

ABSTRACT
Efficient directed migration requires tight regulation of chemoattractant signal transduction pathways in both space and time, but the mechanisms involved in such regulation are not well understood. Here, we investigated the role of protein kinase A (PKA) in controlling signaling of the chemoattractant cAMP in Dictyostelium discoideum. We found that cells lacking PKA display severe chemotaxis defects, including impaired directional sensing. Although PKA is an important regulator of developmental gene expression, including the cAMP receptor cAR1, our studies using exogenously expressed cAR1 in cells lacking PKA, cells lacking adenylyl cyclase A (ACA) and cells treated with the PKA-selective pharmacological inhibitor H89, suggest that PKA controls chemoattractant signal transduction, in part, through the regulation of RasG, Rap1 and TORC2. As these pathways control the ACA-mediated production of intracellular cAMP, they lie upstream of PKA in this chemoattractant signaling network. Consequently, we propose that the PKA-mediated regulation of the upstream RasG, Rap1 and TORC2 signaling pathways is part of a negative feedback mechanism controlling chemoattractant signal transduction during Dictyostelium chemotaxis.
KEY WORDS: Chemotaxis, Migration, PKA, Negative feedback, Directional sensing, Chemotactic signaling
Highlighted Article: Protein kinase A is necessary for gradient sensing and chemotaxis towards cAMP in Dictyostelium, in part, through the proper spatiotemporal regulation of key upstream chemoattractant signal transduction pathways.
INTRODUCTION
Chemotaxis, the ability of cells to detect and migrate up chemical (chemoattractant) gradients, is a conserved cellular behavior that is not only essential to normal human physiology but is also implicated in the onset and progression of diseases (Bravo-Cordero et al., 2012; Kolaczkowska and Kubes, 2013; Richardson and Lehmann, 2010; Sadik and Luster, 2012; Theveneau and Mayor, 2012; Zernecke and Weber, 2010). The detection of chemoattractants by cells triggers an intracellular network of signal transduction pathways that coordinately control the ability of cells to interpret the gradient, polarize and move (Artemenko et al., 2014). Moreover, these processes are often coupled to the paracrine release of chemoattractants that relay the original signal to promote group cell migration (Afonso et al., 2012; Garcia and Parent, 2008; Wyckoff et al., 2004). In order for cells to efficiently perform chemotaxis, chemoattractant signal transduction pathways are tightly regulated in both space and time, and adapt to persistent stimulation (Charest and Firtel, 2006; Hoeller et al., 2014; Wang, 2009). Adaptation is believed to be necessary for cells to sense and respond to increasing chemoattractant concentrations as they migrate up the gradient (Chang and Levchenko, 2013). However, very little is understood about the mediators of adaptation and the regulatory mechanisms involved in the spatiotemporal control of chemotactic signaling.
In the established chemotaxis experimental model Dictyostelium discoideum, Ras-family GTPases, including RasC, RasG and Rap1, are critical mediators of the chemotactic response to cAMP (Charest and Firtel, 2007). Rap1 controls cell–substrate adhesion and cell polarity by promoting remodeling of both actin and myosin, in part, through phosphoinositide 3-kinase (PI3K), Rac, the serine/threonine kinase Phg2, as well as through the regulation of the Target of Rapamycin Complex 2 (TORC2) (Jeon et al., 2007a; Khanna et al., 2016; Kortholt et al., 2006, 2010; Mun and Jeon, 2012; Plak et al., 2013). RasG activates PI3K, thereby controlling the site of F-actin polymerization and the direction of migration (Bolourani et al., 2006; Sasaki et al., 2004; Zhang et al., 2008). Tight spatiotemporal regulation of RasG is critical for gradient sensing during chemotaxis (Takeda et al., 2012; Zhang et al., 2008). Finally, RasC directly promotes TORC2 activation, thereby modulating F-actin dynamics and controlling cAMP production by adenylyl cyclase A (ACA, encoded by acaA) (Bolourani et al., 2006; Cai et al., 2010; Charest et al., 2010; Khanna et al., 2016). In addition, we have found that a negative-feedback mechanism promotes adaptation of chemoattractant-stimulated RasC activity (Charest et al., 2010). Downstream from RasC, TORC2-activated Akt/protein kinase B (PKB) and PKB-related PKBR1 (also known as PKGB) phosphorylate the scaffold protein Sca1, which assembles a RasC-activating complex containing the RasC-specific guanine exchange factor (GEF) Aimless (also known as RasGEFA). Phosphorylation of Sca1 by PKB kinases inhibits the localization of the Sca1 complex to the plasma membrane, thereby preventing further RasC activation. Interestingly, phosphorylation of Sca1 by PKB kinases is elevated in cells that lack the PKA catalytic subunit (PKA-C; pkaC null cells) (Charest et al., 2010). Since the RasC–TORC2–PKB-kinase pathway controls the ACA-mediated intracellular production of cAMP, this pathway promotes PKA activation in response to extracellular cAMP chemoattractant stimulation (Charest et al., 2010; Lee et al., 2005; Lim et al., 2001). Thus, these previous observations indicate that PKA might regulate the RasC–TORC2–PKB-kinase signaling pathway during chemotaxis through a negative-feedback loop.
cAMP in Dictyostelium plays two different and completely separate roles: that of an extracellular chemoattractant and that of an intracellular signaling molecule (Reymond et al., 1995). Since cAMP is membrane impermeable, the extracellular cAMP acting as chemoattractant for Dictyostelium does not directly activate PKA, it only acts through stimulation of specific seven-transmembrane cAMP chemoattractant receptors (cARs; Insall et al., 1994). In response to the chemoattractant stimulation, intracellular cAMP is produced by ACA, and part of this cAMP is used to activate PKA and part is used for relaying the chemoattractant signal to neighboring Dictyostelium cells by being actively exported outside of the cells through ABC transporters (Garcia and Parent, 2008; Miranda et al., 2015).
The role of PKA in Dictyostelium development and morphogenesis is well characterized (Loomis, 1998), whereas its role in chemotaxis is not understood. PKA is required for the starvation-induced aggregation of Dictyostelium cells (Mann and Firtel, 1991; Mann et al., 1997), a process driven by chemotaxis, and was found to be involved in controlling the directional extension of pseudopods during migration (Stepanovic et al., 2005; Zhang et al., 2003). In mammalian cells, PKA has been shown to play a central role in actin-based cell migration through the differential regulation of Rac, Rho and Rap1 GTPases, as well as of VASP, PI3K, PAK and LIM kinases, at the leading edge of migrating cells (Chen et al., 2005; Howe, 2004; Howe et al., 2005; Jones and Sharief, 2005; Lim et al., 2008; Nadella et al., 2009; Paulucci-Holthauzen et al., 2009; Takahashi et al., 2013; Toriyama et al., 2012; Zimmerman et al., 2013).
The present study was undertaken to investigate the role of PKA in controlling chemotactic signaling pathways and directed cell migration in Dictyostelium. We performed a detailed characterization of the effect that the lack of PKA activity in Dictyostelium has on chemotaxis in response to cAMP and on the regulation of known cAMP-induced responses in Dictyostelium. We found that, in cells lacking PKA function, many early cAMP-induced chemotactic responses, including RasG, Rap1, PI3K, TORC2, PKB and PKBR1 activation, are upregulated and fail to be spatially restricted upon exposure to cAMP gradients. Therefore, our results suggest that PKA is necessary for proper spatiotemporal regulation of early chemoattractant signal transduction pathways, which is critical to Dictyostelium chemotaxis. Although this regulatory mechanism might include the PKA-mediated transcriptional control of additional involved proteins, our study suggests that direct control of the signaling pathways by PKA could explain the observed effects.
RESULTS
pkaC null cells are unable to perform chemotaxis
To investigate the role of PKA in Dictyostelium chemotaxis along a cAMP gradient, we started by characterizing the chemotaxis phenotypes of Dictyostelium cells that lacked PKA-C (pkaC null) and compared these phenotypes to those of wild-type cells. pkaC null cells are viable and, although they have been shown to lack expression of ACA (Mann et al., 1997), pkaC and acaA null cells express key aggregative genes when provided with exogenous cAMP pulses (Mann et al., 1992, 1997; Pitt et al., 1993). Hence, in such conditions, the use of pkaC null cells should be informative as to the role of PKA in chemotaxis. Using cells that were responsive to the chemoattractant cAMP (pulsed with exogenous cAMP for 5.5 h), we found that pkaC null cells exhibit severe chemotaxis defects (Fig. 1). Developed wild-type cells placed in an exponential cAMP gradient polarize and migrate efficiently, whereas pkaC null cells do not polarize and they extend pseudopods in random directions, often in directions opposite to the gradient (Fig. 1A; Movies 1 and 2). As a consequence, pkaC null cells display poor persistence of movement (0.16±0.07 versus 0.79±0.09 for wild-type cells; mean±s.d.) and completely fail to migrate towards the chemoattractant source, in this case a micropipette filled with 150 μM cAMP (chemotactic index of 0.03±0.07 versus 0.76±0.12 for wild-type cells) (Fig. 1B,C). However, the pkaC null cells are motile and display an averaged displacement speed of 4.9±1.4 µm/min compared to 5.8±0.9 µm/min for wild-type cells (Fig. 1C). To verify that this phenotype is not unique to this pkaC null strain, we tested another strain, in which PKA-C had been independently disrupted by another group (HBW1; Primpke et al., 2000). However, we found that these cells are a little heterogeneous and unstable as the phenotypes changed with passages in cell culture. Nevertheless, we found that young HBW1 cells, compared to their control cells (HBW2), have severe chemotaxis defects and have cAMP-induced responses similar to those of the other pkaC null cells (Fig. S1).
Fig. 1.
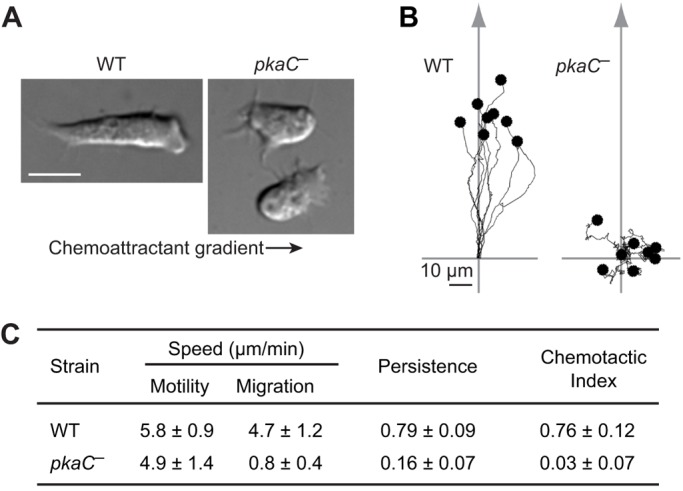
The chemotaxis phenotype of pkaC null cells. (A) Morphology of wild-type (WT) and pkaC null cells (pkaC—) exposed to a cAMP gradient. The direction of the gradient is indicated by the arrow. Scale bar: 10 μm. (B) Traces of representative cells migrating in an exponential cAMP gradient created by a point source. The starting point of each cell was apposed to the axis origin. The arrow indicates the direction of the gradient. (C) Quantitative analysis of the chemotactic behavior of cells (13 WT and eight pkaC null cells) from three independent experiments. Motility speed represents the total path length divided by time; migration speed represents the total linear displacement (end point–starting point) divided by time; persistence indicates path linearity and was calculated as the linear displacement divided by total path length; chemotactic index represents the directionality of the cells' movements relative to the gradient. Data are mean±s.d.
Taken together, our analyses suggests that, although pkaC null cells are unable to perform chemotaxis, they do extend protrusions and move. These observations then suggest that PKA is not required for cell motility in Dictyostelium but is essential for the establishment of cell polarity and chemotaxis towards cAMP.
pkaC null cells fail to properly localize and temporally regulate F-actin and myosin
To explore the underlying causes for the strong chemotaxis defects of pkaC null cells, we first assessed the spatiotemporal dynamics of F-actin in cells using the fluorescent reporter Lifeact-GFP (Riedl et al., 2008). As previously described (Chung and Firtel, 1999), we observe that migrating wild-type cells display cortical F-actin and F-actin-rich pseudopods at the leading edge (Fig. 2A; Movie 3). Cells lacking PKA also display cortical F-actin, but, consistent with the observed pkaC null chemotaxis phenotype, these cells exhibit many small and randomly localized F-actin-rich protrusions (Fig. 2A; Movie 4). Lifeact-GFP labeling of F-actin allowed us to observe that these protrusions, although they sometimes produce a pseudopod that correlates with random movement of the cell, often form an endocytic cup or are just retracted. In addition, in cells that were pulsed with cAMP, washed and then left to rest instead of being exposing to a cAMP gradient, we observed that, in contrast to wild-type cells displaying low basal levels of F-actin, most pkaC null cells display elevated basal levels of F-actin enriched within numerous spontaneous protrusions that often look like membrane ruffles rather than pseudopods (Fig. 2B,D; Movies 5 and 6).
Fig. 2.

Spatiotemporal dynamics of F-actin and myosin in pkaC null cells. Live imaging of F-actin (Lifeact-GFP) and Myosin II (GFP-labeled myosin light chain, GFP–MyoII) fluorescent reporters in cells exposed to an exponential gradient of cAMP (A, arrows indicate the direction of the gradient) and in cells uniformly stimulated with 5 μM cAMP for the indicated times (B). Scale bars: 10 μm. (C) Relative fluorescence intensity of Lifeact-GFP in cells uniformly stimulated with cAMP for the indicated times. Data represent the mean fluorescence intensity±s.e.m. of 71 wild-type (WT) T and 48 pkaC null (pkaC—) cells. (D) cAMP-induced F-actin polymerization measured by phalloidin binding, expressed as a fold over the basal level. (Inset) Basal F-actin levels measured according to protein mass, expressed as a fold over the WT basal levels. Data represent the mean±s.e.m. of five different experiments. *P<0.05 in a two-tailed t-test compared to corresponding wild-type samples. (E) Relative cytosolic fluorescence intensity of GFP–MyoII in cells that had been uniformly stimulated with cAMP for the indicated times. Data represent the mean fluorescence intensity±s.e.m. of 91 WT and 50 pkaC null cells. Imaging data are representative of at least three independent experiments.
cAMP stimulation of wild-type Dictyostelium cells produces a biphasic F-actin polymerization response, with a sharp first peak at 5 s, which is linked to actin cytoskeleton reorganization (cringe response), and a lower but wider peak at 30-60 s post stimulation associated with pseudopod protrusion (Fig. 2B–D; Movies 5 and 6) (Hall et al., 1989). By contrast, pkaC null cells display considerably higher basal F-actin levels (near significant difference with P=0.07) and, as a likely consequence, a blunted F-actin polymerization response, with a markedly smaller first peak followed by limited depolymerization and prolonged elevated levels of F-actin (Fig. 2B–D; Movies 5 and 6). Taken together, these results suggest that F-actin levels are upregulated in resting pkaC null cells and that F-actin is both temporally and spatially misregulated in migrating cells that lack PKA-C.
Similarly, we investigated the spatiotemporal profile of myosin II in cells that had been exposed to cAMP using GFP-fused myosin heavy chain A (GFP–MyoII) (Chung and Firtel, 1999). As reported previously, GFP–MyoII is excluded from the leading edge and localizes to the sides and posterior of migrating wild-type cells (Fig. 2A) (Chung and Firtel, 1999). Resting, unstimulated wild-type cells display basal levels of GFP–MyoII at the cell cortex, which rapidly delocalizes to the cytosol upon uniform cAMP stimulation (Fig. 2B) (Jeon et al., 2007a). This delocalization is then followed by an increase in cortical GFP–MyoII, peaking at ∼30-40 s, and a return to basal levels by 60 s post stimulus (Fig. 2B,E). In pkaC null cells, we observed some heterogeneity in the spatiotemporal dynamics of GFP–MyoII. On average, however, we found that pkaC null cells that were either unstimulated or had been exposed to a cAMP gradient display relative cortical levels of GFP–MyoII similar to those of wild-type cells, with GFP–MyoII being excluded from extended protrusions and enriched in retracting protrusions (Fig. 2A,B). Consistent with the observed random extension of membrane protrusions in migrating pkaC null cells, the cortical localization of GFP–MyoII in these cells was not restricted to the side of the cells that faced the lowest cAMP concentrations but was observed on any side of the cells, changing frequently and displaying no directional bias (Fig. 2A). In addition, we observed that, upon uniform cAMP stimulation, GFP–MyoII at the cell cortex of pkaC null cells translocates to the cytosol with delayed kinetics compared to those in wild-type cells and then only slowly returns to basal cortical levels (Fig. 2B,E). These results suggest that, in addition to F-actin, the timing and localization of myosin II assembly in cells that lack PKA-C are misregulated during chemotaxis.
Cells lacking PKA function have elevated PI3K, TORC2, PKB and PKBR1 activity
We have previously found that the phosphorylation of Sca1 mediated by PKB kinases is elevated in pkaC null cells (Charest et al., 2010). To then investigate the mechanism by which PKA controls cytoskeletal remodeling and chemotaxis, we assessed the kinase activity of PKB and PKBR1 in cells that lacked PKA-C. We observed that the basal levels of PKB and PKBR1 kinase activity are elevated and the cAMP-induced stimulation of PKB and PKBR1 activity is prolonged in pkaC null cells compared to that in wild-type cells (Fig. 3A). We also observed that in cells lacking ACA, PKB and PKBR1 display similarly elevated basal activity levels and prolonged cAMP-induced activation, although these effects are less pronounced than those observed in pkaC null cells (Fig. 3A). This finding is not surprising because, although ACA is responsible for most of the chemoattractant-induced cAMP production in Dictyostelium, some cAMP is produced in acaA null cells and, thus, these cells are expected to have residual PKA activity (Bagorda et al., 2009; Kim et al., 1998; Söderbom et al., 1999). Consistent with the observed elevated PKB and PKBR1 activity levels in pkaC null cells, these cells also display higher basal phosphorylation levels and extended cAMP-induced phosphorylation of cellular substrates of PKB kinases compared to those found in wild-type cells (Fig. 3B), reminiscent of the Sca1 phosphorylation profile previously shown in these cells (Charest et al., 2010). Thus, these results suggest that PKB and PKBR1 activities are upregulated in cells that lack PKA function.
Fig. 3.

Activity of chemotactic effectors PKB, PKBR1, PI3K and TORC2 in cells lacking PKA function. (A) cAMP-induced PKB and PKBR1 kinase activity in wild-type (WT) and pkaC null (pkaC—) cells, and in cells lacking adenylyl cyclase A (acaA—). The kinase activity of immunopurified PKB and PKBR1 was assessed using H2B as a substrate. H2B phosphorylation was detected by autoradiography, and PKB and PKBR1 were revealed by immunoblotting. (B) cAMP-induced phosphorylation of PKB and PKBR1 substrates was detected using an antibody against PKB-phosphorylated substrates (P-PKBS). (C) Localization of the PI(3,4,5)P3 reporter PHcrac–GFP in developed resting cells. (D) Live imaging of PHcrac–GFP in cells exposed to an exponential gradient of cAMP. The direction of the gradient is indicated by the arrow. (E) Live imaging of PHcrac–GFP in cells that had been uniformly stimulated with 5 μM cAMP for the indicated times, and relative cytosolic fluorescence intensity of PHcrac–GFP measured in cells that had been uniformly stimulated with cAMP. Graph data represent the mean fluorescence intensity±s.e.m. of 20 WT and 26 pkaC null cells, expressed as a fold over the basal level. (F) TORC2-mediated phosphorylation of PKB and PKBR1 at their respective hydrophobic motifs (HMP) was assessed in WT, pkaC— and acaA— cells, and in WT cells treated with the PKA pharmacological inhibitor H89 or the vehicle (DMSO). CB, Coomassie blue staining. Scale bars: 10 μm. Data are representative of at least three independent experiments.
Two upstream pathways co-regulate the activity of PKB and PKBR1 during cAMP chemotaxis in Dictyostelium: (1) the PI3K-dependent pathway that leads to the phosphoinositide-dependent protein kinase (PDK)-mediated phosphorylation of the activation loop in PKB and PKBR1; and (2) the TORC2 pathway leading to phosphorylation of the hydrophobic motif in PKB and PKBR1 (Kamimura and Devreotes, 2010; Liao et al., 2010). We asked whether the elevated activity of PKB and PKBR1 in pkaC null cells results from upregulated PI3K and/or TORC2 activity. To assess PI3K activity, we expressed a fluorescent phosphatidylinositol (3,4,5)-trisphosphate [PI(3,4,5)P3] reporter comprising the pleckstrin homology domain of the cytosolic regulator of adenylyl cyclase fused to GFP (PHcrac–GFP) (Dormann et al., 2002; Parent et al., 1998). We observed that PHcrac–GFP mostly localized to the cytosol of resting wild-type cells with occasional enrichment to the plasma membrane of extended protrusions, whereas many unstimulated pkaC null cells displayed membrane-localized PHcrac–GFP at structures corresponding to protrusions, membrane ruffles and endocytic cups (Fig. 3C). During chemotaxis in response to cAMP, PHcrac–GFP localizes to the front of chemotaxing wild-type cells, as previously described (Dormann et al., 2002; Parent et al., 1998), whereas in pkaC null cells exposed to the cAMP gradient, the PI(3,4,5)P3 reporter localizes randomly around the cell cortex, corresponding to the numerous randomly extended protrusions (Fig. 3D), reminiscent of the localization of the F-actin reporter Lifeact-GFP described above.
As previously reported, uniform cAMP stimulation induces the temporary translocation of PHcrac–GFP from the cytosol to the plasma membrane, peaking at ∼8 s, reflecting PI3K activation and transient PI(3,4,5)P3 accumulation at this site (Fig. 3E; Movie 7). After peaking at ∼8 s post-cAMP stimulation, the levels of PHcrac–GFP at the membrane of wild-type cells decrease below pre-stimulus levels, consistent with previous observations (Tang et al., 2014). In contrast, pkaC null cells display a reduced PHcrac–GFP translocation response, when normalized to their respective basal levels, with a slightly delayed and broader peak that reaches a maximum at ∼10-12 s before returning to pre-stimulus levels (Fig. 3E; Movie 8). Therefore, these observations indicate that PI(3,4,5)P3 production is misregulated both temporally and spatially in cells that lack PKA.
As a measure of TORC2 activity, we assessed the TORC2-mediated phosphorylation of PKB and PKBR1 at their hydrophobic motifs (HM) before and upon cAMP stimulation, as previously described (Kamimura et al., 2009). In contrast to wild-type cells in which the TORC2 phosphorylation of PKB and PKBR1 HMs is very transient, peaking at 5-10 s and returning to basal levels by 40 s post-stimulus, pkaC null cells display considerably elevated and extended HM phosphorylation in PKB and PKBR1 (Fig. 3F). Similarly, we observed that the TORC2-mediated phosphorylation of the PKB and PKBR1 HMs is elevated in cells that lacked ACA, as well as in cells that had been treated with the PKA-selective pharmacological inhibitor H89 (Fig. 3F; Fig. S2) (Zeng et al., 2001). Hence, these results suggest that loss of PKA function leads to an increased activation of TORC2. Taken together, our observations indicate that the elevated PKB and PKBR1 activity levels in cells that lack PKA function are likely to result from the combination of upregulated PI3K and TORC2 pathways in these cells.
Lack of PKA function has different effects on RasC, RasG and Rap1
In Dictyostelium, the Ras protein RasG is the main mediator of PI3K activation in response to cAMP stimulation (Bolourani et al., 2006; Sasaki et al., 2004), RasC is the main activator of TORC2 (Cai et al., 2010; Charest et al., 2010), and Rap1 has been shown to regulate the activity of both PI3K and TORC2 (Khanna et al., 2016; Kortholt et al., 2010) in addition to other Rap1 effectors. To then investigate potential mechanisms underlying the elevated activity levels of TORC2 and PI3K in cells that lack PKA function, we examined the activity of RasC, RasG and Rap1 under such conditions. First, we assessed the activity of Flag-tagged RasC in pkaC null cells and in wild-type cells treated with H89. Interestingly, we observed that cAMP-induced RasC activation is considerably decreased in cells lacking PKA activity (Fig. 4A). We also found that Sca1 fails to translocate from the cytosol to the plasma membrane in pkaC null cells that had been stimulated with cAMP and that Sca1 translocation is considerably reduced in acaA null cells compared to that in wild-type cells (Fig. 4B,C). These observations are consistent with our previous findings that phosphorylation of Sca1 mediated by PKB kinases is elevated in pkaC null cells and that this phosphorylation inhibits the translocation of the Sca1 complex to the plasma membrane and RasC activation (Charest et al., 2010). These results show that RasC activity is downregulated in cells that lack PKA function and, therefore, suggest that PKA is necessary for optimal RasC activation. Consequently, this PKA-mediated positive regulation of RasC cannot explain the elevated TORC2 activity observed in pkaC null cells.
Fig. 4.

RasC pathway activation in cells lacking PKA function. (A) cAMP-induced activation of Flag-tagged RasC expressed in wild-type (WT) and pkaC null (pkaC—) cells, and in WT cells treated with the PKA pharmacological inhibitor H89 or the vehicle (DMSO). Active RasC (RasC–GTP) was pulled down with GST–Byr2(RBD). RasC–GTP and total RasC were revealed by immunoblotting for Flag. (B) Live imaging of GFP–Sca1 in cells that had been uniformly stimulated with 5 μM cAMP for the indicated times. Scale bars: 10 μm. (C) Relative cytosolic fluorescence intensity of GFP–Sca1 in cells that had been uniformly stimulated with cAMP. Data represent the mean fluorescence intensity±s.e.m. of 150 WT, 33 pkaC null cells and 147 acaA null cells. Data represent or are representative of at least three independent experiments.
In contrast to the decrease in RasC activation observed in cells that lack PKA activity, we found that both RasG and Rap1 display elevated basal activity levels, and elevated and extended cAMP-induced activation, in pkaC null cells compared to those in wild-type cells (Fig. 5A). To image RasG activity dynamics, we used the Ras activity reporter that comprises the Ras-binding domain (RBD) of human Raf1 fused to GFP [Raf1(RBD)–GFP], which binds to RasG and not to RasC (Kae et al., 2004; Kortholt et al., 2013; Sasaki et al., 2004). Although Raf1(RBD)–GFP also binds to the active forms of Dictyostelium RasB and RasD (Zhang et al., 2008), RasG is believed to be the predominant Ras protein expressed in cAMP-responsive aggregation-stage Dictyostelium cells (at 5–8 h after onset of development) (Rot et al., 2009), and thus Raf1(RBD)–GFP is believed to be a good reporter of active RasG. Raf1(RBD)–GFP is mostly found in the cytosol of resting wild-type cells, whereas many resting pkaC null cells display cortex-localized Raf1(RBD)–GFP – at membrane protrusions, membrane ruffles and endocytic cups – which is similar to the observed localization of both the F-actin and PI(3,4,5)P3 reporters in these cells (Fig. 5B). Also reminiscent of the observed F-actin and PI(3,4,5)P3 dynamics in pkaC null cells, Raf1(RBD)–GFP is highly enriched in the randomly extended membrane protrusions of pkaC null cells exposed to a cAMP gradient (Fig. 5C). Consistent with the profile of the PI(3,4,5)P3 response, which lies downstream from RasG, the Ras activity reporter exhibits a reduced translocation to the cell cortex in pkaC null cells upon uniform cAMP stimulation compared to that in wild-type cells, when normalized to their respective basal levels (Fig. 5D; Movies 9 and 10).
Fig. 5.

Spatiotemporal dynamics of RasG and Rap1 activities in pkaC null cells. (A) cAMP-induced RasG and Rap1 activation. Active RasG (Ras–GTP) and Rap1 (Rap1–GTP) were pulled down with GST–Raf1(RBD) and GST–RalGDS(RBD), respectively, and revealed by immunoblotting for pan-Ras and Rap1. (B,E) Localization of Raf1(RBD)–GFP (B) and RalGDS(RBD)–GFP (E) in resting cells. (C,F) Live imaging of Raf1(RBD)–GFP (C) and RalGDS(RBD)–GFP (F) in cells that had been exposed to an exponential gradient of cAMP. (D,G) Live imaging of Raf1(RBD)–GFP (D) and RalGDS(RBD)–GFP (G) in cells that had been uniformly stimulated with 5 μM cAMP for the indicated times. Relative reporter cytosolic fluorescence intensity is shown on the right, expressed as a fold over the basal level. Scale bars: 10 μm. Quantified data represent the mean fluorescence intensity±s.e.m. of 20 Raf1(RBD)–GFP-expressing WT cells, nine Raf1(RBD)–GFP-expressing pkaC null cells, 109 RalGDS(RBD)–GFP-expressing WT cells and 135 RalGDS(RBD)–GFP-expressing pkaC null cells. Data represent or are representative of at least three independent experiments.
To image Rap1 activity dynamics, we used the Rap1 activity reporter comprising the RBD of human RalGDS [RalGDS(RBD)–GFP] (Jeon et al., 2007b). In resting cells, although some pkaC null cells are found with slightly more RalGDS(RBD)–GFP localized to the membrane, for the most part, RalGDS(RBD)–GFP displays localization patterns more similar to those in wild-type cells (Fig. 5E). However, RalGDS(RBD)–GFP is clearly enriched in the randomly extended membrane protrusions of pkaC null cells exposed to a cAMP gradient (Fig. 5F). In addition, the Rap1 activity reporter displays increased translocation to and prolonged localization at the cell cortex in pkaC null cells in response to uniform cAMP stimulation compared to that in wild-type cells (Fig. 5G; Movies 11 and 12). Therefore, together, our observations suggest that, in contrast to the PKA-dependent positive regulation of RasC, PKA has a negative regulatory effect on RasG and Rap1 activation.
pkaC null cells have severe gradient-sensing defects
Spatiotemporal regulation of RasG activity is required for directional sensing of cAMP gradients in Dictyostelium (Zhang et al., 2008). Our findings that pkaC null cells are motile but fail to migrate along a cAMP gradient and that RasG activity is upregulated in the absence of PKA suggest that PKA may be necessary for gradient sensing. To test whether PKA plays a role in cAMP gradient sensing, we assessed the Ras response, using Raf1(RBD)–GFP, in cells that had been treated with the F-actin inhibitor Latrunculin B (LatB). LatB treatment generates cells that are unable to polymerize F-actin and are, thus, paralyzed, symmetrical and spherical (Parent et al., 1998). LatB-treated cells can then be used to interrogate cAMP gradient-sensing capabilities in the absence of feedback from the actin cytoskeleton (Zhang et al., 2008). As previously reported (Zhang et al., 2008), LatB-treated wild-type cells respond to the cAMP gradient and accumulate Raf1(RBD)–GFP in the form of a crescent at the cell cortex on the side closest to the chemoattractant source, a cAMP-filled micropipette (Fig. 6A). Upon repositioning of the micropipette to the opposite side of the cell, Raf1(RBD)–GFP is quickly delocalized from its previous site and accumulates at the cortex on the side closest to the new position of the micropipette within 10-15 s, reflecting the rapid deactivation and activation of Ras at each site, respectively (Fig. 6A,B; Movie 13). In wild-type cells, Raf1(RBD)–GFP accumulation is highly restricted to the side closest to the cAMP chemoattractant source and never accumulates at the side opposite to the micropipette or the lateral sides, illustrating the highly spatially regulated Ras response.
Fig. 6.

Gradient sensing in pkaC null cells. (A) Live imaging of Raf1(RBD)–GFP in cells, treated with 10 μM LatB, upon changes of cAMP gradient orientation. Two examples of pkaC null cells are shown (cell 1 and cell 2). Data are representative of at least three independent experiments. * indicates the position of the cAMP-filled micropipette. Scale bars: 10 μm. (B) Quantification of the relative fluorescence intensity of membrane-localized Raf1(RBD)–GFP in cells shown in A, for the sides alternatively closest and opposite to the positioned micropipette (sides A and B; top graphs) and the lateral sides (sides 1 and 2; bottom graphs), as indicated in the graph key.
In contrast to the rapid and restricted accumulation of Raf1(RBD)–GFP at the cell cortex of LatB-treated wild-type cells upon cAMP gradient sensing, accumulation of the Ras reporter to the cortex of LatB-treated pkaC null cells does not as strongly correlate with the position of the chemoattractant source (Fig. 6; Movies 14 and 15). In pkaC null cells, accumulation of Raf1(RBD)–GFP on the side of the cell closest to the chemoattractant does become enriched, but with very slow kinetics (∼ 1 min) compared to that for wild-type cells (within 15 s), and localization of the reporter is not restricted there. Raf1(RBD)–GFP is often observed to be located at the lateral sides and even sometimes on the opposite side from that facing the micropipette, which is never observed for wild-type cells. In pkaC null cells, the Ras reporter appears to be moving around the cell cortex as if the Ras signal is propagating, reminiscent of the previously reported self-organized waves of PI(3,4,5)P3 that are generated spontaneously on the membrane in the absence of the chemoattractant (Arai et al., 2010). These observations suggest that, although pkaC null cells respond to the cAMP gradient, PKA is necessary for cells to properly restrict Ras activation to the side closest to the chemoattractant source, which is likely to underlie the inability of cells that lack PKA to accurately determine the direction of the gradient during the chemotaxis response to cAMP.
cAR1 expression in pkaC null cells
Our results using the pkaC null cells as well as acaA null cells and the H89 inhibitor suggest a role for PKA in regulating the Ras and Rap1 pathways in response to the cAMP chemoattractant in Dictyostelium. However, it is also possible that some of the observed effects associated with the lack of PKA are indirect and due to its role in controlling gene expression. We determined that, at least, Ras, Rap1, Pia (an essential TORC2 component), PKB and PKBR1 are well expressed in pkaC null cells (Fig. 7A; Fig. S3). Interestingly, as previously reported for wild-type cells (Stajdohar et al., 2015), the observed regulated decrease in PKB expression in 5.5-h cAMP-pulsed cells indicate that some developmental gene expression is properly conserved in pkaC null cells (Fig. 7A). By contrast, we failed to detect the expression of the cAMP chemotactic receptor cAR1 (also known as carA) by immunoblotting. However, the fact that pkaC null cells respond well to cAMP stimulation, whereas cells lacking cAR1 (carA null cells) mostly don't, suggests that some cAR1 is likely to be present in pkaC null cells (Fig. 7D,E) (Liao et al., 2013). In order to test whether the shortage of cAR1 expression in pkaC null cells is responsible for the pkaC null phenotypes, we assessed the ability of exogenously expressed cAR1–GFP to rescue the chemotaxis, RasG, Rap1 and TORC2 activity defects of pkaC null cells. We found that, at similar cAR1–GFP expression levels in carA null and pkaC null cells, cAR1–GFP rescued the ability of carA null cells to undergo chemotaxis and only very partially rescued the chemotactic defects of pkaC null cells (Fig. 7C; Movies 16 and 17). Indeed, some of the cAR1–GFP-expressing pkaC null cells closest to the chemoattractant source did find the way to the tip of the micropipette, albeit inefficiently. Nonetheless, this result suggests that part of the chemotaxis phenotype of pkaC null cells is due to lack of cAR1 expression.
Fig. 7.

The expression of cAR1 and its role in RasG, Rap1 and TORC2 activation in pkaC null cells. (A) Expression of cAR1, Ras, Rap1, Pia, PKB and PKBR1 in wild-type (WT) and pkaC null (pkaC—) vegetative cells (0 h) and in cells pulsed with cAMP for 5.5 h, detected by immunoblotting using protein-specific antibodies. (B) cAR1–GFP expression in carA and pkaC null cells, revealed by GFP immunoblotting. (C) Chemotaxis phenotype of cAR1–GFP-expressing carA and pkaC null cells migrating in an exponential cAMP gradient created by a point source. Differential interference contrast (DIC) and fluorescence (GFP) images showing expressed cAR1–GFP are shown. (Right) Traces of the migration path for a subset of the migrating cells. *, position of the cAMP-filled micropipette. Scale bars: 50 μm. (D) cAMP-induced RasG and Rap1 activation. Active RasG (Ras–GTP) and Rap1 (Rap1–GTP) were pulled down with GST–Raf1(RBD) and GST–RalGDS(RBD), respectively, and revealed by immunoblotting for pan-Ras and Rap1. (E) cAMP-induced phosphorylation of the hydrophobic motif (HMP; TORC2 site) of PKB and PKBR1. CB, Coomassie Blue staining. Data are representative of at least three independent experiments.
We next assessed the cAMP-induced activation of RasG, Rap1 and TORC2 in carA null cells compared to those in pkaC null cells, and tested the effect of expressing exogenous cAR1–GFP at similar levels in both strains, as controlled by immunoblot and fluorescence measurements. As previously reported (Kae et al., 2004), the basal level of Ras activity, as well as that of Rap1 activity, is elevated in carA null cells and, by contrast with the pkaC null cells, the carA null cells fail to respond to a cAMP stimulus by further increasing the activity of RasG and Rap1 (Fig. 7D). In addition, although the cAMP-induced TORC2-mediated phosphorylation of PKB and PKBR1 is elevated and prolonged in pkaC null cells, cAMP stimulation produces minimal TORC2 activation in carA null cells (Fig. 7E), which is likely to be mediated by the cAMP receptor 3, which is expressed at low levels in aggregation-competent cells (Insall et al., 1994). As expected, cAR1–GFP expression in carA null cells rescued the cAMP-induced activation of Ras, Rap1 and TORC2 in this strain (Fig. 7D,E). Interestingly, pkaC null cells expressing cAR1–GFP, at levels similar to those rescuing the carA null cell phenotypes, displayed the same elevated and extended cAMP-induced RasG, Rap1 and PKB and PKBR1 phosphorylation responses as in pkaC null cells (Fig. 7D,E). Taken together, these observations suggest that (i) some cAR1 is expressed in pkaC null cells; (ii) little cAR1 is sufficient to fully activate the RasG, Rap1 and TORC2 pathways, whereas high levels of cAR1 are necessary for chemotaxis; and finally, (iii) the lack of cAR1 is not responsible for the observed upregulated signaling pathways in pkaC null cells.
DISCUSSION
It is widely appreciated that specific kinetics and localization of chemotactic signaling pathways, as well as their adaptation to the chemoattractant signal, are required for the directional response of cells to chemoattractant gradients. However, how chemoattractant signaling is regulated and how signaling pathway dynamics and polarity are achieved is not understood. Our study suggests that PKA is essential to chemotaxis and plays an important role in the control of both the localized activation and adaptation of key cAMP-induced chemotactic signaling pathways in Dictyostelium, involving RasG, Rap1, PI3K, TORC2, PKB and PKBR1, as well as the spatiotemporal regulation of the cytoskeleton (Fig. 8). The observed effect of lack of PKA function on chemotaxis is likely to result from a combination of roles in gene regulation and signaling. Indeed, although it is hard to separate these two cellular functions of PKA, our experiments using acaA null cells – which produce a lot less cAMP and, thereby, have little PKA activity (Meima and Schaap, 1999) – as well as our results with the PKA-selective pharmacological inhibitor H89, strongly suggest that PKA plays a signaling role in controlling the chemotactic pathways in Dictyostelium. Although we found that pkaC null cells lack detectable cAR1 expression, as assessed by immunoblotting, they are responsive to cAMP stimulation, which suggests that some cAR1 is present. Interestingly, vegetative cells, which express little cAR1, also produce a full cAMP-induced TORC2 activation response (Liao et al., 2013). This previous observation and our present study suggest that only small amounts of cAR1 are necessary to activate, at least some, cAMP-induced signaling pathways, whereas more cAR1 is necessary for efficient chemotaxis towards cAMP. In fact, the partial rescue of chemotaxis of pkaC null cells by the exogenous expression of cAR1 suggests that lack of cAR1 is partly responsible for the chemotaxis phenotype of these cells. However, we found that lack of cAR1 expression in pkaC null cells is not responsible for the upregulated cAMP signaling pathways in these cells.
Fig. 8.
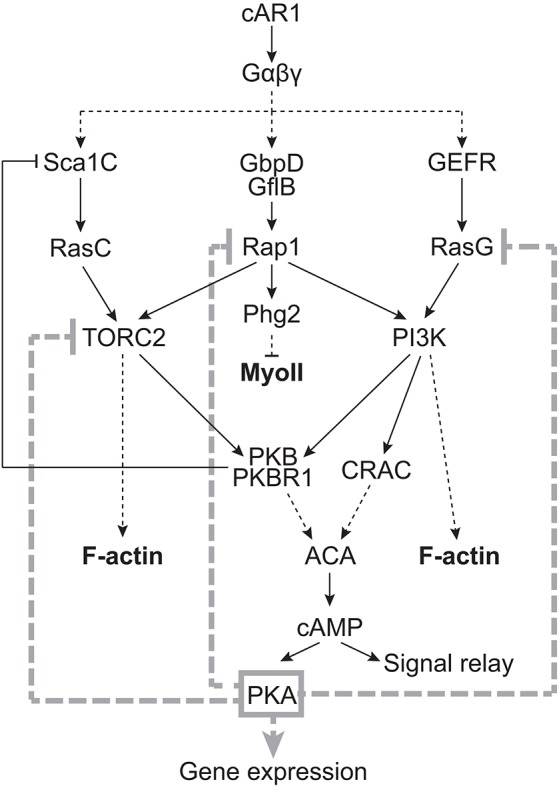
Proposed model for the role of PKA in regulating the chemoattractant signal transduction pathways in Dictyostelium. Although PKA controls gene expression during development, including that of cAR1, and this probably explains part of the observed severe chemotaxis phenotypes of pkaC null cells, our study suggests that PKA is also likely to play a direct role in controlling the directional migration of cells. In light of our findings, we propose that PKA controls chemotaxis, in part, by spatially and temporally regulating the activation of RasG, Rap1 and TORC2.
Consistent with a role for cAMP in the spatiotemporal control of signaling pathways, acaA null cells are unable to suppress the formation of lateral pseudopods, leading to directionality defects during chemotaxis (Stepanovic et al., 2005). We propose that this intracellular role of ACA and cAMP in Dictyostelium is mediated, in part, by the PKA-dependent regulation of the RasG and Rap1 pathways in space and time during chemotaxis. In addition, as the PI3K, TORC2, PKB and PKBR1 signaling pathways contribute to the cAMP-stimulated activation of ACA and cAMP production (Charest et al., 2010; Comer and Parent, 2006; Lee et al., 2005; Lim et al., 2001) and, consequently, to the downstream activation of PKA, we further propose that PKA indirectly controls its own level of activity during chemotaxis in a negative-feedback fashion. This hypothesis is consistent with previous observations that cAMP-stimulated ACA activity fails to adapt in ACA-expressing pkaC null cells (Mann et al., 1997) and that cells with elevated PKA activity display reduced intracellular levels of cAMP compared to those in wild-type cells (Abe and Yanagisawa, 1983; Anjard et al., 1992).
Our analysis of the chemotaxis and cAMP gradient-sensing phenotypes of pkaC null cells suggests that PKA is not necessary for basic cell motility in Dictyostelium but that it is crucial to cAMP chemotaxis and plays a key role in restricting the extent, as well as the site, of chemotactic pathway activation and, thereby, pseudopod protrusion. Our finding that PKA is essential to cAMP chemotaxis is surprising because, although pkaC null cells are unable to aggregate by themselves with or without the ability to produce cAMP (Mann et al., 1997), they have been found to co-aggregate with wild-type cells (Mann and Firtel, 1991). We propose that, in such cases, cell–cell contacts between pkaC null and wild-type cells may have enabled their co-aggregation. By contrast, the chemotaxis phenotype of pkaC null cells is consistent with the considerable reduction in chemotaxis observed for cells with decreased PKA activity due to the expression of PKA regulatory subunit (PKA-R) mutants (Harwood et al., 1992). Intriguingly, cells that lack the PKA-R, in which PKA-C is constitutively active, also display reduced polarity, reduced directionality, the inability to repress the formation of lateral pseudopods and, consequently, severely impaired chemotaxis to cAMP (Zhang et al., 2003). However, given the promiscuous nature of PKA, whose specificity is normally tightly regulated through anchoring complexes (Greenwald and Saucerman, 2011; Pidoux and Taskén, 2010), careful consideration must be given to the phenotype of cells with constitutive PKA activity. Nonetheless, the similarity of pkaC and pkaR null cell phenotypes could support the idea of an auto-regulatory circuit, which is linked by feedback loops that function to tightly regulate PKA activity during chemotaxis, such as that previously proposed by Soll and colleagues (Stepanovic et al., 2005). Disruption of any one component of such a circuit destroys the circuit, and is likely to yield a common phenotype that reflects the role of the circuit. In the case of cAMP–PKA signaling, we propose that the wiring of this circuit allows the coordinated control of directional pseudopod extension and signal relay (Fig. 8).
The cAMP chemotaxis phenotype of pkaC null cells is also highly reminiscent of cells that lack the RasG GAP DdNF1 (nfaA null cells), which exhibit elevated levels of chemoattractant-induced RasG activity (Zhang et al., 2008), as well as of cells expressing a constitutively active Rap1 mutant (Rap1G12V; Jeon et al., 2007a). nfaA null cells display increased phagocytosis and macropinocytosis, are motile but fail to properly sense cAMP gradients, and extend multiple lateral pseudopods leading to severe chemotaxis defects (Bloomfield et al., 2015; Zhang et al., 2008). Rap1G12V-expressing cells also produce lateral pseudopods during migration, and undergo chemotaxis towards cAMP with little directionality (Jeon et al., 2007a). Consequently, we propose that the elevated and spatially misregulated RasG and Rap1 activities in pkaC null cells underlie the inability of these cells to properly sense gradients, to define a leading front and to migrate along a cAMP gradient. Hence, we suggest that PKA regulates the cAMP gradient-sensing abilities of cells mainly by controlling RasG and Rap1 activation. The PKA-mediated regulation of RasG and Rap1 can explain, at least in part, the effects of PKA on the downstream activation of PI3K, TORC2, PKB and PKBR1, as well as F-actin polymerization and MyoII assembly (Fig. 8). Indeed, both RasG and Rap1 mediate PI3K activation, which, in turn, promotes F-actin polymerization, and Rap1 regulates TORC2, as well as MyoII assembly through a parallel pathway involving its effector Phg2 (Khanna et al., 2016; Kortholt et al., 2010; Swaney et al., 2010). Similar to pkaC null cells, cells with elevated RasG activity display elevated PI3K activity and F-actin dynamics (Zhang et al., 2008), and cells with elevated Rap1 activity exhibit elevated TORC2 activity and reduced MyoII assembly (Jeon et al., 2007a; Khanna et al., 2016). Of course, we cannot exclude the possibility that PKA directly regulates PI3K, PKB and PKBR1, F-actin and MyoII, in addition to RasG, Rap1 and TORC2, and that PKA may have additional roles in chemotaxis to cAMP, such as in regulating Rac GTPases and various actin modulators (Howe, 2004).
In addition to the PKA-dependent regulation of Rap1, which is likely to affect downstream TORC2 activity, we propose that TORC2 is also regulated by PKA independently of Rap1. We have recently shown that RasC is essential for TORC2 activation while Rap1 regulates the RasC-mediated TORC2 activity (Khanna et al., 2016). Since RasC activity in cells that lack PKA is strongly reduced, we propose that the highly elevated TORC2 activity in cells that lack PKA results from the PKA-dependent regulation of both Rap1 and TORC2. In mammalian cells, PKA has been found to associate with mTOR (Mavrakis et al., 2006) and to mediate inhibition of both mTORC1 and mTORC2 by inducing dissociation of the complexes (Xie et al., 2011). However, whether PKA directly phosphorylates mTOR or components of mTOR complexes is unknown. By contrast, the observed PKA-dependent positive regulation of RasC is likely to be indirect and to result from the PKA-dependent inhibition of TORC2, as well as of RasG, Rap1 and PI3K signaling, leading to reduced PKB and PKBR1 activity. This reduced activity of PKB and PKBR1, which normally inhibit the Sca1 complex-mediated RasC activation through a negative-feedback loop (Charest et al., 2010), will consequently lead to greater RasC activation (Fig. 8).
In mammalian cells, including neutrophils, PKA activity has been shown to be enriched at the front of migrating cells and to promote actin cytoskeleton polarization (Jones and Sharief, 2005; Paulucci-Holthauzen et al., 2009). One of the suggested mechanisms through which PKA controls mammalian cell migration involves its direct phosphorylation of Rap1, thereby controlling Rap1 membrane localization and activity cycle (Takahashi et al., 2013). Whether this is the mechanism through which PKA controls Rap1 (and/or RasG) activity in Dictyostelium remains to be determined, but this is certainly a possibility. It is also conceivable that PKA targets specific RasG and Rap1 GEFs and/or GAPs, instead of or in addition to directly targeting the GTPases. In any case, our findings highlight a previously unappreciated role of PKA in chemotaxis, through the spatial and temporal regulation of key chemotactic signaling pathways.
MATERIALS AND METHODS
Reagents
cAMP sodium salt monohydrate and anti-Flag M2 were from Sigma-Aldrich, and H2B was from Roche-Genentech. Antibodies against phosphorylated p70 S6 kinase (Thr389; clone 1A5; 1:1000; used to detect PKB and PKBR1 HM phosphorylation), phosphorylated Akt substrates (clone 110B7; 1:1000) and phosphorylated (Ser/Thr) PKA substrates were from Cell Signaling Technology. A pan-Ras antibody (Ab-3; clone RAS10; 1:500) was from Calbiochem – EMD Millipore. H89 and DyLightTM secondary antibodies were purchased from Thermo Fisher Scientific. The Living Colors® GFP monoclonal antibody was purchased from Clontech (now Takara Bio USA Inc.). Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories. The Rap1 (directed against amino acids 169-182 of Dictyostelium Rap1) and Pia antibodies (directed against amino acids 138-159 of Dictyostelium Pianissimo) were custom-made by ProSci Incorporated (Poway, CA, USA). Antibodies against PKB and PKBR1, and Lifeact-GFP, GFP–MyoII and PHcrac–GFP constructs were gifts from Rick Firtel (University of California–San Diego, La Jolla, CA) and have been previously described (Bastounis et al., 2011; Jeon et al., 2007a; Meili et al., 1999; Sasaki et al., 2004). The cAR1 antibody was a generous gift from Peter Devreotes (Johns Hopkins, Baltimore, MD) and is described elsewhere (Hereld et al., 1994). Raf1(RBD)–GFP and RalGDS(RBD)–GFP constructs were kindly provided by Arjan Kortholt (University of Groningen, Groningen, Netherlands) and have been previously described (Kortholt et al., 2010, 2013). GFP–Sca1 has been described previously (Charest et al., 2010). Flag-tagged RasC was generated by fusing the Flag tag nucleotide sequence, GATTATAAAGATGATGATGATAAA, in frame at the N-terminus of the RasC sequence and cloned into the extra-chromosomal vector pDM304, obtained through the Dicty Stock Center, deposited by Douwe Veltman (Veltman et al., 2009). cAR1–GFP was generated by cloning cAR1 into the GFP-containing extra-chromosomal vector pDM323 (Veltman et al., 2009) and was a gift from Chris Janetopoulos (University of the Sciences, Philadelphia, PA).
Cell culture and strains used
Dictyostelium cells were grown attached to substrate in axenic HL5 medium (ForMedium, Hunstanton, Norfolk, UK) at 22°C, and transformants were generated by electroporation. Transformed cells were selected in 10 or 20 µg/ml geneticin, or 50 µg/ml hygromycin B (both from Life Technologies), and expression was confirmed by western blotting. Cells were developed by pulsing with 30 nM cAMP every 6 min for 5.5 h in 12 mM Na2HPO4 and KH2PO4 buffer, pH 6.1, at a confluence of 5×106 cells/ml for wild-type cells and at 7×106 cells/ml for pkaC null cells, which are much smaller, in order to have equivalent densities. The wild-type cells used were AX2 and AX3, depending on the background of mutant used. ScaA null cells are described elsewhere (Charest et al., 2010). pkaC null and acaA null cells were obtained from the Dicty Stock Center, where they were deposited by Rick Firtel and Bill Loomis, respectively; both strains are described elsewhere (Mann and Firtel, 1991; Mann et al., 1992; Stepanovic et al., 2005). The HBW1 and HBW2 cells were a kind gift from Robert Kay (MRC Laboratory of Molecular Biology, Cambridge, UK) and have been described previously (Primpke et al., 2000). The carA null cells (AX2 background) were generously provided by Alan Kimmel (NIH-NIDDK, Bethesda, MD) and have been previously described (Sun and Devreotes, 1991).
Biochemical assays
F-actin measurements using phalloidin staining, PKB and PKBR1 kinase assays, and Ras–GTP and Rap1–GTP pulldown assays were performed as previously described (Insall et al., 1996; Jeon et al., 2007a; Meili et al., 1999; Sasaki et al., 2004, 2007; Zhang et al., 2008). TORC2 activity was assessed by evaluating the TORC2-mediated phosphorylation of PKB and PKBR1, as described previously (Kamimura et al., 2009). Detection of phosphorylated PKB and PKA substrates by western blotting, using the antibodies described above, was performed on whole cell lysates according to the manufacturers’ protocols. For H89 treatments, cells that had been pulsed with cAMP for 5.5 h as described above were treated either with 5% DMSO (control) or 50 µM of DMSO-dissolved H89 for 30 min before cAMP stimulation.
Chemotaxis and imaging
Assessment of chemotaxis, gradient sensing, global responses of fluorescent reporters to cAMP stimulation, and image acquisition were performed as previously described (Charest et al., 2010; Chung and Firtel, 1999; Sasaki et al., 2007; Zhang et al., 2008). Chemotaxis parameters were determined as described previously (Cai et al., 2010). Images were acquired using a Marianas spinning disk confocal workstation (Intelligent Imaging Innovations, Inc., Denver, CO) equipped with an Evolve™ 512 EMCCD camera (Photometrics, Tucson, AZ), and image analysis was performed using the Slidebook software (Intelligent Imaging Innovations, Inc., Denver, CO). Analysis of fluorescent reporter translocations between the cytosol and the cell cortex was performed by measuring changes in fluorescence intensities in the cytosol as described previously (Takeda et al., 2012), normalized to basal levels in each cell.
Acknowledgements
We thank Rick Firtel, Arjan Kortholt, Peter Devreotes, Alan Kimmel and Chris Janetopoulos, as well as the Dicty Stock Center for providing reagents.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
M.S. and A.R.P. contributed equally to the work; P.G.C. conceptualized and designed the study; M.S., A.R.P., R.R., V.F.T., P.L. and P.G.C. performed and interpreted the experiments; M.S., A.R.P. and P.G.C. prepared the manuscript.
Funding
M.S. was supported by National Institutes of Health T32 grant GM008804; and P.L. was supported by a U.S. Public Health Service grant GM037830. The work was supported by a Research Scholar Grant 127940-RSG-15-024-01-CSM from the American Cancer Society to P.G.C. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.177170.supplemental
References
- Abe K. and Yanagisawa K. (1983). A new class of rapidly developing mutants in Dictyostelium discoideum: implications for cyclic AMP metabolism and cell differentiation. Dev. Biol. 95, 200-210. 10.1016/0012-1606(83)90018-0 [DOI] [PubMed] [Google Scholar]
- Afonso P. V., Janka-Junttila M., Lee Y. J., McCann C. P., Oliver C. M., Aamer K. A., Losert W., Cicerone M. T. and Parent C. A. (2012). LTB(4) is a signal-relay molecule during neutrophil chemotaxis. Dev. Cell 22, 1079-1091. 10.1016/j.devcel.2012.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjard C., Pinaud S., Kay R. R. and Reymond C. D. (1992). Overexpression of Dd PK2 protein kinase causes rapid development and affects the intracellular cAMP pathway of Dictyostelium discoideum. Development 115, 785-790. [DOI] [PubMed] [Google Scholar]
- Arai Y., Shibata T., Matsuoka S., Sato M. J., Yanagida T. and Ueda M. (2010). Self-organization of the phosphatidylinositol lipids signaling system for random cell migration. Proc. Natl. Acad. Sci. USA 107, 12399-12404. 10.1073/pnas.0908278107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artemenko Y., Lampert T. J. and Devreotes P. N. (2014). Moving towards a paradigm: common mechanisms of chemotactic signaling in Dictyostelium and mammalian leukocytes. Cell. Mol. Life Sci. 71, 3711-3747. 10.1007/s00018-014-1638-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagorda A., Das S., Rericha E. C., Chen D., Davidson J. and Parent C. A. (2009). Real-time measurements of cAMP production in live Dictyostelium cells. J. Cell Sci. 122, 3907-3914. 10.1242/jcs.051987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastounis E., Meili R., Alonso-Latorre B., del Alamo J. C., Lasheras J. C. and Firtel R. A. (2011). The SCAR/WAVE complex is necessary for proper regulation of traction stresses during amoeboid motility. Mol. Biol. Cell 22, 3995-4003. 10.1091/mbc.E11-03-0278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield G., Traynor D., Sander S. P., Veltman D. M., Pachebat J. A. and Kay R. R. (2015). Neurofibromin controls macropinocitosis and phagocytosis in Dictyostelium. Elife 4, e04940 10.7554/eLife.04940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolourani P., Spiegelman G. B. and Weeks G. (2006). Delineation of the roles played by RasG and RasC in cAMP-dependent signal transduction during the early development of Dictyostelium discoideum. Mol. Biol. Cell 17, 4543-4550. 10.1091/mbc.E05-11-1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Cordero J. J., Hodgson L. and Condeelis J. (2012). Directed cell invasion and migration during metastasis. Curr. Opin. Cell. Biol. 24, 277-283. 10.1016/j.ceb.2011.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H., Das S., Kamimura Y., Long Y., Parent C. A. and Devreotes P. N. (2010). Ras-mediated activation of the TORC2-PKB pathway is critical for chemotaxis. J. Cell Biol. 190, 233-245. 10.1083/jcb.201001129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H. and Levchenko A. (2013). Adaptive molecular networks controlling chemotactic migration: dynamic inputs and selection of the network architecture. Philos. Trans. R. Soc. B Biol. Sci. 368, 20130117 10.1098/rstb.2013.0117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charest P. G. and Firtel R. A. (2006). Feedback signaling controls leading-edge formation during chemotaxis. Curr. Opin. Genet. Dev. 16, 339-347. 10.1016/j.gde.2006.06.016 [DOI] [PubMed] [Google Scholar]
- Charest P. G. and Firtel R. A. (2007). Big roles for small GTPases in the control of directed cell movement. Biochem. J. 401, 377-390. 10.1042/BJ20061432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charest P. G., Shen Z., Lakoduk A., Sasaki A. T., Briggs S. P. and Firtel R. A. (2010). A Ras signaling complex controls the RasC-TORC2 pathway and directed cell migration. Dev. Cell 18, 737-749. 10.1016/j.devcel.2010.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Wang Y., Yu H. and Xu W. (2005). The cross talk between protein kinase A- and RhoA-mediated signaling in cancer cells. Exp. Biol. Med. 10, 731-741. 10.1177/153537020523001006 [DOI] [PubMed] [Google Scholar]
- Chung C. Y. and Firtel R. A. (1999). PAKa, a putative PAK family member, is required for cytokinesis and the regulation of the cytoskeleton in Dictyostelium discoideum cells during chemotaxis. J. Cell Biol. 147, 559-576. 10.1083/jcb.147.3.559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer F. I. and Parent C. A. (2006). Phosphoinositide 3-kinase activity controls the chemoattractant-mediated activation and adaptation of adenylyl cyclase. Mol. Biol. Cell 17, 357-366. 10.1091/mbc.E05-08-0781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D., Weijer G., Parent C. A., Devreotes P. N. and Weijer C. J. (2002). Visualizing PI3 kinase-mediated cell-cell signaling during Dictyostelium development. Curr. Biol. 12, 1178-1188. 10.1016/S0960-9822(02)00950-8 [DOI] [PubMed] [Google Scholar]
- Garcia G. L. and Parent C. A. (2008). Signal relay during chemotaxis. J. Microsc. 231, 529-534. 10.1111/j.1365-2818.2008.02066.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald E. C. and Saucerman J. J. (2011). Bigger, better, faster: principles and models of AKAP anchoring protein signaling. J. Cardiovasc. Pharmacol. 58, 462-469. 10.1097/FJC.0b013e31822001e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A., Warren V., Dharmawardhane S. and Condeelis J. (1989). Identification of actin nucleation activity and polymerization inhibitor in amoeboid cells: Their regulation by chemotactic stimulation. J. Cell. Biol. 109, 2207-2213. 10.1083/jcb.109.5.2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood A. J., Hopper N. A., Simon M.-N., Bouzid S., Veron M. and Williams J. G. (1992). Multiple roles for cAMP-dependent protein kinase during Dictyostelium development. Dev. Biol. 149, 90-99. 10.1016/0012-1606(92)90266-J [DOI] [PubMed] [Google Scholar]
- Hereld D., Vaughan R., Kim J. Y., Borleis J. and Devreotes P. (1994). Localization of ligand-induced phosphorylation sites to serine slusters in the C-terminal domain of the Dictyostelium cAMP receptor, cAR1. J. Biol. Chem. 269, 7036-7044. [PubMed] [Google Scholar]
- Hoeller O., Gong D. and Weiner O. D. (2014). How to understand and outwit adaptation. Dev. Cell 28, 607-616. 10.1016/j.devcel.2014.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe A. K. (2004). Regulation of actin-based cell migration by cAMP/PKA. Biochim. Biophys. Acta-Mol. Cell Res. 1692, 159-174. 10.1016/j.bbamcr.2004.03.005 [DOI] [PubMed] [Google Scholar]
- Howe A. K., Baldor L. C. and Hogan B. P. (2005). Spatial regulation of the cAMP-dependent protein kinase during chemotactic cell migration. Proc. Natl. Acad. Sci. USA 102, 14320-14325. 10.1073/pnas.0507072102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insall R. H., Soede D., Schaap P. and Devreotes P. N. (1994). Two cAMP receptors activate common signaling pathways in Dictyostelium. Mol. Biol. Cell 5, 703-711. 10.1091/mbc.5.6.703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insall R. H., Borleis J. and Devreotes P. N. (1996). The aimless RasGEF is required for processing of chemotactic signals through G-protein-coupled receptors in Dictyostelium. Curr. Biol. 6, 719-729. 10.1016/S0960-9822(09)00453-9 [DOI] [PubMed] [Google Scholar]
- Jeon T. J., Lee D.-J., Merlot S., Weeks G. and Firtel R. A. (2007a). Rap1 controls cell adhesion and cell motility through the regulation of myosin II. J. Cell Biol. 176, 1021-1033. 10.1083/jcb.200607072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon T. J., Lee D.-J., Lee S., Weeks G. and Firtel R. A. (2007b). Regulation of Rap1 activity by RapGAP1 controls cell adhesion at the front of chemotaxing cells. J. Cell Biol. 179, 833-843. 10.1083/jcb.200705068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S. L. and Sharief Y. (2005). Asymmetrical protein kinase A activity establishes neutrophil cytoskeletal polarity and enables chemotaxis. J. Leukoc. Biol. 78, 248-258. 10.1189/jlb.0804459 [DOI] [PubMed] [Google Scholar]
- Kae H., Lim C. J., Spiegelman G. B. and Weeks G. (2004). Chemoattractant-induced Ras activation during Dictyostelium aggregation. EMBO Rep. 5, 602-606. 10.1038/sj.embor.7400151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura Y. and Devreotes P. N. (2010). Phosphoinositide-dependent protein kinase (PDK) activity regulates phosphatidylinositol 3,4,5-triphosphate-dependent and -independent protein kinase B activation and chemotaxis. J. Biol. Chem. 285, 7938-7946. 10.1074/jbc.M109.089235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura Y., Tang M. and Devreotes P. (2009). Assays for chemotaxis and chemoattractant-stimulated TorC2 activation and PKB substrate phosphorylation in dictyostelium. Meth. Mol. Biol. 571, 255-270. 10.1007/978-1-60761-198-1_17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna A., Lotfi P., Chavan A. J., Montano N. M., Bolourani P., Weeks G., Shen Z., Briggs S. P., Pots H., Van Haastert P. J. M. et al. (2016). The small GTPases Ras and Rap1 bind to and control TORC2 activity. Sci. Rep. 6, 25823 10.1038/srep25823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.-J., Chang W.-T., Meima M., Gross J. D. and Schaap P. (1998). A novel adenylyl cyclase detected in rapidly developing mutants of Dictyostelium. J. Biol. Chem. 273, 30859-30862. 10.1074/jbc.273.47.30859 [DOI] [PubMed] [Google Scholar]
- Kolaczkowska E. and Kubes P. (2013). Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159-175. 10.1038/nri3399 [DOI] [PubMed] [Google Scholar]
- Kortholt A., Rehmann H., Kae H., Bosgraaf L., Keizer-Gunnink I., Weeks G., Wittinghofer A. and Van Haastert P. J. M. (2006). Characterization of the GbpD-activated Rap1 pathway regulating adhesion and cell polarity in dictyostelium discoideum. J. Biol. Chem. 281, 23367-23376. 10.1074/jbc.M600804200 [DOI] [PubMed] [Google Scholar]
- Kortholt A., Bolourani P., Rehmann H., Keizer-Gunnink I., Weeks G., Wittinghofer A. and Van Haastert P. J. M. (2010). A Rap/phosphatidylinositol 3-kinase pathway controls pseudopod formation [corrected]. Mol. Biol. Cell 21, 936-945. 10.1091/mbc.E09-03-0177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortholt A., Keizer-Gunnink I., Kataria R. and Van Haastert P. J. M. (2013). Ras activation and symmetry breaking during Dictyostelium chemotaxis. J. Cell Sci. 126, 4502-4513. 10.1242/jcs.132340 [DOI] [PubMed] [Google Scholar]
- Lee S., Comer F. I., Sasaki A., McLeod I. X., Duong Y., Okumura K., Yates J. R. III, Parent C. A. and Firtel R. A. (2005). TOR complex 2 integrates cell movement during chemotaxis and signal relay in Dictyostelium. Mol. Biol. Cell 16, 4572-4583. 10.1091/mbc.E05-04-0342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X.-H., Buggey J. and Kimmel A. R. (2010). Chemotactic activation of Dictyostelium AGC-family kinases AKT and PKBR1 requires separate but coordinated functions of PDK1 and TORC2. J. Cell Sci. 123, 983-992. 10.1242/jcs.064022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X.-H., Buggey J., Lee Y. K. and Kimmel A. R. (2013). Chemoattractant stimulation of TORC2 is regulated by receptor/G protein-targeted inhibitory mechanisms that function upstream and inpdependently of an essential GEF/Ras activation pathway in Dictyostelium. Mol. biol. Cell. 24, 2146-2155. 10.1091/mbc.E13-03-0130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C. J., Spiegelman G. B. and Weeks G. (2001). RasC is required for optimal activation of adenylyl cyclase and Akt/PKB during aggregation. EMBO J. 20, 4490-4499. 10.1093/emboj/20.16.4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C. J., Kain K. H., Tkachenko E., Goldfinger L. E., Gutierrez E., Allen M. D., Groisman A., Zhang J. and Ginsberg M. H. (2008). Integrin-mediated protein kinase A activation at the leading edge of migrating cells. Mol. Biol. Cell 19, 4930-4941. 10.1091/mbc.E08-06-0564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis W. F. (1998). Role of PKA in the timing of developmental events in Dictyostelium cells. Microbiol. Mol. Biol. Rev. 62, 684-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann S. K. O. and Firtel R. A. (1991). A developmentally regulated, putative serine/threonine protein kinase is essential for development in Dictyostelium. Mech. Dev. 35, 89-101. 10.1016/0925-4773(91)90060-J [DOI] [PubMed] [Google Scholar]
- Mann S. K., Yonemoto W. M., Taylor S. S. and Firtel R. A. (1992). DdPK3, which plays essential roles during Dictyostelium development, encodes the catalytic subunit of cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 89, 10701-10705. 10.1073/pnas.89.22.10701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann S. K. O., Brown J. M., Briscoe C., Parent C., Pitt G., Devreotes P. N. and Firtel R. A. (1997). Role of cAMP-dependent protein kinase in controlling aggregation and postaggregative development in Dictyostelium. Dev. Biol. 183, 208-221. 10.1006/dbio.1996.8499 [DOI] [PubMed] [Google Scholar]
- Mavrakis M., Lippincott-Schwartz J., Stratakis C. A. and Bossis I. (2006). Depletion of type IA regulatory subunit (RI) of protein kinase A (PKA) in mammalian cells and tissues activates mTOR and causes autophagic deficiency. Hum. Mol. Genet. 15, 2962-2971. 10.1093/hmg/ddl239 [DOI] [PubMed] [Google Scholar]
- Meili R., Ellsworth C., Lee S., Reddy T. B., Ma H. and Firtel R. A. (1999). Chemoattractant-mediated transient activation and membrane localization of Akt/PKB is required for efficient chemotaxis to cAMP in Dictyostelium. EMBO J. 18, 2092-2105. 10.1093/emboj/18.8.2092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meima M. and Schaap P. (1999). Dictyostelium development-socializing through cAMP. Sem. Cell. Dev. Biol. 10, 567-576. 10.1006/scdb.1999.0340 [DOI] [PubMed] [Google Scholar]
- Miranda E. R., Nam E. A., Kuspa A. and Shaulsky G. (2015). The ABC transporter, AbcB3, medaites cAMP export in D. discoideum development. Dev. Biol. 397, 203-211. 10.1016/j.ydbio.2014.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun H. and Jeon T. J. (2012). Regulation of actin cytoskeleton by Rap1 binding to RacGEF1. Mol. Cells 34, 71-76. 10.1007/s10059-012-0097-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadella K. S., Saji M., Jacob N. K., Pavel E., Ringel M. D. and Kirschner L. S. (2009). Regulation of actin function by protein kinase A-mediated phosphorylation of Limk1. EMBO Rep. 10, 599-605. 10.1038/embor.2009.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent C. A., Blacklock B. J., Froehlich W. M., Murphy D. B. and Devreotes P. N. (1998). G protein signaling events are activated at the leading edge of chemotactic cells. Cell 95, 81-91. 10.1016/S0092-8674(00)81784-5 [DOI] [PubMed] [Google Scholar]
- Paulucci-Holthauzen A. A., Vergara L. A., Bellot L. J., Canton D., Scott J. D. and O'Connor K. L. (2009). Spatial distribution of protein kinase A activity during cell migration is mediated by A-kinase anchoring protein AKAP Lbc. J. Biol. Chem. 284, 5956-5967. 10.1074/jbc.M805606200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidoux G. and Taskén K. (2010). Specificity and spatial dynamics of protein kinase a signaling organized by A-kinase-anchoring proteins. J. Mol. Endocrinol. 44, 271-284. 10.1677/JME-10-0010 [DOI] [PubMed] [Google Scholar]
- Pitt G. S., Brandt R., Lin K. C., Devreotes P. N. and Schaap P. (1993). Extracellular cAMP is sufficient to restore developmental gene expression and morphogenesis in Dictyostelium cells lacking the aggregation adenylyl cyclase (ACA). Genes Dev. 7, 2172-2180. 10.1101/gad.7.11.2172 [DOI] [PubMed] [Google Scholar]
- Plak K., Veltman D., Fusetti F., Beeksma J., Rivero F., Van Haastert P. J. M. and Kortholt A. (2013). GxcC connects Rap and Rac signaling during Dictyostelium development. BMC Cell. Biol. 14, 6 10.1186/1471-2121-14-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primpke G., Iassonidou V., Nellen W. and Wetterauer B. (2000). Role of cAMP-dependent protein kinase during growth and early development of Dictyostelium discoideum. Dev. Biol. 221, 101-111. 10.1006/dbio.2000.9662 [DOI] [PubMed] [Google Scholar]
- Reymond C. D., Schaap P., Véron M. and Williams J. G. (1995). Dual role of cAMP during Dictyostelium development. Experientia 51, 1166-1174. 10.1007/BF01944734 [DOI] [PubMed] [Google Scholar]
- Richardson B. E. and Lehmann R. (2010). Mechanisms guiding primordial germ cell migration: strategies from different organisms. Nat. Rev. Mol. Cell. Biol. 11, 37-49. 10.1038/nrm2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J., Crevenna A. H., Kessenbrock K., Yu J. H., Neukirchen D., Bista M., Bradke F., Jenne D., Holak T. A., Werb Z. et al. (2008). Lifeact: a versatile marker to visualize F-actin. Nat. Meth. 5, 605-607. 10.1038/nmeth.1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rot G., Parikh A., Curk T., Kuspa A., Shaulsky G. and Zupan B. (2009). dictyExpress: a Dictyostelium discoideum gene expression database with an explorative data analysis web-based interface. BMC Bioinformatics 10, 265 10.1186/1471-2105-10-265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadik C. D. and Luster A. D. (2012). Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J. Leukoc. Biol. 91, 207-215. 10.1189/jlb.0811402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A. T., Chun C., Takeda K. and Firtel R. A. (2004). Localized Ras signaling at the leading edge regulates PI3K, cell polarity, and directional cell movement. J. Cell Biol. 167, 505-518. 10.1083/jcb.200406177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A. T., Janetopoulos C., Lee S., Charest P. G., Takeda K., Sundheimer L. W., Meili R., Devreotes P. N. and Firtel R. A. (2007). G protein-independent Ras/PI3K/F-actin circuit regulates basic cell motility. J. Cell Biol. 178, 185-191. 10.1083/jcb.200611138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderbom F., Anjard C., Iranfar N., Fuller D. and Loomis W. F. (1999). An adenylyl cyclase that functions during late development of Dictyostelium. Development 126, 5463-5471. [DOI] [PubMed] [Google Scholar]
- Stajdohar M., Jeran L., Kokosar J., Blenkus D., Janez T., Kuspa A., Shaulsky G. and Zupan B. (2015). dictyExpress: visual analytics of NGS gene expression in Dictyostelium. February 26, 2015, URL: https://www.dictyexpress.org. [DOI] [PMC free article] [PubMed]
- Stepanovic V., Wessels D., Daniels K., Loomis W. F. and Soll D. R. (2005). Intracellular role of adenylyl cyclase in regulation of lateral pseudopod formation during Dictyostelium chemotaxis. Eukaryot. Cell 4, 775-786. 10.1128/EC.4.4.775-786.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T. J. and Devreotes P. N. (1991). Gene targeting of the aggregation stage cAMP receptor cAR1 in Dictyostelium. Genes Dev. 5, 572-582. 10.1101/gad.5.4.572 [DOI] [PubMed] [Google Scholar]
- Swaney K. F., Huang C.-H. and Devreotes P. N. (2010). Eukaryotic chemotaxis: a network of signaling pathways controls motility, directional sensing, and polarity. Annu. Rev. Biophys. 39, 265-289. 10.1146/annurev.biophys.093008.131228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M., Dillon T. J., Liu C., Kariya Y., Wang Z. and Stork P. J. S. (2013). Protein kinase A-dependent phosphorylation of Rap1 regulates its membrane localization and cell migration. J. Biol. Chem. 288, 27712-27723. 10.1074/jbc.M113.466904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K., Shao D., Adler M., Charest P. G., Loomis W. F., Levine H., Groisman A., Rappel W.-J. and Firtel R. A. (2012). Incoherent feedforward control governs adaptation of activated ras in a eukaryotic chemotaxis pathway. Sci. Signal. 5, ra2 10.1126/scisignal.2002413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M., Wang M., Changji S., Iglesias P. A., Devreotes P. N. and Huang C.-H. (2014). Evolutionarily conserved coupling of adaptive and excitable networks mediates eukaryotic chemotaxis. Nat. Commun. 5, 5175 10.1038/ncomms6175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E. and Mayor R. (2012). Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol. 366, 34-54. 10.1016/j.ydbio.2011.12.041 [DOI] [PubMed] [Google Scholar]
- Toriyama M., Mizuno N., Fukami T., Iguchi T., Toriyama M., Tago K. and Itoh H. (2012). Phosphorylation of doublecortin by protein kinase A orchestrates microtubule and actin dynamics to promote neuronal progenitor cell migration. J. Biol. Chem. 287, 12691-12702. 10.1074/jbc.M111.316307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltman D. M., Akar G., Bosgraaf L. and Van Haastert P. J. M. (2009). A new set of extrachromosomal expression vectors for Dictyostelium discoideum. Plasmid 61, 110-118. 10.1016/j.plasmid.2008.11.003 [DOI] [PubMed] [Google Scholar]
- Wang F. (2009). The signaling mechanisms underlying cell polarity and chemotaxis. Cold Spring Harb. Perspect. Biol. 1, a002980 10.1101/cshperspect.a002980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff J., Wang W., Lin E. Y., Wang Y., Pixley F., Stanley E. R., Graf T., Pollard J. W., Segall J. and Condeelis J. (2004). A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer. Res. 64, 7022-7029. 10.1158/0008-5472.CAN-04-1449 [DOI] [PubMed] [Google Scholar]
- Xie J., Ponuwei G. A., Moore C. E., Willars G. B., Tee A. R. and Herbert T. P. (2011). CAMP inhibits mammalian target of rapamycin complex-1 and -2 (mTORC1 and 2) by promoting complex dissociation and inhibiting mTOR kinase activity. Cell. Signal. 23, 1927-1935. 10.1016/j.cellsig.2011.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C., Anjard C., Wille S., Nellen W., Primpke G. and Wetterauer B. (2001). Interaction of gdt1 and protein kinase A (PKA) in the growth-differentiation-transition in Dictyostelium. Differentiation 67, 25-32. 10.1046/j.1432-0436.2001.067001025.x [DOI] [PubMed] [Google Scholar]
- Zernecke A. and Weber C. (2010). Chemokines in the vascular inflammatory response of atherosclerosis. Cardiovasc. Res. 86, 192-201. 10.1093/cvr/cvp391 [DOI] [PubMed] [Google Scholar]
- Zhang H., Heid P. J., Wessels D., Daniels K. J., Pham T., Loomis W. F. and Soll D. R. (2003). Constitutively active protein kinase A disrupts motility and chemotaxis in Dictyostelium discoideum. Eukaryot. Cell 2, 62-75. 10.1128/EC.2.1.62-75.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Charest P. G. and Firtel R. A. (2008). Spatiotemporal regulation of Ras activity provides directional sensing. Curr. Biol. 18, 1587-1593. 10.1016/j.cub.2008.08.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman N. P., Roy I., Hauser A. D., Wilson J. M., Williams C. L. and Dwinell M. B. (2013). Cyclic AMP regulates the migration and invasion potential of human pancreatic cancer cells. Mol. Carcinog. 54, 203-215. 10.1002/mc.22091 [DOI] [PMC free article] [PubMed] [Google Scholar]