

Abstract
Type I interferons (IFNs) induce a detrimental response during Listeria monocytogenes (L. monocytogenes) infection. We were interested in identifying mechanisms linking IFN signaling to negative host responses against L. monocytogenes infection. Herein, we found that infection of myeloid cells with L. monocytogenes led to a coordinated induction of type I IFNs and activation of the integrated stress response (ISR). Infected cells did not induce Xbp1 splicing or BiP upregulation, indicating that the unfolded protein response was not triggered. CHOP (Ddit3) gene expression was upregulated during the ISR activation induced by L. monocytogenes. Myeloid cells deficient in either type I IFN signaling or PKR activation had less upregulation of CHOP following infection. CHOP‐deficient mice showed lower expression of innate immune cytokines and were more resistant than wild‐type counterparts following L. monocytogenes infection. These findings indicate that L. monocytogenes infection induces type I IFNs, which activate the ISR through PKR, which contributes to a detrimental outcome in the infected host.
Keywords: CHOP, Integrated stress response, Listeriolysin O, Listeria, Mouse, Type I interferon
Introduction
Listeria monocytogenes (L. monocytogenes) is a Gram‐positive facultative intracellular pathogen responsible for listeriosis 1. Experimental infections with L. monocytogenes have led to profound insights into the cell biological and immunological responses of the infected host. Listeriolysin O (LLO) is an acid‐dependent pore‐forming toxin 2 that is both the major virulence factor and immunogenic protein of L. monocytogenes 3. LLO is required by intracellular L. monocytogenes to escape from the phagolysosome toward the cytosol allowing bacterial survival, replication, and cell‐to‐cell spread. LLO is cytotoxic, triggering programmed cell death at low nanomolar concentrations and cell lysis at higher concentrations 4, 5. It is also responsible for inducing the death of L. monocytogenes infected cells 6. LLO induces multiple cellular changes, which include loss of membrane integrity 7, mobilization of intracellular calcium storage 8, autophagy 9, and apoptosis 10, 11. Our laboratory has been interested in the cellular death and early innate response to L. monocytogenes infection, with a focus on LLO and type I interferon (IFN). This work led us to examine the role of the unfolded protein response (UPR) and integrated stress response (ISR) in L. monocytogenes pathogenesis.
The canonical response to endoplasmic reticulum (ER) stress is the result of a complex integrated signaling transduction pathway known as the UPR. The UPR stress sensors ATF6, PERK, and IRE1α transduce information about the folding status of the ER to the cytosol and nucleus in order to restore protein‐folding capacity. Depending on the time and strength of the stimulus, the UPR either restores homeostasis or induces programmed cell death 12, 13. The UPR is activated by a variety of cellular insults that include microbes and their proinflammatory products. Viral infections with dengue fever virus 14, hepatitis C 15, 16, 17, influenza A 18, and lymphocytic choriomeningitis virus 19 trigger the UPR. Infections with the parasite T. gondii 20, and the bacteria Pseudomonas aeruginosa, 21 and M. tuberculosis 22 also trigger UPR. So do microbial products such as LPS 23, 24 and cholera toxin 25. Nevertheless, the mechanisms leading to activation of the UPR pathways and its role under in vivo infection remain incompletely defined. The UPR may affect a number of cellular responses such as calcium homeostasis, lipid metabolism, redox state, protein glycosylation, and autophagy 26. The detailed relationship between the different UPR responses and the specific cellular processes that drive inflammation is an emerging field.
The kinase PERK can autoactivate in response to perturbations of the ER such as the presence of unfolded proteins, i.e. the UPR. Activated PERK phosphorylates and inactivates the eukaryotic translation‐initiation factor 2α (eIF2α) 27. Phosphorylation of eIF2α negatively affects translation in a dosage‐dependent manner, but it leads to the translation of the cap‐independent transcription factor ATF4 28, 29. ATF4 triggers the transcription of the proapoptotic transcription factor CHOP (Ddit3) and the phosphatase regulatory protein GADD34 (Ppp1r15a) 30. There are two general outcomes of PERK activation: (i) low level of activation leads to reduction in ER protein load, dephosphorylation of P‐eIF2α, and cellular survival and (2) high level of CHOP expression leads to death of the affected cell (12). Besides PERK, other cytosolic kinases can also phosphorylate eIF2α. For example, the hem‐regulated inhibitor kinase (HRI; involved in erythropoiesis), the general control nonderepressible‐2 (GCN2; senses amino acid starvation), and the IFN‐induced double‐stranded RNA‐activated protein kinase (PKR; senses infections) are all eIF2α kinases 31. The term ISR is used to refer to the pathway of eIF2α‐mediated stress that is triggered by any of the eIF2α kinases 32.
The main mode of PKR activation is through binding of virally derived dsRNA to its N‐terminal dsRNA binding domain and subsequent autophosphorylation. Two additional mechanisms of PKR activation have been proposed recently: the formation of endogenous microRNA‐derived dsRNAs and protein ISGylation 33, 34. Both mechanisms can be triggered by type I IFN signaling pathways. Type I IFNs induce the expression of PKR and high concentrations of PKR transcripts have been proposed as an endogenous source of dsRNA 33. High concentrations of PKR protein allow it to homodimerize and autophosphorylate in the absence of dsRNA 34. Furthermore, type I IFNs trigger the expression of ISG15, an activator of PKR in the absence of viral infections 34. ISG15 modifies the lysine residues at positions 69 and 159 of PKR allowing its dimerization and autophosphorylation 34.
During L. monocytogenes infection, type I IFNs induce a detrimental response in the host that includes apoptosis of lymphocytes and myeloid cells, and the secretion of immunosuppresive cytokines, such as IL‐10 35. Mice deficient in the type I IFN receptor (IFNAR KO) are resistant to L. monocytogenes infection 36, 37, 38. Listeria monocytogenes invades the cytosol in an LLO‐dependent manner and this biological activity is required for the induction of type I IFNs 39, 40. These findings led us to hypothesize that either the presence of cytosolic bacteria or LLO could trigger cellular stress mechanisms. For this reason, we examined whether cells infected with L. monocytogenes or treated with LLO protein developed a UPR or ISR. Both infection and LLO treatment increased the expression of the transcription factor Ddit3 (CHOP), as well as other markers of the PERK pathway. PERK was activated early after L. monocytogenes infection, and its downstream signaling was amplified by type I IFN, all in the absence of detectable activity by other two members of the UPR. We found that induction of type I IFN by L. monocytogenes infection led to PKR activation, which in turn enhanced the PERK pathway‐associated response. Our results suggest that L. monocytogenes activated the ISR. The biological consequence of ISR activation was examined in Ddit3−/− (CHOPKO) mice that showed increased resistance to L. monocytogenes infection. We suggest that induction of ISR is as a novel mechanism that helps to explain the detrimental response of the host to type I IFN signaling during L. monocytogenes infection.
Results
Listeria monocytogenes infection triggers the ISR
To determine if L. monocytogenes infection induces ER stress in APCs, we evaluated mRNA expression levels of canonical ER stress markers 47. These markers allowed the identification of which pathway(s) of the UPR were activated. To assess general activation of the UPR, we measured the ER chaperones BIP (Hspa5) and Wfs1 48, 49. For monitoring IRE1α activation, we measured Xbp1 spliced‐form (Xbp1s) levels 50. To measure PERK pathway activation, we evaluated CHOP (Ddit3) expression levels 13. We evaluated two strains of L. monocytogenes, a wild‐type (WT) strain that secretes listeriolysin (LLO) (LM LLO) and another that does not secrete LLO, the Δhly (LM ΔLLO mutant). As a positive control for the induction of ER stress, we used the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) inhibitor thapsigargin (TG). Two APCs were used: GM‐CSF‐derived bone marrow dendritic cells (BMDC) or M‐CSF‐derived bone marrow macrophages (BMM).
Listeria monocytogenes infection of BMDC induced less than a twofold upregulation in Hspa5 and Wfs1 expression relative to the untreated condition (Fig. 1A and B). Xbp1 splicing was not detected (Fig. 1C). These findings indicate that the complete canonical UPR pathway was not induced following infection with L. monocytogenes. Listeria monocytogenes infection induced an ∼10‐ to 15‐fold upregulation in the expression of Ddit3 (CHOP) (Fig. 1D) that was time and bacterial load dependent. It has been shown that Ddit3 expression is upregulated by the phosphorylation of eIF2α by PERK and other members of the eIF2α kinase family of enzymes, including, PKR, GCN2, and HRI (12‐13). PERK pathway activation was confirmed by the finding of an eightfold increase in the expression of the CHOP‐inducible gene Ppp1r15a (GADD34) (Fig. 1E). Ddit3 upregulation was also observed with high multiplicity of infection (MOI) of the Δhly L. monocytogenes strain. After 24 h of infection, Ddit3 expression increased ∼10‐fold in BMDC relative to the untreated cells (Fig. 1D). Other downstream ISR markers were also upregulated: ∼5‐fold for Ppp1r15a and ∼2‐fold for Atf4 (Fig. 1E and F). Similar results were seen in BMM (Supporting Information Fig. 1). We also found that PERK phosphorylation increased with higher bacterial loads of WT L. monocytogenes as early as 3 h of in vitro infection in BMDC (Fig. 1G). Finally, we also detected protein‐level induction of ATF4, as infection of BMM and BMDC allowed for its detection, as assayed by immunoblotting (Supporting Information Fig. 2A and B). Listeria monocytogenes infection selectively triggered the PERK pathway in APCs in the absence of the canonical ER stress response. Therefore, we concluded L. monocytogenes triggered the ISR.
Figure 1.
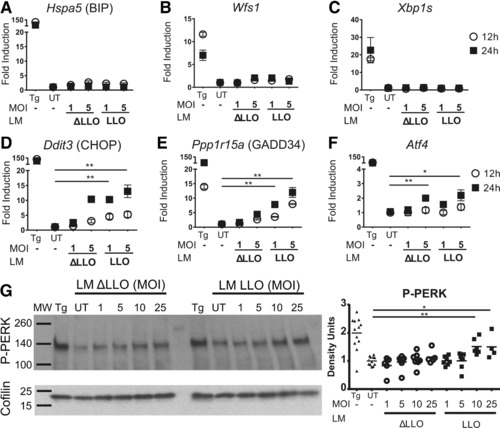
The ISR is triggered in BMDCs infected with L. monocytogenes. A–C. C57BL/6 BMDCs were infected with two different strains of L. monocytogenes, WT L. monocytogenes (LM LLO) and L. monocytogenes Δhly that lack LLO secretion (LM ΔLLO). Two different MOIs were tested. After infection, BMDCs were incubated with indicated L. monocytogenes strain for 12 or 24 h. cDNA was synthesized from extracted mRNA and gene expression measured by qRT‐PCR. (A) Hspa5 (BIP); (B) Wfs1; (C) Xbp1s; (D) Ddit3 (CHOP); (E) Ppp1r15a (GADD34); and (F) Atf4 were amplified with specific primers by qRT‐PCR. The fold change in gene expression was calculated using 2−ΔΔCT, where ΔΔ C T = (C T, Target − C T, Actin)Treated x ‐ (mean CT, Target − mean CT, Actin)Untreated. TG was used as a positive control. Symbols represent mean ± SEM. Results are pooled from two independent experiments performed in triplicates. (G) C57BL/6 BMDCs were infected with L. monocytogenes with the indicated MOIs. The two strains of L. monocytogenes were tested. Whole cell extracts were analyzed by western blot for phosphorylated‐PERK (P‐PERK) and Cofilin as a loading control. Mean of densitometric quantification is plotted. Symbols represent densitometry on 4–12 biological replicates performed in six independent experiments. MW, molecular weight; UT, untreated condition. Thapsigargin (TG) was used as a positive control. p value was calculated with Mann–Whitney test *p < 0.05, **p < 0.001.
This finding led us to evaluate phagocytosis as a possible triggering factor of the ISR activation. Uptake of latex beads by BMDC did not induce an increase in Ddit3 expression (Supporting Information Fig. 2C). We concluded that endocytosis was not sufficient to trigger CHOP. Therefore, in addition to LLO, other L. monocytogenes virulence factors or pathogen‐associated molecular patterns must be involved in early ISR activation.
LLO induces the ISR
To evaluate the direct effect of LLO in the ISR activation in the absence of other L. monocytogenes inflammatory molecules, we pulsed BMDCs with WT recombinant LLO (LLOWT) 46. We used concentrations previously established to be nonlethal to myeloid cells 4, 10, 11. After treatment with LLO, the ISR was activated (Fig. 2A–C). Expression of Ddit3 increased sixfold and Ppp1r15a increased twofold relative to the untreated condition. We did not find significant changes in Hspa5 or Wsf1 expression, and Xbp1 splicing was not detected (Fig. 2D–F). We also tested a mutant variant of LLO, LLOLT, that lacks the hemolytic activity of LLOWT and that allowed us to test it at 10 and 100‐times higher concentrations. LLOLT differs from LLOWT only in an amino acid inversion T515L‐L516T 51. LLOLT triggered the ISR with an upregulation of Ddit3 of ∼20‐fold, Pppr15a1 of ∼10‐fold, and Atf4 of ∼10‐fold (Fig. 2G–I). At higher concentration, we detected upregulation of ∼2‐fold for Hspa5, ∼2‐fold for Wsf1, and ∼2‐fold for Xbp1s (Fig. 2J–L). Thus, LLO protein triggered the ISR unrelated to its cytolytic activity.
Figure 2.
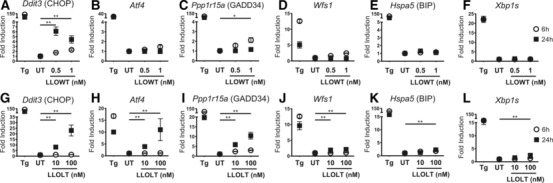
The ISR is triggered by recombinant LLO protein. To avoid cell death, C57BL/6 BMDCs were pulsed with low concentrations of recombinant WT LLO protein (LLOWT) (A–F) or high concentrations of a nonlytic LLO mutant protein (LLOLT) (G–L) and incubated with indicated strains 6 and 24 h. cDNA was synthesized from extracted mRNA and gene expression measured by qRT‐PCR. (A, G) Ddit3 (CHOP); (B, H). Atf4; (C, I) Ppp1r15a (GADD34); (D, J). Wfs1; (E, K) Hspa5 (BIP); (F, L) Xbp1s were amplified with specific primers by qRT‐PCR. The fold change in gene expression was calculated using 2−ΔΔCT. Dots are mean ± SEM of six biological replicates. UT, untreated condition; TG was used as a positive control. p value was calculated with Mann–Whitney test *p < 0.05, **p < 0.001. Results are pooled from two independent experiments performed in triplicates.
To exclude an LPS contamination in our LLO preparations as a confounding trigger of the ISR, we measured Ddit3 expression induced by LLOWT in the presence of the LPS‐binding compound polymixin B (PMB) (Supporting Information Fig. 2D). Purified LPS induced a ∼20‐fold increase in expression of Ddit3 and this response was abolished by pretreatment of the LPS with PMB. LLOWT upregulation of Ddit3 was unaffected by the presence of PMB. Therefore, the increase in CHOP expression induced by LLOWT was not a result of LPS contamination.
Listeria monocytogenes induced activation of the ISR leads to inflammatory cytokine gene expression
To establish whether the ISR activation played a role in the cytokine response induced by L. monocytogenes infection, we examined BMDC and BMM from Ddit3−/− mice (CHOP KO). We tested the CHOP‐deficient APCs to avoid the aberrations in ER morphology and physiology described in PERK‐deficient cells 52, and the hematopoietic system abnormalities in ATF4 KO mice 53. We measured the expression of the type I IFNs (Pan‐Ifna and Ifnb1), IL‐12α (Il12a), IL‐12β (Il12b), and IL‐1β (Il1b) following infection of CHOP KO BMDC and BMM. In general, we found decreased expression of inflammatory cytokine genes in BMDC and BMM from CHOP KO compared to cells from normal mice, with the exception for Il12 that increased only in BMDC (Fig. 3). The overall cytokine responses peaked at 12 h of infection in WT APC and decreased by 24 h. After 12 h, expression levels of Il12a, Il12b, and Il1b, were ∼200‐fold, ∼3500‐fold, and ∼1500‐fold higher in controls versus CHOP KO BMDC (Fig. 3A). For Il12b and Il1b expression changes were detectable even after 24 h. There was induction of IFN‐α and IFN‐β gene expression at the 12 h time. This response was reduced by 1/2 or 1/3 in the BMDC from CHOP KO mice. Also to note is that LM ΔLLO triggered IFN gene expression but required higher MOI compared to WT L. monocytogenes. CHOP KO BMM also showed a statistically significant decrease of Ifna, Ifnb1, and Il1b (Fig. 3B). We concluded that ISR activation regulated the innate immune cytokine induction of APCs following L. monocytogenes infection.
Figure 3.
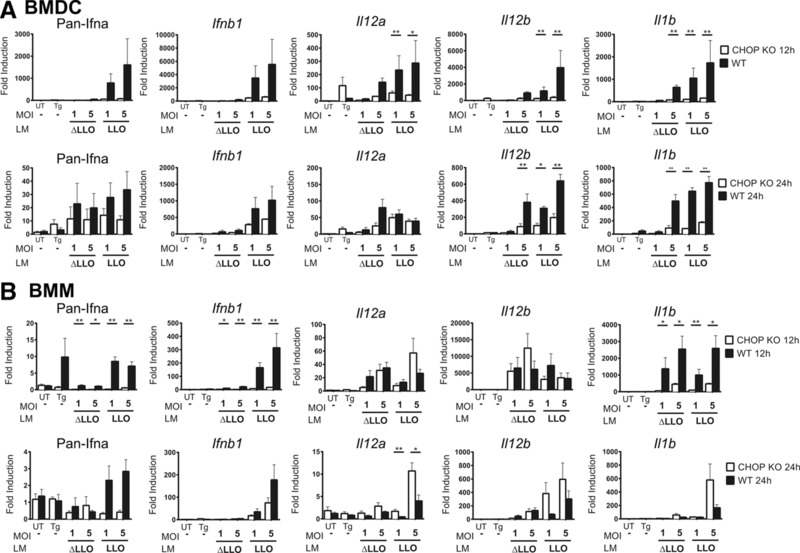
The role of the ISR impairment in innate immune cytokines response from L. monocytogenes infected APC. C57BL/6 and PERK pathway impaired, CHOP knockout mice (CHOP KO) BMDC or BMM were infected with WT L. monocytogenes (LM LLO) and L. monocytogenes Δhly (LM ΔLLO). Two different MOIs were tested. (A) BMDCs or (B) BMMs were incubated with indicated L. monocytogenes strains for 12 and 24 h. cDNA was synthesized from extracted mRNA. Then specific primers were used to amplify Pan‐Ifna; Ifn‐β; Il‐12α; Il‐12β; and Il‐1β by qRT‐PCR. The fold change in gene expression was calculated using 2−ΔΔCT. Bars are mean ± SEM of six biological replicates. UT, untreated condition; Tg, thapsigargin 0.1 μM as a positive control. p‐value was calculated with Mann–Whitney test *p < 0.05, **p < 0.001. Results are pooled from two independent experiments performed in triplicates.
We also evaluated the expression of other ER stress‐related genes in CHOP KO APCs after infection (Supporting Information Fig. 3). We found less than twofold change in expression of Hspa5 and Wfs1 in both BMDC and BMM. Additionally, Pppr15a1 was decreased to half its normal level of expression in BMDC (Supporting Information Fig. 3A). Thus, the impairment of the PERK pathway in CHOP KO APCs did not trigger other compensatory ER stress responses following L. monocytogenes infection.
Type I IFN acting through PKR enhances L. monocytogenes activation of the ISR
Listeria monocytogenes invades the cytosol and initiates a type I IFN response 54. One outcome of type I IFN receptor signaling is the activation of PKR, which in turn induces activation of the ISR, in addition to PERK phosphorylation 32. Hence, we hypothesized that we may have a feed‐forward transcription loop in which type I IFN acting through PKR activates the ISR that enhances PERK‐associated signaling following L. monocytogenes infection.
We evaluated the expression of Ddit3 (CHOP) as a marker of the ISR activation. In APCs derived from mice deficient in the type I IFN receptor (IFNAR KO), Ddit3 transcription was decreased significantly when compared to WT controls (Fig. 4A and B). Blockade of PKR activation using the chemical inhibitor C16 also led to a significant inhibition of the ISR activation at 12 h (Fig. 4A and B) 55, 56. The effect of C16 was not seen at 24 h postinfection in BMM suggesting that the effect of the inhibitor was not absolute or was reversible.
Figure 4.
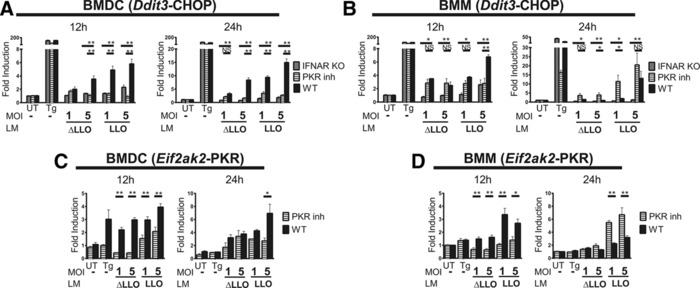
Type I IFN triggers the ISR through PKR activation. (A) BMDCs or (B) BMMs from CHOP KO, IFNAR KO, C57BL/6 mice, and WT APCs in which PKR phosphorylation was inhibited with C16 (PKR inh) were infected with WT L. monocytogenes (LM LLO) and L. monocytogenes Δhly (LM ΔLLO) for 12 and 24 h. Two different MOIs where tested. cDNA was synthesized from extracted mRNA. Then specific primers were used to amplify CHOP (Ddit3) by qRT‐PCR. (C) BMDCs or (D) BMMs were incubated with or without the PKR phosphorylation inhibitor C16 at 2 μM and infected with LM LLO and LM ΔLLO. Then specific primers were used to amplify Eif2ak2 (PKR) by qRT‐PCR. The fold change in gene expression was calculated using 2−ΔΔCT. Bars are mean ± SEM of six biological replicates. UT, untreated condition. TG was used as a positive control. p‐value was calculated with Mann–Whitney test *p < 0.05, **p < 0.001. Results are pooled from two independent experiments performed in triplicates.
Human PKR has been better characterized than mouse PKR. Based on its DNA sequence the molecular mass is about 62 kDa, meanwhile, in the mouse it is 59 kDa. Human PKR has 14 phosphorylation sites, among them, the residue T451 (p‐T451) is required to generate an active kinase. This corresponds p‐T414 of mouse PKR. Conventional western blotting techniques made to identify active PKR (p‐T451) detect diverse molecular weights of PKR in a range of 50–75 kDa 57. Cytoplasmic and nuclear PKR forms also have multiple isoelectric points due to different phosphorylation states. PKR also has diverse posttranslational modifications. There is no monoclonal antibody available for mouse p‐PKR detection and the available regents have been made for human PKR, which cross‐react with mouse PKR. Recent studies showed that human PKR mRNA has no alternate splicing products 57. Furthermore, upregulation of PKR activity evaluated by kinase assays in mouse models is also accompanied with an increase in PKR protein and mRNA levels 58. This is consistent with the reported autoregulation of PKR expression upon stimulation of its activity 59. For these reasons, we measured Eif2ak2 (the PKR gene) transcription as a marker of PKR activation and found ∼3‐fold increase in its expression of following L. monocytogenes infection of both BMDC and BMM (Fig. 4C and D). Additionally, treatment of APCs with the PKR inhibitor C16 prevented more than 50% the upregulation of Eif2ak2 expression detected after infection. (In BMDC, PKR inhibition was detected up to 24 h postinfection. In BMM, PKR inhibition was found at 12 h but not 24 h postinfection.) Therefore, we concluded that L. monocytogenes infection enhances the ISR activation through PKR phosphorylation triggered by type I IFN receptor signaling.
BMM and BMDC were treated with 1 μM GSK2656157 for 1 h and then infected with L. monocytogenes or treated with TG. At this dosage, GSK2656157 is reported to inhibit phosphorylation of eIF2α, and protein expression of ATF4 and CHOP downstream of both tunicamycin and TG treatment, while maintaining PKR activity.
We evaluated ISR induction at 12 and 24 h postinfection. As in the rest of the manuscript, we used TG as a positive control for the induction of ISR and as a benchmark for the inhibitory effects of GSK2656157. In the TG‐treated cells, the PERK inhibitor reduced the expression of Ddit3 (CHOP), albeit not completely (Supporting Information Fig. 4). This result confirmed that our dosage and treatment times for GSK2656157 were able to reduce PERK‐induced Ddit3 expression.
The PERK inhibitor led to an increase in the upregulation of the level of Ddit3 expression following L. monocytogenes infection. We attribute the paradoxical Ddit3 upregulation induced by PERK inhibition during L. monocytogenes infection to NRF2/Keap1 pathway inhibition. NRF2/Keap1 pathway is activated by PERK and antagonizes the expression of Ddit3. Thus, when PERK is blocked, but PKR is still active, the ISR response prevails and Ddit3 (CHOP) is upregulated
The ISR activation modulates host handling of L. monocytogenes infection
To establish the role of the ISR activation during L. monocytogenes infection in vivo, we examined C57BL/6 CHOP KO mice during the early phase of the infection. We infected CHOP KO and WT mice with L. monocytogenes and followed them for survival (Fig. 5A). At an LD50, 60% of WT mice in contrast to 17% of CHOP KO mice died within the first 10 days of infection. CHOP KO mice displayed an average of ten times lower bacterial burden in spleens and livers than WT mice at day 3 postinfection (Fig. 5B).
Figure 5.

Impairing the ISR silencing CHOP transcription factor protects against L. monocytogenes infection in vivo. C57BL/6 and CHOP KO mice were infected WT L. monocytogenes. (A) For survival assay C57BL/6 (n = 30 mice) and CHOP KO (n = 20 mice) were followed for 13 days. p value was calculated with log‐rank (Mantel–Cox) test **p < 0.005. (A–D) For colony counts in spleens and livers, mice were sacrificed after (B) 3 days (n = 10 WT, n = 8 CHOP KO mice) and (C) 7 days (n = 8 WT, n = 9 CHOP KO mice). For histological evaluation mice were infected with 104 CFU (D) spleens and (E) livers from infected mice at day 3 were isolated, fixed, and stained with H&E. Magnification 10×. Scale bar represents 100 μm. Pictures are representative of n = 4 WT and n = 4 CHOP KO mice. (F–G) For serum cytokines evaluation, mice were infected with 104 CFU, n = 8 mice per group. (F) IFN‐γ, (G) IL‐6, and (H) MCP‐1 were quantified by CBA after 24 h. Dots represent biological replicates and the mean of each condition is plotted. Data are pooled from two to three independent experiments with four to ten mice per experiment. p‐value was calculated with Mann–Whitney test *p < 0.05, **p < 0.001.
CHOP KO mice contained, on average, no detectable bacterial burden in their spleens and livers at day 7 postinfection (Fig. 5C). In contrast, WT mice still had a detectable burden in livers at day 7. The differences in bacterial burden were reflected in the tissue pathology. The extensive splenic white pulp apoptosis and necrosis reported in the WT mice during the first 72 h of infection was reduced in the CHOP KO mice 35 (Fig. 5D). The number and size of liver microabscesses was also decreased in CHOP KO mice (Fig. 5E). Finally, CHOP KO mice showed lower innate immune cytokine levels in serum than WT mice (Fig. 5F–H), similar to what was shown in CHOP KO BMDC and BMM (see above). At 24 h after infection, we found statistically significant decrease in IFN‐γ, IL‐6, and MCP‐1 production in CHOP KO versus WT mice. [We detected no difference in TNF‐α production following L. monocytogenes infection of WT versus CHOP KO macrophages in vitro or mice in vivo (Supporting Information Fig. 5).] Together our findings show that CHOP KO mice are more resistant to L. monocytogenes infection than WT mice. Thus, we concluded that the ISR activation is detrimental to host handling of L. monocytogenes infection.
Discussion
Our results identify a novel and important mechanism that explains part of the detrimental response induced by type I IFN during L. monocytogenes infection. We show that L. monocytogenes triggers the ISR and the main component of this response is driven by type I IFN signaling. Type I IFN induces a feed‐forward transcription loop, the consequence of which is the activation of CHOP resulting in further IFN production as surmised by the effects of CHOP KO cells. The ISR activation is detrimental to L. monocytogenes infected mice leading to increased morbidity and mortality.
Our findings indicate that PKR is one of the cytosolic enhancers of the ISR during L. monocytogenes infection. Although high MOI of L. monocytogenes infection trigger phosphorylation of PERK as early as 3 h, a second signal is required to maintain and/or enhance the ISR in infected cells at lower doses. PKR acts as a link between the ISR activation and type I IFN response. Indeed, cells from the IFNAR KO mice had markedly decreased transcription of PKR. It is known that type I IFN induces PKR activation increasing its synthesis and autophosphorylation 34, 59. When we inhibited PKR phosphorylation, Ddit3 expression was decreased. The whole mechanism for activation suggests that the release of type I IFN after L. monocytogenes internalization activates PKR to phosphorylate eIF2α triggering ATF4‐CHOP transcriptional program.
Our results showed that L. monocytogenes triggers PERK phosphorylation 3 h after infection, before the transcriptional markers are activated. PERK phosphorylation not only triggers a cell death program but also is an autoregulated pathway that allows cell survival 13, 60. The survival branch described as part of the PERK pathway is an ISR‐independent response that is also triggered by p‐PERK, which directly activates NRF2/Keap1 pathway and elicits an antioxidant program 61. Both pathways share ATF4 activation but NRF2/Keap1 pathway inhibits CHOP expression. Thus, we speculate that L. monocytogenes could activate both PERK pathway branches. In the presence of CHOP, as part of the ISR, PERK‐eIF2α‐CHOP branch predominates sending the cell to oxidative stress and death 62, 63. In the absence of CHOP, as is the case of CHOP KO mice, the PERK‐NRF2‐Keap1 activation could be promoting cell survival in the same way as previously described for CHOP inhibition in a cecum‐ligation induced sepsis model 64. We also found that a specific p‐PERK inhibitor 65 is able to inhibit both pathways but when PKR is still active, the ISR response prevails and Ddit3 (CHOP) is upregulated in APC. Future studies will be required to clarify the cross‐talk between both PERK pathway branches and the mechanism of this early PERK phosphorylation induced by L. monocytogenes.
Ddit3 (CHOP) has been reported as highly upregulated gene after L. monocytogenes infection 39, but its role as a component of the ISR and its association with type I IFN signature response was unknown. Here, we found that L. monocytogenes infection induced upregulation of Atf4, Ddit3 (CHOP), and Ppp1r15a (GADD34). Accordingly, we confirmed the ISR activation by L. monocytogenes infection as a result of Type I IFN signaling. Also, the absence of increased expression of other ER stress markers, such as Hspa5 (BIP), Wfs1, and spliced Xbp1s (XBP1s), allowed us to exclude this PERK pathway activation as part of the canonical UPR. Previous studies showed that low MOI of L. monocytogenes induce XBP1s at 8 h 23. In contrast, our model of infection tested 10–50 times higher MOIs in later time points 12 and 24 h. XBP1s has been proposed as an early adaptive response to stressors 13. Based on both findings, we suggest that at low bacterial MOIs the adaptive cellular response dominates. Meanwhile, at high MOIs and after long infection times the adaptive response is not sustained and the cell death fate triggered by the ISR predominates.
CHOP is one of the major effector transcription factors of the ISR. CHOP induces a number of effects such as oxidative stress 62, ER‐calcium release 66, inflammasome activation 67, proinflammatory cytokines secretion 68, and cell death 63, 69, 70. Listeria monocytogenes is a well‐known inductor of different forms of cell death including apoptosis 35 and pyroptosis 71. On the other hand, CHOP KO mice have decreased apoptotic phenotypes 66, 72. In this study, we found CHOP KO mice infected with L. monocytogenes had a decrease in splenic cell death, fewer liver microabscesses, decreased bacterial proliferation, and a better survival than WT mice. Additionally, CHOP KO mice had a reduced release of systemic inflammatory cytokines. We also confirmed lower expression of inflammatory cytokines in both BMDC and BMM from CHOP KO mice, with the exception for IL12 that increased in CHOP KO BMM 38. Together our findings suggest that in the absence of CHOP, the earliest DCs and macrophages that phagocytose the bacteria may be more resistant to cell death. Consequently, the local control of the infection at early steps, when DCs and macrophages are viable, leads to an immune response that avoids a strong proinflammatory cytokine release and the recruitment of more immune cells. As a result, tissue damage in spleen and liver was decreased.
In summary, we described an alternative pathway that allows a better understanding of the detrimental response induced by type I IFN release during L. monocytogenes infection, the ISR activation. The specific role of the ISR in the pathogenesis of other bacterial infections remained unexplored. However, there is evidence that M. tuberculosis activates PERK‐pathway markers and IRE1α in macrophage rich granulomas 22. Also, homoserine lactone (HSL‐C12), a membrane permeant molecule secreted by P. aeruginosa, is able to trigger an inflammatory response through activation of PERK pathway 21. Based on the benign phenotype of CHOP KO mice, this study opens the possibility of exploring the use of CHOP inhibitors as an alternative or a coadjuvant to antibiotic therapies, either to enhance host responses during bacterial infections, or to decrease the extensive organ damage associated with antibacterial immune responses.
Materials and methods
Mice
WT C57BL/6 and C57BL/6 CHOP knock out (B6.129S(Cg)‐Ddit3tm2.1Dron/J) mice were purchased from Jackson Laboratory (Bar Harbor, ME). The CHOP KO mouse was made in 129S6/SvEvTac‐derived W4 embryonic stem cells. The resulting mice were backcrossed and are on a mixed C57BL/6J‐C57BL/6N background. After backcrossing, by single nucleotide polymorphism analysis only one marker of 32 was found to be derived from 129S6 (Jackson Laboratory). C57BL/6 IFN‐αβR knock out (IFNAR KO) were obtained from Dr. Robert Schreiber (Washington University School of Medicine, St. Louis, MO). All the experiments were performed with sex and age matched mice at 8–20 weeks of age. Mice were maintained in our specific pathogen‐free animal facility according to institutional guidelines and protocols approved by the Division of Comparative Medicine of Washington University School of Medicine (Association for Assessment and Accreditation of Laboratory Animal Care [AAALAC] accreditation number A3381‐01). All mice were bred and maintained in the same room and on the same rack within our facility. Only mice bred in our facility were used for all of the experiments reported herein.
Cell cultures
DCs were derived from BMDC as previously described with modifications 41. BM cells were cultured at 106 cells per well in 6‐well plates for 6 days in GM‐CSF–containing medium (pyrogen‐free DMEM [Gibco, Thermo‐Fisher, Waltham, MA] with 20 UI/mL GM‐CSF from conditioned supernatants, 10% FCS [Hyclone, GE Healthcare, Logan, UT], 1× nonessential aminoacids [Gibco], 1 mM Na pyruvate [Gibco], 1× GlutaMAX™[Gibco], 50μM β‐ME [Fluka, Sigma‐Aldrich, St. Louis, MO], and kanamycin 0.1 mg/mL [Gibco]). Cultures media was replaced on day 4 with conditioned media without antibiotic.
Macrophages were derived from BMM as previously described with modifications 42. BM cells were cultured at 10–15 × 106 cells in 150 mm Petri dishes for 7 days in M‐CSF–containing medium (pyrogen‐free DMEM with 20% l‐cell conditioned media, 10% FCS, 5% Horse serum [Sigma], 1 mM Na pyruvate, 1.5 mM l‐glutamine [Corning, Corning, NY], 1× penicillin/streptomycin [Gibco]). BMM were harvested on day 7 and replated at 106 cells per well in 6‐well plates in conditioned media without antibiotic.
Listeria monocytogenes infection
Infections were performed as previously described 43. We used L. monocytogenes strain EGD (LM LLO) and LM ΔLLO strain EJL1. For histological examination, small samples of spleens and livers were fixed in 10% formalin and stained with hematoxylin and eosin (H&E). Cytokines assays were performed on sera of infected mice by cytometric bead array (CBA, BD Biosciences, San Jose, CA) analysis. Mouse Inflammation Kit (BD Biosciences) was followed according to the manufacturer's instructions. CBA data were analyzed on a FACSCalibur™ with CELLQuest™ software and the CBA analysis software package.
For cell cultures infection, BMDC or BMM were infected for 3 to 24 h. In brief, L. monocytogenes strains were grown overnight on Brain‐Heart‐Infusion broth at 37°C. Bacteria concentration was estimated with OD560 then indicated bacterial dilutions were made in D10F (DMEM with 10%FCS) or D10F plus the Imidazolo‐oxindole PKR inhibitor C16 (Sigma). Half of the initial cells culture volume was replaced with bacteria‐containing media and centrifuged at 2000 rpm per 10 min. Plates were incubated at 37°C for 30 min. Finally, cell cultures were replaced with D10F or D10F‐C16 plus gentamicin at 5 μg/mL [Gibco]to kill non‐phagocytized bacteria.
RNA isolation and real‐time PCR
For infected BMDC and BMM, total RNA was isolated with Ambion RNAqueous‐Micro Kit (Life Technologies, Carslbad, CA) following manufacturer's instructions. RNA was quantified by OD260 using Nanodrop (Thermo Fisher Scientific). cDNA was obtained from total RNA using First Strand Synthesis Protocol with Reverse Transcriptase (New England BioLabs). TaqMan PCR was performed using TaqMan Fast Universal PCR Master Mix. Primers for quantitative RT‐PCR were designed using the PrimeTime predesigned qPCR assays (IDT DNA, Coralville, IA), except for Pan‐Ifna, which transcripts were made based on previous work 44. PrimeTime primers employed 5’‐nuclease detection. Xbp1s was performed using SYBR® green detection technology. To amplify the spliced form of mouse XBP‐1, we used mXBP1.7S: CAG CAC TCA GAC TAT GTG CA and mXBP1.10AS: GTC CAT GGG AAG ATG TTC TGG as previously described 45. StepOne 2.1 software was used to perform quality control and the relative expression quantification.
Recombinant LLOWT and LLOLT purification
The expression vector pET29b containing His‐tagged LLO sequence (LLOWT) was provided by Dr. Daniel Portnoy (University of California, Berkeley, CA). LLOWT was purified as previously described using Ni‐NTA Sepharose (Qiagen, Valencia, CA) and dialyzed extensively against storage buffer (50 mM phosphate/acetate [pH 6.0], 1 M NaCl, 1 mM EDTA, and 5 mM DTT) 10, 46. For LLOLT, LLOWT was modified with an inversion T515L‐L516T using Quickchange Site‐Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA) following the manufacturer's protocol. Mutagenesis was confirmed by sequencing of constructs at the Washington University Nucleic Acids Core facility. Proteins purity was confirmed by SDS‐PAGE.
Western blots
Infected BMDCs were lysed on ice in 1× Laemmli buffer (for P‐PERK), with Complete Protease Inhibitor Cocktail (Roche Diagnostic Systems, Indianapolis, IN), and PhosStop Phosphatase Inhibitor Cocktail (Roche Diagnostic Systems). Lysates were resolved on a 4–15% Mini‐PROTEAN® TGX™ precast polyacrylamide gel (Bio‐Rad) and transferred to a PVDF membrane. Rabbit mAb Phospo‐PERK (Thr980) (16F8), Cofilin (D3F9) XP™ and the secondary Ab Goat anti‐rabbit‐HRP were purchased from Cell Signaling Technologies (Beverley, MA).
Statistical analysis
Mann–Whitney U test was used to determine significant differences between samples. All data were plotted using GraphPad Prism 6 (GraphPad Software, La Jolla, CA).
Conflict of interest
The authors declare no financial or commercial conflict of interest
Abbreviations
- BMDC
bone marrow derived dendritic cells
- BMM
bone marrow derived macrophages
- CBA
cytometric bead array
- CHOP KO
Ddit3−/− mice
- ER
endoplasmic reticulum
- IFNAR
type 1 interferon receptor
- ISR
integrated stress response
- LLO
listeriolysin O
- LM LLO
wild‐type Listeria monoctytogenes
- LM ΔLLO
Δhly Listeria monocytogenes
- L. monocytogenes
Listeria monocytogenes
- TG
thapsigargin
- UPR
unfolded protein response
Supporting information
Supplementary Material
Supporting Information Figure 1. The ISR is triggered by L. monocytogenes infection in BMM. C57BL/6 BMM (106 cells) were infected with L. monocytogenes (LM LLO) and L. monocytogenes Δhly (LM ΔLLO) per 12h and 24h. Two different MOI where tested. After infection, BMM were incubated per 12h and 24h. cDNA was synthesized from extracted mRNA. Then A. Hspa5 (BIP); B. Wfs1; C. Xbp1s; D. Ddit3 (CHOP); E. Ppp1r15a (GADD34); F. Atf4 were amplified with specific primers by qRT‐PCR. The fold change in gene expression was calculated using 2‐ΔΔCT. Dots are mean ±SEM of 6 biological replicates. UT: Untreated condition, Tg: Thapsigargin 0.1μM as a positive control. P value was calculated with Mann‐Whitney test p<0.05 (*), p<0.001 (**).
Acknowledgments
We thank all the members of our laboratory for many helpful discussions and assistance, and in particular Katherine E. Frederick and Xiaoxiao Wan for technical support. This research was supported by the National Institutes of Health grant R01AI062832 (E.R.U.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1. Stavru, F. , Archambaud, C. and Cossart, P. , Cell biology and immunology of Listeria monocytogenes infections: novel insights. Immunol. Rev. 2011. 240: 160–184. [DOI] [PubMed] [Google Scholar]
- 2. Köster, S. , Van Pee, K. , Hudel, M. , Leustik, M. , Rhinow, D. , Kühlbrandt, W. , Chakraborty, T. et al., Crystal structure of listeriolysin O reveals molecular details of oligomerization and pore formation. Nat. Commun. 2014. 5: 3690. [DOI] [PubMed] [Google Scholar]
- 3. Hamon, M. , Ribet, D. , Stavru, F. and Cossart, P. , Listeriolysin O: the Swiss army knife of Listeria. Trends Microbiol. 2012. 20: 360–368. [DOI] [PubMed] [Google Scholar]
- 4. Carrero, J. , Vivanco‐Cid, H. and Unanue, E. , Listeriolysin o is strongly immunogenic independently of its cytotoxic activity. PLoS One 2012. 7: 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gaillard, J. L. , Berche, P. and Sansonetti, P. , Transposon mutagenesis as a tool to study the role of hemolysin in the virulence of Listeria monocytogenes . Infect. Immun. 1986. 52:50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Portnoy, D. A. , Jacks, P. S. and Hinrichs, D. J. , Role of hemolysin for the intracellular growth of Listeria monocytogenes . J. Exp. Med. 1988. 167: 1459–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Czuczman, M. , Fattouh, R. , Van Rijn, J. , Canadien, V. , Osborne, S. , Muise, A. , Kuchroo, V. et al., Listeria monocytogenes exploits efferocytosis to promote cell‐to‐cell spread. Nature 2014. 509: 230–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gekara, N. , Westphal, K. , Ma, B. , Rohde, M. , Groebe, L. and Weiss, S. , The multiple mechanisms of Ca2+ signalling by listeriolysin O, the cholesterol‐dependent cytolysin of Listeria monocytogenes . Cell. Microbiol. 2007. 9: 2008–2021. [DOI] [PubMed] [Google Scholar]
- 9. Lam, G. , Cemma, M. , Muise, A. , Higgins, D. and Brumell, J. , Host and bacterial factors that regulate LC3 recruitment to Listeria monocytogenes during the early stages of macrophage infection. Autophagy 2013. 9: 985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carrero, J. , Calderon, B. and Unanue, E. , Listeriolysin O from Listeria monocytogenes is a lymphocyte apoptogenic molecule. J. Immunol. 2004. 172: 4866–4874. [DOI] [PubMed] [Google Scholar]
- 11. Guzmán, C. A. , Domann, E. , Rohde, M. , Bruder, D. , Darji, A. , Weiss, S. , Wehland, J. et al., Apoptosis of mouse dendritic cells is triggered by listeriolysin, the major virulence determinant of Listeria monocytogenes. Mol. Microbiol. 1996. 20: 119–126. [DOI] [PubMed] [Google Scholar]
- 12. Walter, P. and Ron, D. , The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011. 334: 1081–1086. [DOI] [PubMed] [Google Scholar]
- 13. Hetz, C. , The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012. 13: 89–102. [DOI] [PubMed] [Google Scholar]
- 14. Peña, J. and Harris, E. , Dengue virus modulates the unfolded protein response in a time‐dependent manner. J. Biol. Chem. 2011. 286: 14226–14236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Merquiol, E. , Uzi, D. , Mueller, T. , Goldenberg, D. , Nahmias, Y. , Xavier, R. , Tirosh, B. et al., HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS One 2011. 6: e24660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tardif, T. , Mori, K. , Kaufman, R. and Siddiqui, A. , Hepatitis C virus suppresses the IRE1‐XBP1 pathway of the unfolded protein response. J. Biol. Chem. 2004. 279: 17158–17164. [DOI] [PubMed] [Google Scholar]
- 17. Pavio, N. , Romano, P. , Graczyk, T. , Feinstone, S. and Taylor, D. , Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2 alpha kinase PERK. J. Virol. 2003. 77: 3578–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hassan, I. , Zhang, M. , Powers, L. , Shao, J. , Baltrusaitis, J. , Rutkowski, D. , Legge, K. et al., Influenza A viral replication is blocked by inhibition of the inositol‐requiring enzyme 1 (IRE1) stress pathway. J. Biol. Chem. 2012. 287: 4679–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pasqual, G. , Burri, D. , Pasquato, A. , De la Torre, J. and Kunz, S. , Role of the host cell's unfolded protein response in arenavirus infection. J. Virol. 2011. 85: 1662–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamamoto, M. , Ma, J. , Mueller, C. , Kamiyama, S. , Saiga, H. , Kubo, E. , Kimura, T. et al., ATF6beta is a host cellular target of the Toxoplasma gondii virulence factor ROP18. J. Exp. Med. 2011. 208: 1533–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grabiner, M. , Fu, Z. , Wu, T. , Barry, K. , Schwarzer, C. and Machen, T. , Pseudomonas aeruginosa quorum‐sensing molecule homoserine lactone modulates inflammatory signaling through PERK and eI‐F2a. J. Immunol. 2014. 193: 1459–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seimon, T. , Kim, M. , Blumenthal, A. , Koo, J. , Ehrt, S. , Wainwright, H. , Bekker, L. et al., Induction of ER stress in macrophages of tuberculosis granulomas. PLoS One 2010. 5: e12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martinon, F. , Chen, X. , Lee, A. and Glimcher, L. , TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010. 11: 411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Woo, C. , Cui, D. , Arellano, J. , Dorweiler, B. , Harding, H. , Fitzgerald, K. , Ron, D. et al., Adaptive suppression of the ATF4‐CHOP branch of the unfolded protein response by toll‐like receptor signalling. Nat. Cell Biol. 2009. 11: 1473–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cho, J. , Lee, A. , Platzer, B. , Cross, B. , Gardner, B. , De Luca, H. , Luong, P. et al., The unfolded protein response element IRE1α senses bacterial proteins invading the ER to activate RIG‐I and innate immune signaling. Cell Host Microbe 2013. 13: 558–569. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26. Bettigole, S. and Glimcher, L. , Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 2015. 33: 107–138. [DOI] [PubMed] [Google Scholar]
- 27. Scheuner, D. , Song, B. , McEwen, E. , Liu, C. , Laybutt, R. , Gillespie, P. , Saunders, T. et al., Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 2001. 7: 1165–1176. [DOI] [PubMed] [Google Scholar]
- 28. Harding, H. , Zhang, Y. , Bertolotti, A. , Zeng, H. and Ron, D. , Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000. 5: 897–904. [DOI] [PubMed] [Google Scholar]
- 29. Vattem, K. and Wek, R. , Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 2004. 101: 11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Novoa, I. , Zeng, H. , Harding, H. and Ron, D. , Feedback inhibition of the unfolded protein response by GADD34‐mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001. 153: 1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dabo, S. and Meurs, E. , dsRNA‐dependent protein kinase PKR and its role in stress, signaling and HCV infection. Viruses 2012. 4: 2598–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harding, H. , Zhang, Y. , Zeng, H. , Novoa, I. , Lu, P. , Calfon, M. , Sadri, N. et al., An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003. 11: 619–633. [DOI] [PubMed] [Google Scholar]
- 33. Ishibashi, O. , Ali, M. M. , Luo, S.‐S. , Ohba, T. , Katabuchi, H. , Takeshita, T. and Takizawa, T. , Short RNA duplexes elicit RIG‐I‐mediated apoptosis in a cell type‐ and length‐dependent manner. Sci. Signal. 2011. 4: ra74. DOI: 10.1126/scisignal.2001614. [DOI] [PubMed] [Google Scholar]
- 34. Okumura, F. , Okumura, A. , Uematsu, K. , Hatakeyama, S. , Zhang, D. and Kamura, T. , Activation of double‐stranded RNA‐activated protein kinase (PKR) by interferon‐stimulated gene 15 (ISG15) modification down‐regulates protein translation. J. Biol. Chem. 2013. 288: 2839–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Carrero, J. and Unanue, E. , Mechanisms and immunological effects of apoptosis caused by Listeria monocytogenes . Adv. Immunol. 2012. 113: 157–174. [DOI] [PubMed] [Google Scholar]
- 36. Carrero, J. , Calderon, B. and Unanue, E. , Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J. Exp. Med. 2004. 200: 535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. O'Connell, R. M. , Saha, S. K. , Vaidya, S. A. , Bruhn, K. W. , Miranda, G. A. , Zarnegar, B. , Perry, A. K. et al., Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J. Exp. Med. 2004. 200: 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Auerbuch, V. , Brockstedt, D. G. , Meyer‐Morse, N. , O'Riordan, M. and Portnoy, D. A. , Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes . J. Exp. Med. 2004. 200: 527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McCaffrey, R. , Fawcett, F. , O'Riordan, M. , Lee, K. , Havell, E. , Brown, P. and Portnoy, D. , A specific gene expression program triggered by Gram‐positive bacteria in the cytosol. Proc. Natl. Acad. Sci. U. S. A. 2004. 101: 11386–11391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stockinger, S. , Materna, T. , Stoiber, D. , Bayr, L. , Steinborn, R. , Kolbe, T. , Unger, H. et al., Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes . J. Immunol. 2002. 169: 6522–6529. [DOI] [PubMed] [Google Scholar]
- 41. Harding, H. , Zeng, H. , Zhang, Y. , Jungries, R. , Chung, P. , Plesken, H. , Sabatini, D. et al., Diabetes mellitus and exocrine pancreatic dysfunction in PERK‐/‐ mice reveals a role for translational control in secretory cell survival. Mol. Cell 2001. 7: 1153–1163. [DOI] [PubMed] [Google Scholar]
- 42. Masuoka, H. C. and Townes, T. M. , Targeted disruption of the activating transcription factor 4 gene results in severe fetal anemia in mice. Blood 2002. 99: 736–745. [DOI] [PubMed] [Google Scholar]
- 43. O'Riordan, M. , Yi, C. , Gonzalez, R. , Lee, K. and Portnoy, D. , Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc. Natl. Acad. Sci. U. S. A. 2002. 99: 13861–13866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ingrand, S. , Barrier, L. , Lafay‐Chebassier, C. , Fauconneau, B. , Page, G. and Hugon, J. , The oxindole/imidazole derivative C16 reduces in vivo brain PKR activation. FEBS Lett. 2007. 581: 4473–4478. [DOI] [PubMed] [Google Scholar]
- 45. Farrand, A. , LaChapelle, S. , Hotze, E. , Johnson, A. and Tweten, R. , Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc. Natl. Acad. Sci. U. S. A. 2010. 107: 4341–4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jammi, N. , Whitby, L. and Beal, P. , Small molecule inhibitors of the RNA‐dependent protein kinase. Biochem. Biophys. Res. Commun. 2003. 308: 50–57. [DOI] [PubMed] [Google Scholar]
- 47. Blalock, W. L. , Bavelloni, A. , Piazzi, M. , Tagliavini, F. , Faenza, I. , Martelli, A. M. , Follo, M. Y. et al., Multiple forms of PKR present in the nuclei of acute leukemia cells represent an active kinase that is responsive to stress. Leukemia 2011. 25: 236–245. [DOI] [PubMed] [Google Scholar]
- 48. Nakamura, T. , Furuhashi, M. , Li, P. , Cao, H. , Tuncman, G. , Sonenberg, N. , Gorgun, C. Z. et al., Double‐stranded RNA‐dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 2010. 140: 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gusella, G. , Musso, T. , Rottschafer, S. , Pulkki, K. and Varesio, L. , Potential requirement of a functional double‐stranded RNA‐dependent protein kinase (PKR) for the tumoricidal activation of macrophages by lipopolysaccharide or IFN‐alpha beta, but not IFN‐gamma. J. Immunol. 1995. 154: 345–354. [PubMed] [Google Scholar]
- 50. Cullinan, S. and Diehl, A. , Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006. 38: 317–332. [DOI] [PubMed] [Google Scholar]
- 51. Cullinan, S. and Diehl, J. , PERK‐dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004. 279: 20108–20117. [DOI] [PubMed] [Google Scholar]
- 52. Han, J. , Back, S. , Hur, J. , Lin, Y. , Gildersleeve, R. , Shan, J. , Yuan, C. et al., ER‐stress‐induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013. 15: 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tabas, I. and Ron, D. , Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011. 13: 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ferlito, M. , Wang, Q. , Fulton, W. , Colombani, P. , Marchionni, L. , Fox‐Talbot, K. , Paolocci, N. et al., Hydrogen sulfide [corrected] increases survival during sepsis: protective effect of CHOP inhibition. J. Immunol. 2014. 192: 1806–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Atkins, C. , Liu, Q. , Minthorn, E. , Zhang, S.‐Y. , Figueroa, D. J. , Moss, K. , Stanley, T. B. et al., Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013. 73: 1993–2002. [DOI] [PubMed] [Google Scholar]
- 56. Li, G. , Mongillo, M. , Chin, K. , Harding, H. , Ron, D. , Marks, A. and Tabas, I. , Role of ERO1‐alpha‐mediated stimulation of inositol 1,4,5‐triphosphate receptor activity in endoplasmic reticulum stress‐induced apoptosis. J. Cell Biol. 2009. 186: 783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Endo, M. , Mori, M. , Akira, S. and Gotoh, T. , C/EBP homologous protein (CHOP) is crucial for the induction of caspase‐11 and the pathogenesis of lipopolysaccharide‐induced inflammation. J. Immunol. 2006. 176: 6245–6253. [DOI] [PubMed] [Google Scholar]
- 58. Allagnat, F. , Fukaya, M. , Nogueira, T. , Delaroche, D. , Welsh, N. , Marselli, L. , Marchetti, P. et al., C/EBP homologous protein contributes to cytokine‐induced pro‐inflammatory responses and apoptosis in β‐cells. Cell Death Differ. 2012. 19: 1836–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Oyadomari, S. and Mori, M. , Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004. 11: 381–389. [DOI] [PubMed] [Google Scholar]
- 60. Hiramatsu, N. , Chiang, W. , Kurt, T. , Sigurdson, C. and Lin, J. , Multiple mechanisms of unfolded protein response‐induced cell death. Am. J. Pathol. 2015. 185: 1800–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sauer, J. , Witte, C. , Zemansky, J. , Hanson, B. , Lauer, P. and Portnoy, D. , Listeria monocytogenes triggers AIM2‐mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 2010. 7: 412–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zinszner, H. , Kuroda, M. , Batchvarova, N. , Lightfoot, R. , Remotti, H. , Stevens, J. and Ron, D. , CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998. 12: 982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Inaba, K. , Inaba, M. , Romani, N. , Aya, H. , Deguchi, M. , Ikehara, S. , Muramatsu, S. et al., Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony‐stimulating factor. J. Exp. Med. 1992. 176: 1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Celada, A. , Gray, P. , Rinderknecht, E. and Schreiber, R. , Evidence for a gamma‐interferon receptor that regulates macrophage tumoricidal activity. J. Exp. Med. 1984. 160: 55–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Edelson, B. , Bradstreet, T. , Hildner, K. , Carrero, J. , Frederick, K. , KC, W. , Belizaire, R. et al., CD8α(+) dendritic cells are an obligate cellular entry point for productive infection by Listeria monocytogenes. Immunity 2011. 35: 236–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stockinger, S. , Kastner, R. , Kernbauer, E. , Pilz, A. , Westermayer, S. , Reutterer, B. , Soulat, D. et al., Characterization of the interferon‐producing cell in mice infected with Listeria monocytogenes . PLoS Pathog. 2009. 5: e1000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Allen, J. R. , Nguyen, L. X. , Sargent, K. E. G. , Lipson, K. L. , Hackett, A. and Urano, F. , High ER stress in beta‐cells stimulates intracellular degradation of misfolded insulin. Biochem. Biophys. Res. Commun. 2004. 324: 166–170. [DOI] [PubMed] [Google Scholar]
- 68. Wheeler, M. , Rizzi, M. , Sasik, R. , Almanza, G. , Hardiman, G. and Zanetti, M. , KDEL‐retained antigen in B lymphocytes induces a proinflammatory response: a possible role for endoplasmic reticulum stress in adaptive T cell immunity. J. Immunol. 2008. 181: 256–264. [DOI] [PubMed] [Google Scholar]
- 69. Kozutsumi, Y. , Segal, M. , Normington, K. , Gething, M. J. and Sambrook, J. , The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose‐regulated proteins. Nature 1988. 332: 462–464. [DOI] [PubMed] [Google Scholar]
- 70. Fonseca, S. G. , Fukuma, M. , Lipson, K. L. , Nguyen, L. X. , Allen, J. R. , Oka, Y. and Urano, F. , WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta‐cells. J. Biol. Chem. 2005. 280: 39609–39615. [DOI] [PubMed] [Google Scholar]
- 71. Calfon, M. , Zeng, H. , Urano, F. , Till, J. H. , Hubbard, S. R. , Harding, H. P. , Clark, S. G. et al., IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP‐1 mRNA. Nature 2002. 415: 92–96. [DOI] [PubMed] [Google Scholar]
- 72. Glomski, I. , Gedde, M. , Tsang, A. , Swanson, J. and Portnoy, D. , The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. J. Cell Biol. 2002. 156: 1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supporting Information Figure 1. The ISR is triggered by L. monocytogenes infection in BMM. C57BL/6 BMM (106 cells) were infected with L. monocytogenes (LM LLO) and L. monocytogenes Δhly (LM ΔLLO) per 12h and 24h. Two different MOI where tested. After infection, BMM were incubated per 12h and 24h. cDNA was synthesized from extracted mRNA. Then A. Hspa5 (BIP); B. Wfs1; C. Xbp1s; D. Ddit3 (CHOP); E. Ppp1r15a (GADD34); F. Atf4 were amplified with specific primers by qRT‐PCR. The fold change in gene expression was calculated using 2‐ΔΔCT. Dots are mean ±SEM of 6 biological replicates. UT: Untreated condition, Tg: Thapsigargin 0.1μM as a positive control. P value was calculated with Mann‐Whitney test p<0.05 (*), p<0.001 (**).