [Google Scholar]
- Schmidt N., Akaaboune M., Gajendran N., Martinez-Pena y Valenzuela I., Wakefield S., Thurnheer R. and Brenner H. R. (2011). Neuregulin/ErbB regulate neuromuscular junction development by phosphorylation of alpha-dystrobrevin. J. Cell Biol. 195, 1171-1184. 10.1083/jcb.201107083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub V. and Campbell K. P. (1997). Muscular dystrophies and the dystrophin-glycoprotein complex. Curr. Opin. Neurol. 10, 168-175. 10.1097/00019052-199704000-00016 [DOI] [PubMed] [Google Scholar]
- Turney S. G., Culican S. M. and Lichtman J. W. (1996). A quantitative fluorescence-imaging technique for studying acetylcholine receptor turnover at neuromuscular junctions in living animals. J. Neurosci. Methods 64, 199-208. 10.1016/0165-0270(95)00135-2 [DOI] [PubMed] [Google Scholar]
- Yoon J. H., Johnson E., Xu R., Martin L. T., Martin P. T. and Montanaro F. (2012). Comparative proteomic profiling of dystroglycan-associated proteins in wild type, mdx, and Galgt2 transgenic mouse skeletal muscle. J. Proteome Res. 11, 4413-4424. 10.1021/pr300328r [DOI] [PMC free article] [PubMed] [Google Scholar]