

ABSTRACT
Focal adhesions (FAs) are macromolecular complexes that regulate cell adhesion and mechanotransduction. By performing fluorescence recovery after photobleaching (FRAP) and fluorescence loss after photoactivation (FLAP) experiments, we found that the mobility of core FA proteins correlates with their function. Structural proteins such as tensin, talin and vinculin are significantly less mobile in FAs than signaling proteins such as FAK (also known as PTK2) and paxillin. The mobilities of the structural proteins are directly influenced by substrate stiffness, suggesting that they are involved in sensing the rigidity of the extracellular environment. The turnover rates of FAK and paxillin, as well as kindlin2 (also known as FERMT2), are not influenced by substrate stiffness. By using specific Src and FAK inhibitors, we reveal that force-sensing by vinculin occurs independently of FAK and paxillin phosphorylation. However, their phosphorylation is required for downstream Rac1-driven cellular processes, such as protrusion and cell migration. Overall, we show that the FA is composed of different functional modules that separately control mechanosensing and the cellular mechano-response.
KEY WORDS: Vinculin, Talin, Paxillin, FAK, Mechanotransduction, Focal adhesion
Summary: Quantitative fluorescence microscopy shows that focal adhesions are composed of functional protein modules, which control distinct aspects of mechanotransduction.
INTRODUCTION
Fundamental cellular processes such as adhesion and migration require precise communication between cells and their environment (Geiger et al., 2001). Focal adhesions (FAs) are major sites of cell–extracellular matrix (ECM) crosstalk (Yamada and Geiger, 1997). These structures mature under actomyosin tension from focal complexes (FXs), which originally form at the cell edge in a tension-independent manner (Geiger et al., 2001). At these sites, the integrin transmembrane receptors connect ECM proteins to the cytoplasmic FA plaque complex, providing a link to the contractile actomyosin machinery (Hynes, 2002). While the overall molecular architecture of FAs has been established (Kanchanawong et al., 2010; Liu et al., 2015), much less is known about the dynamic processes that occur within FAs, and their functional relevance to mechanotransduction, the conversion of a mechanical signal into a cellular response.
Talin, vinculin and tensin (which has several isoforms) are three FA structural proteins that provide a physical link between the integrins and the actin cytoskeleton (Atherton et al., 2016; Calderwood et al., 2013). Talin binds to and activates integrins and is essential for cell adhesion and spreading (Atherton et al., 2015), while absence of vinculin compromises FA function and force transduction (Dumbauld et al., 2013; Xu et al., 1998). Tensin, depending on the isoform, can vary in its localization pattern (Clarke et al., 2015). While tensin2 appeared predominantly in FAs, tensin3 was almost exclusively in fibrillar adhesions (FBs). Tensin1 localizes to both FAs and FBs (Clarke et al., 2015). Despite these findings, little is known about the activation and regulation of tensin family proteins. Talin and vinculin activation requires tension-dependent conformational changes (Atherton et al., 2015; Carisey et al., 2013; del Rio et al., 2009; Hirata et al., 2014a), suggesting that they are involved in sensing force-generated signals from the environment. When active, talin and vinculin bind both to each other and to the actomyosin machinery, leading to the maturation and stabilization of the FA, a process that is dependent on actomyosin tension (Humphries et al., 2007). There is evidence to suggest that the actin crosslinker α-actinin may also be required for tension-dependent FA maturation (Roca-Cusachs et al., 2013). This could be due to the transmission of forces through α-actinin (Roca-Cusachs et al., 2013), or via the establishment of an actin stress fiber template (Oakes et al., 2012).
Kindlin2 (also known as FERMT2), FAK (also known as PTK2) and paxillin also contribute to adhesion regulation and mechanotransduction. Kindlin2 is essential for integrin activation and is involved in the recruitment of paxillin (Theodosiou et al., 2016). FAK and paxillin phosphorylation is increased on rigid substrates (Bae et al., 2014). Additionally, FAK phosphorylation is required for cells to respond to substrate stiffness during cell spreading (Swaminathan et al., 2016). FAK forms a complex with Src kinase in FAs (Parsons, 2003), which also seems to play a key role in mechanotransduction because Src kinase activity (Sai et al., 1999) and subsequent FAK phosphorylation (Wang et al., 2001) are required for the cellular response to cyclic stretching. Phosphorylation, mainly driven by the FAK–Src complex, also dictates the localization of paxillin at cell–ECM adhesions (Zaidel-Bar et al., 2007). A phosphomimetic form of paxillin, where two key tyrosine residues (Y31 and Y118) are mutated to glutamate (denoted Y2E), preferentially localized to newly formed FXs, whereas the phospho-null mutation where these two residues were mutated to phenylalanine (denoted Y2F) localized to FBs. It is not known, however, whether these proteins are involved in the initial sensing of substrate stiffness, or are required to orchestrate the cellular response.
FA proteins do not remain stationary; rather, they are continually turned over within the complex (Lele et al., 2006). Here, we used the photokinetic microscopy techniques fluorescence recovery after photobleaching (FRAP) and fluorescence loss after photoactivation (FLAP) to measure how the dynamics of 12 core FA proteins vary within the FA. We found that adhesion proteins can be separated into distinct ‘modules’ based on their turnover, which is representative of protein function. The turnover rates of the ‘structural module’ proteins talin, vinculin and tensin are stabilized by increased extracellular substrate stiffness and appear to be important in mechanosensing. This response is not seen with FAK and paxillin, two proteins in the ‘signaling module’. Furthermore, we show that FAK and paxillin have a role in directing the cellular response by controlling lamellipodial protrusions and cell migration.
These results highlight the distinct roles played by different FA protein modules, allowing a cell to sense and respond to extracellular mechanical signals. Both the structural and signaling protein modules are critical for mechanosensitive cellular processes such as coordinated cell migration.
RESULTS
Differential mobility of FA proteins
A large number of proteins are implicated in the regulation of FAs, but little is known about their dynamic behavior. We analyzed the mobility of 12 GFP-tagged FA proteins using FRAP (Fig. 1A; Movie 1). Two parameters, the half-time (t½) of recovery and the mobile fraction (FM), were assessed as indicators of protein mobility (Carisey et al., 2011).
Fig. 1.

Differential FA protein turnover in FAs. (A) Representative time-lapse images showing FRAP of NIH3T3 cells transfected with GFP-tagged talin and FAK. Targeted FAs are indicated by encircled regions. Images are presented in a color intensity scale. Graphs show the mean fluorescence recovery curves for all recorded FAs (n=32–34 FAs), error bars are s.e.m. Scale bar: 5 μm. (B) Mean t½ FRAP values for the indicated FA proteins in NIH3T3 cells (scatter dots). n=23–83 FAs. The box represents the 25–75th percentiles, and the median is indicated. The whiskers show the range of values. *P<0.05; ****P<0.0001; n.s., no significant difference (Kruskal–Wallis with Dunn's multiple comparisons test). (C,D) Representative time-lapse panels showing FRAP of GFP–β1-integrin. The graph on the right of C,D shows the average curve fits of all recorded FAs (n=20); error bars are s.e.m. The white line across the FA indicates the position of line profile plots shown in D. Scale bar: 5 μm. Representative line profile plots of GFP–β1-integrin. The blue line represents the fluorescence intensity across the line and red dashed line indicates background fluorescence. Note that the fluorescence intensity of the photobleached FA falls below the background fluorescence at time 0 s and only recovers to background levels at 40 s.
FRAP experiments revealed a wide distribution in the t½ of different FA proteins (Fig. 1B). We found tensin1 (t½=59.0 s) and talin (t½=49.4 s) to have the slowest turnover, followed by vinculin (t½=39.8 s), α-actinin (t½=29.6 s), ILK (t½=29.1 s), α-parvin (t½=26.0 s), kindlin2 (t½=23.3 s), paxillin (t½=15.7 s), p130Cas (also known as BCAR1) (t½=14.4 s), VASP (t½=11.1 s), FAK (t½=9.9 s) and zyxin (t½=9.4 s) (Fig. 1B). The statistical analysis of t½ FRAP indicates that these proteins can be broadly split into three groups. Tensin, talin and vinculin are the slowest, α-actinin, ILK, α-parvin and kindlin2 make up the intermediate group and the remaining proteins constitute the most rapidly cycling group of proteins (Fig. 1B). Analyzing mobile fractions (FM) showed talin and tensin to have the lowest FM (48.4% and 57.3%), followed by the other FA proteins analyzed (FM of 60–80%) (Fig. S1A).
To compare the dynamics of the cytoplasmic plaque proteins with transmembrane ECM-bound integrins, we used FRAP to study the turnover of GFP–β1-integrin. Initial observations showed a short t½ in FRAP experiments of 19.4 s, suggesting a surprisingly fast turnover. However, the low FM (36.5%) indicated a high immobile fraction (Fig. 1C). Analyzing line profiles of fluorescence intensities during the course of recovery showed that GFP–β1-integrin only recovered to the levels observed in the background membrane areas outside FAs, which constitutes diffuse GFP–β1-integrin (∼30% of the total initial intensity found in FAs) (Fig. 1D). This suggests that there are two populations of integrins in FAs; one population that is tightly engaged with the ECM and therefore less mobile than a second highly mobile population, which has a similar mobility to unliganded, presumably inactive, integrins in other areas of the membrane.
Taken together, these results reveal the differential dynamics of distinct groups of FA proteins, suggesting a modular organization of proteins with a similar function within the FA.
Structural, but not signaling, proteins modify their turnover in response to extracellular stiffness
FLAP is a complementary method to FRAP for measuring protein turnover (Atherton et al., 2015). FLAP experiments using photoactivatable GFP [PA(GFP)]-tagged talin, vinculin, FAK and paxillin, with mCherry–zyxin used as a FA marker showed a similar protein turnover pattern as found in FRAP experiments, with talin and vinculin displaying a significantly higher t½ than FAK and paxillin (Fig. S1B,C). We found no correlation between initial fluorescence intensity post photoactivation (relating to protein expression level) and the rate of turnover, demonstrating that protein expression levels did not influence the rate of protein turnover (Fig. S1D). This method was used instead of FRAP in subsequent experiments because the analysis of the positive fluorescence signal after photoactivation was less variable and more sensitive.
Of the 12 proteins analyzed previously, we chose tensin1, talin, vinculin, α-actinin, kindlin2, paxillin and FAK to examine further. Vinculin and talin are known mechanosensors, and FAK and paxillin are important signaling components involved in regulating FA dynamics and mechanotransduction (Bae et al., 2014; Dumbauld et al., 2013; Humphries et al., 2007; Webb et al., 2004; Zaidel-Bar et al., 2007). The roles of the other three proteins in mechanotransduction are not well established. To test whether the mechanical properties of the ECM affect protein dynamics, we performed FLAP experiments in NIH3T3 cells cultured on fibronectin (FN)-coated intermediate (∼8 kPa) and stiff (∼100 kPa) polyacrylamide (PAA) gel substrates and glass (∼1 GPa) (Fig. 2A). The rigidities chosen for the PAA gels are within the physiological range (∼8 kPa for muscle and ∼100 kPa for collagenous bone) (Engler et al., 2006). Consistent with previous findings, cells on the three substrates spread well and displayed FAs that increased in size with increasing substrate stiffness (Fig. S2A,B) (Han et al., 2012; Pelham and Wang, 1997). Analyzing turnover rates revealed that the t½ for talin, vinculin and tensin1 significantly increased with substrate stiffness (Fig. 2A,B; Fig. S2C). Conversely, the dynamics of FAK, paxillin and kindlin2 were unaffected (Fig. 2A,B; Fig. S2C). Interestingly kindlin2, while not sensitive to forces, showed a different post-activation pattern than that of FAK and paxillin. Fractions of activated PAGFP–paxillin and –FAK that were not bound to FAs diffused rapidly away from adhesion sites; however, those of PAGFP–kindlin2 produced a halo around adhesions, which seemed to be associated with the membrane (Fig. S2D). For α-actinin, we found no difference in turnover between soft and stiff PAA gels (Fig. 2A,B; Fig. S2C). There was a very small, although significant, decrease in mobility between 8 kPa and glass, which puts forward the possibility that α-actinin is somewhat sensitive to ECM stiffness in larger force ranges. However, since the changes are extremely low in comparison to tensin1, talin and vinculin, the latter appear to be the main mechanosensitive proteins of those analyzed.
Fig. 2.

Extracellular stiffness differentially modulates the turnover rates of several FA proteins. (A) FLAP curves of experiments performed in NIH3T3 cells plated on FN-coated 8 kPa and 100 kPa polyacrylamide (PAA) gels and glass. Results are mean±s.e.m. (B) t½ FLAP graph showing that the turnover rates of talin, vinculin and tensin are stabilized in a step-wise manner as substrate stiffness increases. α-actinin is partially stabilized by increased substrate stiffness, while the dynamics of FAK, paxillin and kindlin2 are unaffected. Data in A and B are pooled from three independent repeats, n=47–118 FAs. The box represents the 25–75th percentiles, and the median is indicated. The whiskers show the range of values. *P<0.05; **P<0.01; ****P<0.001; ****P<0.0001; n.s., no significant difference (Kruskal–Wallis with Dunn's multiple comparisons test).
Overall, these results demonstrate that the turnover of proteins linking integrins to the actin cytoskeleton (vinculin, talin and tensin1) is directly influenced by the physical properties of the ECM. Substrate stiffness had no influence on the turnover rates of paxillin, FAK and kindlin2, which appear to associate more transiently with FAs. α-actinin sits between these groups in both rate of turnover and sensitivity to ECM stiffness. The data suggest a modular composition of FAs with the mobile behavior of protein subsets responding differently when encountering environments of different rigidities.
Phosphorylation state and active vinculin do not affect the dynamics of FAK and paxillin
In order to investigate the molecular mechanisms of mechanical activation in more detail, we focused on talin, vinculin, FAK and paxillin. The activities of vinculin and talin are influenced by the application of forces, and constitutively active forms have significantly reduced turnovers compared to WT (Atherton et al., 2015; Cohen et al., 2006; Humphries et al., 2007). FAK and paxillin, however, are predominantly regulated by tyrosine phosphorylation. To analyze the effect of phosphorylation on protein turnover, we performed FLAP with FAK and paxillin mutated at crucial tyrosine residues (Fig. 3A).
Fig. 3.

Phosphorylation state does not affect turnover of FAK and paxillin. (A) Schematic showing wild-type FAK and paxillin and the mutant forms that were used in the study. (B) Representative time-lapse images showing the loss of fluorescence of the indicated (PA)GFP-tagged FA protein, following photoactivation. Scale bar: 5 μm. Graphs show the mean fluorescence loss over time, error bars are s.e.m. (C,D) Mean t½ FLAP (C) and FM (D) in NIH3T3 cells expressing the indicated FA protein and mutant constructs (scatter dots). Note there is no difference in turnover for the FAK and paxillin mutants. Data in these figures are pooled from three independent repeats, n=71–96 FAs. The box represents the 25–75th percentiles, and the median is indicated. The whiskers show the range of values. n.s., no significant difference (Kruskal–Wallis with Dunn's multiple comparisons test).
Interestingly, for paxillin, neither phosphomimetic (Y2E) nor phospho-null (Y2F) mutations had any effect on the mobility of the protein in FAs (Fig. 3B–D). Similarly, for FAK, neither mutating its autophosphorylation (Y397F) site nor expression of a kinase-dead (K454R) form, which significantly reduces autophosphorylation (Schaller et al., 1999), had any impact on turnover (Fig. 3B-D).
By using FLAP, we have previously shown that talin has a significantly reduced mobility in the presence of a constitutively active form of vinculin (vinT12) (Fig. S3A; Atherton et al., 2015). We found here that the co-expression of vinT12 had no effect on the turnover of FAK and paxillin (Fig. S3A–C).
Taken together, these data demonstrate that the dynamic exchange of FAK and paxillin are not affected by their phosphorylation state or vinculin activity; however, talin is stabilized by active vinculin. Again, results support a modular organization within FAs whereby the activity of one set of proteins can affect the mobility of similar proteins within the same module, but has no influence on the dynamics of others in separate modules.
FAK and paxillin phosphorylation in mechanotransduction
Our above results indicate that the dynamics of paxillin and FAK are unaffected by substrate stiffness, phosphorylation or vinculin activity. Therefore, we investigated whether they contribute to mechanotransduction via an alternative mechanism, such as altered phosphorylation. To this end, we analyzed the ratio of total paxillin to paxillin phosphorylated on Y118 (pY118-paxillin) and the ratio of total paxillin to FAK phosphorylated on Y397 (pY397-FAK) by assessing immunofluorescence of cells on FN-coated PAA gels or glass (Fig. S3D). We found that the percentage of the cell area that is composed of pY118-paxillin or pY397-FAK positive FAs (area fraction) increases with substrate stiffness (Fig. 4A). While total phosphorylation appears to rise with substrate stiffness, the ratio of phosphorylation of pPax and pFAK to total paxillin in FAs does not increase (Fig. 4B).
Fig. 4.

FAK and paxillin phosphorylation are involved in mechanotransduction. (A,B) Graphs showing the percentage of the cell area that is composed of pY118-paxillin- or pY397-FAK-positive FAs (A) and the ratio of pY118-paxillin or pY397-FAK to total paxillin (B) in cells plated on the indicated substrates. Data are pooled from three independent repeats, n=43–50 cells. ***P<0.001 (one-way ANOVA with Tukey post-hoc test). (C) Fluorescence loss curves (mean±s.e.m.) of vinculin and tensin FLAP experiments performed in NIH3T3 cells plated on the indicated substrates and treated with FAKi+Srci. (D) t½ FLAP graph showing that the turnover rates of vinculin and tensin have different responses to substrate stiffness following treatment with FAKi and Srci. Data in C and D are pooled from three independent repeats, n=58–75 FAs. *P<0.05; ***P<0.001; n.s., no significant difference (one-way ANOVA with Tukey post-hoc test). For the box plots in A,B,D, the box represents the 25–75th percentiles, and the median is indicated. The whiskers show the range of values.
The above results suggest that only the dynamics of FA proteins linking integrins to the actin cytoskeleton change in response to ECM stiffness. We next explored whether the varying levels of phosphorylated FAK and paxillin present at FAs in cells on different substrates contributes to the dynamics of these proteins. We inhibited phosphorylation by treating cells with specific FAK and Src kinase inhibitors (denoted FAKi and Srci; AZ13256675 and AZD0530, respectively) (Fig. S4A). These inhibitors have been extensively characterized and lead to a strong inhibition of FAK and paxillin phosphorylation in the FA (Horton et al., 2016, Fig. S4A). Similar results were obtained using an alternative FAK inhibitor (PF-573228) (data not shown). FRAP analysis showed that FAKi and Srci treatment had no effect on the turnover of vinculin or FAK compared to the DMSO control (Fig. S4B).
To test whether FAK and paxillin phosphorylation contributes to mechanosensing, we used FLAP to examine vinculin and tensin mobility on the PAA gels, following treatment with FAKi and Srci. For vinculin, inhibitor-treated cells showed the same previously seen pattern of reduced dynamic exchange as the substrate rigidity increased from 8 kPa to 100 kPa and from 100 kPa to glass (∼1 GPa) (Fig. 4C,D). Conversely, upon inhibition of FAK and Src activity, the turnover of tensin was no longer sensitive to ECM stiffness, with no significant difference in t½ FLAP between the substrates (Fig. 4C,D).
These results indicate that mechanosensing by vinculin and tensin occurs via different mechanisms. Vinculin is mechanosensitive irrespective of FAK and Src kinase activity. Surprisingly, tensin shows the opposite behavior and is no longer mechanosensitive when FAK and Src are inhibited.
FAK and Src kinase activity influence FA dynamics
Our above results showed that FA morphology and phosphorylation of FAK and paxillin at FAs are both influenced by substrate stiffness (Fig. S2; Fig. 4A). Therefore, we wanted to test the impact of FAK and paxillin phosphorylation on FA formation and turnover. Inhibiting FAK and Src activity (combined treatment with FAKi and Srci) led to a reduction in the number of small dot-like FXs at the cell periphery compared to DMSO-treated control cells, but their formation was not completely abolished (Fig. 5A). Furthermore, inhibitor-treated cells had more mature FAs, leading to an increase in mean FA size (Fig. 5A,B).
Fig. 5.

FAK and Src kinase activity influence FX formation and turnover. (A) Representative images showing NIH3T3 cells transfected with GFP–paxillin and treated with DMSO, FAKi or FAKi+Srci. Scale bar: 10 μm. Red boxes indicate the regions that are shown as a magnified view (below). Scale bars: 5 μm. (B) Graph showing the adhesion size in NIH3T3 cells under the indicated conditions. Data are pooled from three independent repeats, n=62–68 cells. The box represents the 25–75th percentiles, and the median is indicated. The whiskers show the range of values. ***P<0.001 (Student's t-test). (C) Representative time-lapse images of DMSO or drug-treated NIH3T3 cells transfected with GFP–paxillin. The colored images show adhesions present at the indicated time points. Overlap in color shows that the GFP–paxillin-containing FA has persisted between the time points (yellow, 0 and 30 min; turquoise, 30 and 60 min; white, all three time points). Note the persistence of FAs following FAKi and Srci treatment. (D) Representative images (above) and zoomed regions (below) of NIH3T3 cells pre-treated in suspension with the indicated drugs and co-stained for paxillin and pY118-paxillin after plating on FN-coated coverslips. Graphs show the fluorescence intensity across the indicated line. Note the presence of paxillin-positive FXs even in the absence of intracellular tension and tyrosine phosphorylation. Scale bar: 10 μm (upper images) and 5 μm (lower images).
To analyze FA dynamics in real time, we acquired time-lapse recordings of cells transfected with GFP–paxillin, with or without inhibitors. In DMSO-treated cells, FAs clearly matured from FXs that localized along the entire edge of protruding lamellipodia. In contrast, cells treated with the combination of FAKi and Srci had a reduced number of FXs, and many FAs appeared to mature from complexes at the base of prominent filopodia (Figs 5C and 6B; Movie 2). Interestingly, these cells displayed extremely stable FAs that persisted for much longer than the dynamic adhesions in control cells (Fig. 5C; Movie 2). Cells treated with the FAKi alone displayed a phenotype in between that of control cells and cells treated with both inhibitors (Fig. 5C).
Fig. 6.

FAK and Src kinase activity are required for lamellipodial protrusions, cell migration and spreading. (A) Representative recordings following the indicated drug treatments of NIH3T3 cells transfected with RFP-lifeact. The color-coded outlines show the position of the cell during the course of the 90 min movie (4 min intervals). Kymographs (below) were taken at the indicated position (red line). Note the dramatic drop in protrusive behavior following the inhibition of FAK and Src. Scale bar: 10 μm. (B) Quantification of the number of filopodia around the cell periphery following treatment with DMSO, FAKi or FAKi+Srci. Graph shows the mean stress of all cells analyzed (scatter dots), n=31–35 cells. **P<0.01 (one-way ANOVA with Tukey post-hoc test). (C) Representative trajectories of cells recorded for 16 h on 10 μm-wide fibronectin stripes; images of NIH3T3 cells plated on fibronectin stripes (inset). Note the decreased migration rate of cells treated with FAKi and FAKi+Srci. (D) Graph showing the increase in cell area over time following plating on FN-coated cover slips. Error bars are ±s.e.m., n=113–233 cells. *P<0.05; **P<0.01; ***P<0.001; n.s., no significant difference (one-way ANOVA with Tukey post-hoc test). (E) Representative cell images and stress maps of traction force microscopy. Stress maps are colored to show intensity. Scale bar: 10 μm. Graph shows the mean stress of each cell analyzed (scatter dots), n=34–45 cells. n.s., no significance (one-way ANOVA with Tukey post-hoc test). For the box plots in B and E, the box represents the 25–75th percentiles, and the median is indicated. The whiskers show the range of values.
Cells treated with FAKi and Srci appeared to form fewer FXs and more large FAs. Conversely, treatment with the Rho kinase (ROCK) inhibitor Y-27632 leads to the disassembly of mature, tension-dependent FAs, but tension-independent FXs are still present (Ballestrem et al., 2001). Therefore, we hypothesized that cells lacking FAK, Src and ROCK activity will not be able to form any cell–matrix adhesion structures. To our surprise, NIH3T3 cells pre-treated in suspension with FAKi, Srci and Y-27632 were still able to spread and form FXs at the cell periphery. These were positive for vinculin and paxillin (Fig. S4C), but contained no tyrosine-phosphorylated paxillin (Fig. 5D).
Taken together, these results suggest that adhesion complexes are extremely robust structures. Furthermore, although important for FA turnover, tyrosine phosphorylation of FAK and paxillin is not necessary for initial FX formation.
FAK and Src kinase activity are required for lamellipodial protrusions, migration and cell spreading
The preceding experiments indicated that there is little, if any, role for FAK and paxillin phosphorylation in FA formation, but that they are involved in adhesion disassembly. Therefore, we hypothesized that the major role of their phosphorylation may lie in the coordination of cellular protrusions. Tracking the cell edge dynamics of NIH3T3 cells expressing RFP-Lifeact using the QuimP plugin for ImageJ (Tyson et al., 2010) revealed that control (DMSO-treated) cells formed large, polarized protrusions, typically associated with 2D directional cell migration. FAKi-treated cells also formed lamellipodia, but these appeared as multiple small and disorganized protrusions (Fig. 6A; Movie 3). In contrast, cells treated with FAKi+Srci together formed no lamellipodia and were largely stationary, but had a large number of prominent filopodia at the cell edge (Fig. 6A,B; Movie 3).
These defects in polarized protrusion formation after drug treatment also translated into defects in polarized, two-dimensional migration. DMSO-treated cells plated on 10 µm-wide FN stripes [which forces cells to adopt an elongated shape, thus assisting cell polarization (Doyle et al., 2009)] migrated readily along the stripes. Conversely, cells treated with FAKi showed a significant reduction in cell migration, which was completely blocked by treatment with FAKi plus Srci (Fig. 6C; Fig. S4D).
Both lamellipodial protrusion and migration are Rac1-dependent processes (Ridley, 2011). To further analyze the role of FAK- and Src-mediated phosphorylation in Rac1-driven cellular functions, we performed cell spreading assays, a process also known to be driven by Rac (Price et al., 1998). While FAKi treatment of NIH3T3 cells barely affected long-term cell spreading (150 min), the process was significantly slower than for control cells. This effect was more pronounced when using both FAKi and Srci (Fig. 6D; Fig. S4E).
From these data, we conclude that phosphorylation of FAK and paxillin is vital for generating Rac1-driven cellular processes such as lamellipodial protrusions, which are critical for polarized cell migration and spreading.
Src-mediated tyrosine phosphorylation of FAK and paxillin has no impact on cellular traction forces
Coordinated cell motility and environmental stiffness sensing requires force exertion by the cell onto the matrix (Plotnikov et al., 2012). To explore whether the observed defects in cell migration and spreading could be due to a reduction in these forces, we measured the traction forces exerted by cells following treatment with FAKi and Srci. This treatment did not lead to any change in the traction stress applied by cells to the substrate (Fig. 6E). Therefore, although vital for initiating cellular protrusions, FAK and paxillin phosphorylation does not contribute to the generation of traction forces required to sense ECM stiffness and to pull the cell body forward during migration.
DISCUSSION
FAs are composed of a complex network of proteins that are integral for ECM stiffness sensing, cell adhesion and migration (Geiger et al., 2001). We have shown that FA proteins are organized into functional modules, and that these modules are each responsible for regulating distinct aspects of mechanotransduction.
Here, we classify the FA proteins that have been previously shown to link ECM-bound integrins to the actin cytoskeleton [including talin (Calderwood et al., 1999; Zhang et al., 2008), vinculin (Case et al., 2015; Menkel et al., 1994) and tensin1 (Haynie, 2014)] as belonging to a ‘structural module’. FA proteins known to be involved in adhesion-based signaling such as FAK (Mitra et al., 2005) and paxillin (Deakin and Turner, 2008) belong to a ‘signaling module’. Some proteins, such as kindlin2 and a-actinin, have been shown to have alternative roles, and belong to an intermediate module (Otey and Carpen, 2004; Roca-Cusachs et al., 2013; Theodosiou et al., 2016). In line with their roles as structural proteins with the FA, those proteins in the structural module have the slowest turnover, as measured by FRAP, whereas the signaling module proteins have a very fast turnover, suggesting a much more transient residency time within the adhesion (Fig. 1B).
Interestingly, the structural module proteins (talin, vinculin and tensin1), but not the signaling module proteins (FAK and paxillin), change their turnover in response to ECM stiffness. This suggests that it is those proteins that are involved in linking integrins to the actin cytoskeleton that are directly involved in sensing ECM stiffness. Our results showing that FAK- and Src-mediated phosphorylation within adhesions is required for protrusion (Fig. 6A), migration (Fig. 6C) and spreading (Fig. 6D), all of which are Rac1-dependent events (Ridley, 2011) that are also affected by substrate stiffness (Lo et al., 2000; Plotnikov et al., 2012; Swaminathan et al., 2016), suggest that these signaling module proteins are involved in generating the intracellular signaling events driving these phenomena. Therefore, the process of mechanotransduction requires the cooperation of both modules to function correctly: the structural proteins are involved in directly sensing mechanical stimuli (mechanosensing), whereas the signaling module proteins are involved in generating the intracellular signaling events in response (mechanosignaling) (Fig. 7).
Fig. 7.
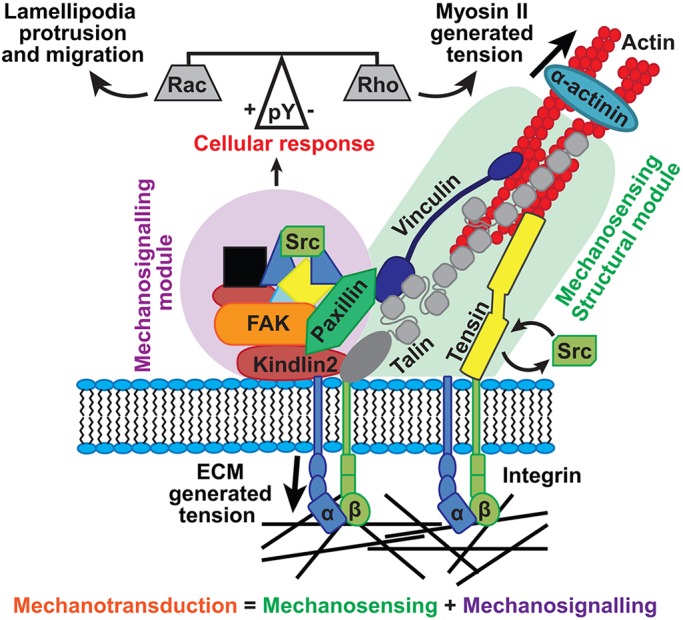
Model of mechanotransduction by the structural and signaling protein modules. Proteins in the structural module, including tensin1, talin and vinculin, are tightly associated with integrin and actin. These proteins sense extracellular mechanical signals by modifying their activation state and rate of turnover. This signal is transferred to the proteins in the signaling module, including FAK and paxillin, by modifying their level of tyrosine phosphorylation (pY). The level of tyrosine phosphorylation on these proteins determines the cellular response to the mechanical signal, activating Rac to promote protrusion and migration or Rho, leading to adhesion growth and stabilization.
The recent finding that FAK Y397 phosphorylation is required for stiffness-dependent cell spreading agrees with this model (Swaminathan et al., 2016). We propose that these cells are still mechanosensitive but are deficient in mechanosignaling. Similarly, perturbing either of these modules blocks polarized cell motility as has been shown for knockout cells, and with mutants or drugs that compromise talin and vinculin function (Atherton et al., 2015; Carisey et al., 2013), or FAK and paxillin function (Ilić et al., 1995; Petit et al., 2000; Swaminathan et al., 2016; Webb et al., 2004).
Differences in protein turnover rates reflect distinct function in cells
The observed differences in turnover rates between FA proteins were striking (Fig. 1). Lele et al. (2008) proposed that the turnover rate of an FA protein may, in part, depend on its number of interaction partners. It appears, however, that the function of the protein has a greater influence. Integrins have the lowest mobility, but can be subdivided into two populations, a more stable ligand-binding fraction and a highly mobile fraction that diffuses in the membrane (Fig. 1C,D). This is supported by super-resolution microscopy experiments, which have found reduced movement of integrins within FAs (Rossier et al., 2012).
After the slowly turned over structural proteins, there appears to be a module of proteins with intermediary turnover rates comprising actin-binding proteins including α-actinin and the ILK–PINCH–parvin complex. These proteins may be more involved in crosslinking the actin cytoskeleton, rather than linking integrins to the actomyosin machinery. However, more work will be required to analyze this subset of proteins further. As well as containing the highly mobile signaling proteins (FAK, paxillin and p130Cas), the third module also comprises proteins known to regulate actin polymerization (e.g. zyxin and VASP) (Beckerle, 1998; Hoffman et al., 2006). Proteins in this latter group localize to actin filament termini and are involved in actin organization and polymerization (Beckerle, 1998; Drees et al., 1999), rather than being an integral part of the mechanical structure between integrins and actin.
Interestingly, classifying FA proteins according to their dynamic behavior correlates well with the compartments identified by super-resolution microscopy (Kanchanawong et al., 2010; Liu et al., 2015): (1) the ‘integrin signaling layer’ (containing FAK and paxillin), (2) the ‘force transduction layer’ (talin and vinculin) and (3) the ‘actin regulatory layer’ (zyxin, VASP and α-actinin). According to our data, proteins residing within these individual layers have very similar rates of turnover. One interesting exception is α-actinin, whose slow turnover is likely due to its role as an actin crosslinker (Otey and Carpen, 2004), as well as its direct association with β-integrin (Roca-Cusachs et al., 2013).
Kindlin2, although turned over rapidly, showed a unique pattern of mobility. In contrast to other proteins, much of the protein diffused along the plasma membrane starting from the initially photoactivated pool of the FA structure (Fig. S2D). These findings are in line with kindlin2 bearing a pleckstrin homology (PH) domain, which binds phosphoinositides and is involved in recruiting proteins to the plasma membrane (Liu et al., 2011).
The relationship between protein dynamics and mechanosensing
Mechanical forces are thought to contribute to the activation of both talin and vinculin (del Rio et al., 2009; Hirata et al., 2014b; Yao et al., 2014). Previously, we have shown that the turnover of talin and vinculin reflects their activation state (Atherton et al., 2015; Carisey et al., 2013). Our observation that the turnover rates of talin, vinculin and tensin1 decreased with increasing ECM stiffness (Fig. 2), suggests that these proteins are more active on rigid substrates. Increasing ECM stiffness leads to elevated Rho activity (Paszek et al., 2005); therefore, it is possible that this, in turn, through a downstream increase of actomyosin tension, leads to increased activation of talin and vinculin in adhesion sites. Indeed, it has been shown by using a talin tension sensor construct, that the amount of tension across talin is modified by substrate stiffness and that vinculin is required for the maximal tension to be applied through talin at FAs (Austen et al., 2015; Kumar et al., 2016).
Tensin1 is present within both FAs and FBs (Clark et al., 2010). Our findings suggest that, within FAs under normal conditions, tensin1 is exposed to forces in a similar manner to talin and vinculin, and possibly acts as a mechanosensor via an analogous mechanism.
Interestingly, the above-mentioned mechanosensitive proteins differ from α-actinin in that α-actinin does not modify its turnover in response to different substrate stiffness on PAA gels (Fig. 2). Therefore, while on one hand α-actinin may be part of the clutch model, whereby talin and vinculin follow actin retrograde flow out of FAs, controlling the transmission of actomyosin tension through FAs (Hu et al., 2007; Thievessen et al., 2013), we propose that it belongs to a different, actin-binding module distinct from the mechanically activated integrin–talin–vinculin complex, in line with the super-resolution microscopy findings showing that α-actinin is in an actin-binding layer.
Crosstalk between the modules and its role in mechanosensing
Interestingly, constitutively active vinculin does not influence the turnover of FAK or paxillin, proteins in the ‘signaling module’ (Fig. S3). This is surprising, given that both of these proteins remain present at FAs after removal of intracellular tension through blebbistatin treatment or Rho kinase inhibition when they are co-expressed with vinT12 (Carisey et al., 2013).
The phosphorylation of the signaling module proteins FAK and paxillin is critical for cell motility (Fig. 6C) (Mitra et al., 2005; Subauste et al., 2004). The overall amount of phosphorylated FAK and paxillin that localized to FAs increased with substrate stiffness (Fig. 4A). This builds on previous biochemical data, which found that the whole-cell levels of phosphorylated FAK and paxillin increased on rigid substrates (Bae et al., 2014). Importantly, inhibiting phosphorylation using a combination of FAKi and Srci did not affect the ability of vinculin to modify its turnover in response to ECM stiffness (Fig. 4C,D). Similarly, in marked contrast to the well-documented roles for vinculin and talin in generating cellular traction forces (Atherton et al., 2015; Dumbauld et al., 2013; Thievessen et al., 2013), which are crucial for mechanosensing (Plotnikov et al., 2012), we find that FAK and Src inhibition has no effect on traction force generation (Fig. 6E). This suggests that this module is not directly involved in sensing environmental mechanical cues. Rather, it appears to control the downstream response to these signals through the activation of Rac1, which is essential for cell edge protrusions and migration (Ballestrem et al., 2001) (Fig. 6).
It has been suggested that tension-dependent paxillin phosphorylation has a role in vinculin recruitment to FAs (Case et al., 2015; Pasapera et al., 2010). However, we observed vinculin in FXs in the absence of both intracellular tension and tyrosine-phosphorylated paxillin (Fig. 5D; Fig. S4C), suggesting that vinculin can be recruited to FXs by several mechanisms.
It is interesting to note that tensin1 lost the ability to modify its turnover in response to ECM stiffness after drug treatment (FAKi and Srci; Fig. 4C,D). Tensin1 binds to the NPxY motif of the β-integrin cytoplasmic tail at the same site as talin, and phosphorylation of the tyrosine motif by Src has been shown to greatly reduce the affinity for talin, thus leading to an increase in tensin1 recruitment (McCleverty et al., 2007). Additionally, we found that the turnover of tensin1 in cells plated on glass is increased after drug treatment [t½=89.7 s (drugs) vs 123.8 s (no drugs)] (compare Fig. 4D with Fig. 2B). Therefore, tensin1 may be insensitive to ECM stiffness changes after FAKi and Srci treatment due to perturbation to its recruitment under these conditions.
Taken together, these findings suggest that a complex feedback mechanism exists between the FA protein modules. Although they themselves do not appear to be directly sensitive to ECM stiffness, proteins in the signaling module of the FA still appear to have a role in influencing the recruitment and mechanosensitive function of FA structural proteins.
Our findings shed further light on the modular organization of FAs and how separate modules contribute to mechanotransduction in distinct ways. Understanding precisely how these FA protein modules communicate with one another requires further research and would be an extremely interesting line of enquiry.
MATERIALS AND METHODS
Antibodies and reagents
Primary antibodies and dilutions used were: anti-vinculin (clone hVIN-1, Sigma), 1:500; anti-paxillin (clone 349, Millipore), 1:400; anti-paxillin pY118 (clone 44-722G, Life Technologies), 1:400; anti-FAK pY397 (clone 141-9, Life Technologies), 1:400 and anti-phosphorylated tyrosine (pY; clone 4G10, Millipore), 1:250. Secondary antibodies conjugated to DyLight-488 or -594 were all from Jackson ImmunoResearch Laboratories (used at 1:500). Texas-Red-X- (1:500) and Alexa Fluor 647-conjugated (1:200) phalloidin were from Life Technologies.
The FAK inhibitor (FAKi; AZ13256675) and Src inhibitor (Srci; AZD0530) were obtained from Astra Zeneca, and are available from the pharmacology toolbox (http://openinnovation.astrazeneca.com/what-we-offer/pharmacology-toolbox/). Y-27632 dihydrochloride was obtained from Tocris Bioscience.
Microscopy
Immunofluorescence microscopy and FRAP experiments were carried out using a Delta Vision (Applied Precision) microscope with a 60×1.42 NA Plan-Apochromat (immunofluorescence) or 100×1.40 NA Uplan S Apochromat (FRAP) objective and a Sedat Quad filter set (Chroma 86000v2), with images collected using a Coolsnap HQ (Photometrics) camera.
FLAP experiments, time-lapse recordings and actin retrograde flow measurements were acquired using a CSU-X1 spinning disc confocal system (Yokagowa) on a Zeiss Axio-Observer Z1 microscope with a 60×1.40 NA oil Plan-Apochromat objective, Evolve EMCCD camera (Photometrics) and motorized xyz stage (ASI). The 405 nm, 488 nm and 561 nm lasers were controlled using an Acousto-optical tunable filter through the laserstack [Intelligent Imaging Innovations (3i)].
For all constructs, we analyzed cells with low to intermediate expression levels. Protein expression level had no effect on the rate of protein turnover (Fig. S1D).
Cell migration experiments were performed using an AS MDW live-cell imaging system (Leica) using a 10×0.30 NA HC Plan Fluotar objective. The system was controlled using Image Pro 6.3 by Media Cybernetics Ltd. Cells were maintained at 37°C and 5% CO2. Images were collected using a Coolsnap HQ CCD camera (Photometrics).
Cell lines and transfection
NIH3T3 fibroblasts were cultured at 37°C in Dulbecco's Modified Eagle Medium (DMEM; Sigma), supplemented with 10% (v/v) fetal calf serum (FCS; Lonza), 1% (v/v) non-essential amino acids and 2 mM L-glutamine (both Sigma), and transfected using Lipofectamine reagent and Plus reagent, according to the manufacturer's instructions (Life Technologies). Cells were plated on glass-bottomed dishes (IBL) coated with 10 µg/ml bovine plasma fibronectin (FN; Sigma). For live-cell imaging experiments, the cells were transferred to carbonate-free Ham's F-12 medium supplemented with L-glutamine, 25 mM HEPES, penicillin-streptomycin (all Sigma) and 2% FCS. Starting solutions of FAKi and Srci were diluted in DMSO (to give a concentration of 3 mM) and Y-27632 was diluted in water (to give a concentration of 100 mM), which were then used to give the final concentrations as stated in subsequent sections.
Immunofluorescence microscopy
Cells were fixed with 4% (w/v) PFA and permeabilized with 0.5% (v/v) Triton X-100 (both Sigma-Aldrich). Antibodies were diluted in 1% BSA (Sigma-Aldrich) and added to the cells for 1 h.
Images were acquired on the Delta Vison system (see above) and processed using the FIJI-ImageJ software (version 1.48u). To analyze FA size and area fraction, images were bandpass filtered and background was subtracted using a rolling ball algorithm. FAs were thresholded manually and particle analysis for particles between 0.1 and 10 μm2 was used to analyze the average adhesion size for each cell.
To determine the ratio of pY118-paxillin or pY397-FAK to total paxillin, images of co-immunostained cells were taken with the same exposure time between channels. FA images were processed as above. The same threshold was used in both the paxillin and pY118-paxillin or pY397-FAK channels. The mean intensity of every adhesion was measured in both channels by using particle analysis, and the ratio of pY118-Pax or pY397-FAK to total paxillin was calculated for all adhesions in the cell.
Flilpodia were quantified using the FiloDetect MATLAB script, developed and made freely available by Theodore Perkins' research group, University of Ottawa (Nilufar et al., 2013).
Y-27632 experiments
NIH3T3 cells were trypsinized and suspended in 2 ml of supplemented DMEM containing 3 µM FAKi+3 µM Srci+50 µM Y-27632, or an equivalent volume of DMSO for 1 h. These concentrations and treatment times are sufficient for maximal FAK and Src inhibition (Horton et al., 2016). Cells were plated on FN-coated coverslips for 4 h prior to fixation and staining. Images were acquired with the DeltaVision system (see above).
Polyacrylamide gel preparation
FN-coated PAA gels [6% (∼8 kPa) or 25% (∼100 kPa) of gel diluted from 30% Protogel in PBS, 37.5:1 fixed ratio of acrylamide:bis-acrylamide; EC-890, National Diagnostics] were prepared in 35 mm glass bottomed dishes (IBL) according to previously published methods, with modifications (Pelham and Wang, 1998; Zhang et al., 2013). The FN coating density was equivalent between PAA gels and glass.
The stiffness of the 6% (8.760±0.209 kPa; n=110) and 25% (113.188±8.04 kPa; n=100) PAA gels were measured with a CellHesion Atomic Force Microscope (Nanowizard, CellHesion 200; JPK Instruments, Berlin, Germany) with tip-less cantilevers (NP-O10, Bruker AFM Probes), modified by attaching 10 mm diameter polystyrene beads (PPS-10.0, Kisker). The gel stiffness values shown are mean±s.e.m.
Live-cell imaging
FRAP
Transfected NIH3T3 fibroblasts were incubated overnight at 37°C. The cells were placed in the microscope chamber at 37°C for 1 h prior to imaging, to ensure they were in equilibrium. For cells treated with FAKi and Srci, medium containing 3 µM FAKi, 3 µM FAKi plus 3 µM Srci or an equivalent volume of DMSO was added to the cells for 1 h.
Images were acquired using the Delta Vision system (above). Five peripheral mature FAs per cell were selected, and photobleaching was achieved with a 7.5 ms burst of the 488 nm laser at 100% power, used to bleach a 2 µm diameter circle within the FAs. Softworks software was used to capture three images prior to photobleaching and then one image every 10 s for 5 min post bleaching. Movies were analyzed using Softworks inbuilt photokinetics analysis and MATLAB, with a script developed in-house, as described previously (Carisey et al., 2011; Humphries et al., 2007).
FLAP
NIH3T3 fibroblasts were co-transfected with the required (PA)GFP-tagged DNA constructs and an mCherry-tagged FA marker protein (mCherry–zyxin, unless otherwise specified). The cells were incubated overnight at 37°C. The medium was replaced and the cells were placed in the microscope chamber at 37°C for 1 h.
Cells were imaged using the CSU-X1 spinning disc confocal system. Approximately five peripheral mature FAs were photoactivated per cell with a 5 ms burst of a 405 nm laser at 100% power. Slidebook software was used to capture three images prior to photoactivation and images were then taken every 10 s for 3–5 min post photoactivation.
Movies were analyzed with MATLAB using a script developed in-house, as described previously (Atherton et al., 2015).
FAKi and Srci experiments
NIH3T3 cells were co-transfected with GFP–paxillin or YFP–dSH2 and plated on FN-coated glass–bottomed dishes (IBL). Cells were treated as described above for 1 h prior to fixation. Cells were imaged using the CSU-X1 spinning disc confocal system.
For time-lapse recordings, the medium was replaced 1 h prior to imaging with supplemented DMEM containing 3 µM FAKi, 3 µM FAKi+3 µM Srci or an equivalent volume of DMSO. Images were acquired every 2 min for 90 min. Cell membrane protrusion was analyzed using the QuimP11b toolbox for ImageJ, kindly provided by Till Bretschneider and Richard Tyson (University of Warwick, Coventry, UK; Tyson et al., 2010).
Migration
Polydimethylsiloxane (PDMS) stamps were generated by coating a glass mask (10 µm stripes) with PDMS (10:1 weight ratio of base:curing agent) and heating to 60°C for 2 h. The PDMS stamps were coated with 50 µg/ml FN for 1 h at room temperature and placed on a glass coverslip for 1 h.
NIH3T3 cells were plated on the patterned coverslips for 1 h at 37°C. Unattached cells were washed away with PBS and the medium was replaced with supplemented DMEM containing 25 mM HEPES as well as 3 µM FAKi, 3 µM FAKi+3 µM Srci or an equivalent volume of DMSO. Cells were incubated for 1 h prior to imaging. Images were acquired every 10 min for 16 h using the AS MDW live-cell imaging system (above). Cell migration was measured using the Manual Tracking plugin, and analyzed by using the chemotaxis and migration tool for FIJI-ImageJ (Ibidi).
Spreading
NIH3T3 cells were trypsinized and suspended in 1 ml supplemented DMEM containing 3 µM FAKi, 3 µM FAKi+3 µM Srci or an equivalent volume of DMSO for 1 h. Treated cells were plated on FN-coated coverslips and fixed after the indicated times and stained for actin. Images were acquired using an AxioObserver Z1 microscope with a 10× objective and a 565 nm LED. Cell area was measured manually using FIJI-ImageJ software.
Traction force microscopy
NIH3T3 cells were transfected with GFP–paxillin and plated on FN-coated ∼8 kPa PAA gels containing 4% (v/v) FluoSpheres carboxylate-modified microspheres [F8810, red fluorescent (580/605); Thermo Fisher]. Cells were treated with 3 µM FAKi, 3 µM FAKi+3 µM Srci or an equivalent volume of DMSO for 1 h prior to imaging. Traction force images were acquired on the CSU-X1 spinning disc confocal system (as above), and analyzed as previously described (Atherton et al., 2015).
Statistical analysis
Graphing and statistical analysis were performed by using GraphPad Prism 6 software. When comparing means, the D'Agostino–Pearson test was used to assess the normality of the data to determine the appropriate statistical tests to use.
Acknowledgements
The C.B. laboratory is part of the Wellcome Trust Centre for Cell-Matrix Research, University of Manchester, which is supported by core funding from the Wellcome Trust (grant number 088785/Z/09/Z). The Bioimaging Facility microscopes were purchased with grants from Biotechnology and Biological Sciences Research Council (BBSRC), Wellcome Trust and the University of Manchester Strategic Fund. Special thanks to Simon T. Barry from AstraZeneca for providing the FAK inhibitor and Professor David Critchley, Professor Martin Humphries and Dr Janet Askari for critical reading of the manuscript. We thank the staff of the Bioimaging facility at the University of Manchester, in particular Dr Peter March and Dr Egor Zindy for their help with imaging and analysis, Dr Kristian Franze for his help with traction force microscopy analysis and Till Bretschneider for providing the QuimP11b software.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
B.S., P.A. and R.T. designed and conducted the experiments and analyzed the data. B.S. and P.A. performed photoactivation experiments and experiments using FAKi and Srci. R.T. performed FRAP experiments and P.A. ran the traction force experiments. D.-Y.W. designed the protocols for PAA gel preparation and traction force microscopy. C.B., B.S., P.S. and R.T. conceived the ideas; C.B. and B.S. wrote the manuscript with contributions from R.T. and P.A. C.B. supervised and directed the project.
Funding
C.B. acknowledges the Biotechnology and Biological Sciences Research Council (BBSRC) and Wellcome Trust for funding of this project. B.S. was supported by Wellcome Trust (099734/Z/12/Z). R.T. was supported by a BBSRC DTP studentship. P.A. was funded by BBSRC (BB/J012254/1) and Bioventus and, since October 2016, is funded by the BBSRC (BB/P000681/1). Deposited in PMC for release after 6 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.195362.supplemental
References
- Atherton P., Stutchbury B., Wang D.-Y., Jethwa D., Tsang R., Meiler-Rodriguez E., Wang P., Bate N., Zent R., Barsukov I. L. et al. (2015). Vinculin controls talin engagement with the actomyosin machinery. Nat. Commun. 6, 10038 10.1038/ncomms10038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton P., Stutchbury B., Jethwa D. and Ballestrem C. (2016). Mechanosensitive components of integrin adhesions: role of vinculin. Exp. Cell Res. 343, 21-27. 10.1016/j.yexcr.2015.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austen K., Ringer P., Mehlich A., Chrostek-Grashoff A., Kluger C., Klingner C., Sabass B., Zent R., Rief M. and Grashoff C. (2015). Extracellular rigidity sensing by talin isoform-specific mechanical linkages. Nat. Cell Biol. 17, 1597-1606. 10.1038/ncb3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae Y. H., Mui K. L., Hsu B. Y., Liu S.-L., Cretu A., Razinia Z., Xu T., Pure E. and Assoian R. K. (2014). A FAK-Cas-Rac-Lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci. Signal. 7, ra57 10.1126/scisignal.2004838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballestrem C., Hinz B., Imhof B. A. and Wehrle-Haller B. (2001). Marching at the front and dragging behind: differential alphaVbeta3-integrin turnover regulates focal adhesion behavior. J. Cell Biol. 155, 1319-1332. 10.1083/jcb.200107107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckerle M. C. (1998). Spatial control of actin filament assembly: lessons from Listeria. Cell 95, 741-748. 10.1016/S0092-8674(00)81697-9 [DOI] [PubMed] [Google Scholar]
- Calderwood D. A., Zent R., Grant R., Rees D. J. G., Hynes R. O. and Ginsberg M. H. (1999). The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 274, 28071-28074. 10.1074/jbc.274.40.28071 [DOI] [PubMed] [Google Scholar]
- Calderwood D. A., Campbell I. D. and Critchley D. R. (2013). Talins and kindlins: partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 14, 503-517. 10.1038/nrm3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carisey A., Stroud M., Tsang R. and Ballestrem C. (2011). Fluorescence recovery after photobleaching. Methods Mol. Biol. 769, 387-402. 10.1007/978-1-61779-207-6_26 [DOI] [PubMed] [Google Scholar]
- Carisey A., Tsang R., Greiner A. M., Nijenhuis N., Heath N., Nazgiewicz A., Kemkemer R., Derby B., Spatz J. and Ballestrem C. (2013). Vinculin regulates the recruitment and release of core focal adhesion proteins in a force-dependent manner. Curr. Biol. 23, 271-281. 10.1016/j.cub.2013.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case L. B., Baird M. A., Shtengel G., Campbell S. L., Hess H. F., Davidson M. W. and Waterman C. M. (2015). Molecular mechanism of vinculin activation and nanoscale spatial organization in focal adhesions. Nat. Cell Biol. 17, 880-892. 10.1038/ncb3180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K., Howe J. D., Pullar C. E., Green J. A., Artym V. V., Yamada K. M. and Critchley D. R. (2010). Tensin 2 modulates cell contractility in 3D collagen gels through the RhoGAP DLC1. J. Cell. Biochem. 109, 808-817. 10.1002/jcb.22460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J. H., Giudici M.-L., Burke J. E., Williams R. L., Maloney D. J., Marugan J. and Irvine R. F. (2015). The function of phosphatidylinositol 5-phosphate 4-kinase gamma (PI5P4Kgamma) explored using a specific inhibitor that targets the PI5P-binding site. Biochem. J. 466, 359-367. 10.1042/BJ20141333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D. M., Kutscher B., Chen H., Murphy D. B. and Craig S. W. (2006). A conformational switch in vinculin drives formation and dynamics of a talin-vinculin complex at focal adhesions. J. Biol. Chem. 281, 16006-16015. 10.1074/jbc.M600738200 [DOI] [PubMed] [Google Scholar]
- Deakin N. O. and Turner C. E. (2008). Paxillin comes of age. J. Cell Sci. 121, 2435-2444. 10.1242/jcs.018044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rio A., Perez-Jimenez R., Liu R., Roca-Cusachs P., Fernandez J. M. and Sheetz M. P. (2009). Stretching single talin rod molecules activates vinculin binding. Science 323, 638-641. 10.1126/science.1162912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle A. D., Wang F.W., Matsumoto K. and Yamada K. M. (2009). One-dimensional topography underlies three-dimensional fibrillar cell migration. J. Cell Biol. 184, 481-490. 10.1083/jcb.200810041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees B. E., Andrews K. M. and Beckerle M. C. (1999). Molecular dissection of zyxin function reveals its involvement in cell motility. J. Cell Biol. 147, 1549-1560. 10.1083/jcb.147.7.1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumbauld D. W., Lee T. T., Singh A., Scrimgeour J., Gersbach C. A., Zamir E. A., Fu J., Chen C. S., Curtis J. E., Craig S. W. et al. (2013). How vinculin regulates force transmission. Proc. Natl. Acad. Sci. USA 110, 9788-9793. 10.1073/pnas.1216209110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler A. J., Sen S., Sweeney H. L. and Discher D. E. (2006). Matrix elasticity directs stem cell lineage specification. Cell 126, 677-689. 10.1016/j.cell.2006.06.044 [DOI] [PubMed] [Google Scholar]
- Geiger B., Bershadsky A., Pankov R. and Yamada K. M. (2001). Transmembrane crosstalk between the extracellular matrix and the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2, 793-805. 10.1038/35099066 [DOI] [PubMed] [Google Scholar]
- Han S. J., Bielawski K. S., Ting L. H., Rodriguez M. L. and Sniadecki N. J. (2012). Decoupling substrate stiffness, spread area, and micropost density: a close spatial relationship between traction forces and focal adhesions. Biophys. J. 103, 640-648. 10.1016/j.bpj.2012.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynie D. T. (2014). Molecular physiology of the tensin brotherhood of integrin adaptor proteins. Proteins 82, 1113-1127. 10.1002/prot.24560 [DOI] [PubMed] [Google Scholar]
- Hirata H., Chiam K.-H., Lim C. T. and Sokabe M. (2014a). Actin flow and talin dynamics govern rigidity sensing in actin-integrin linkage through talin extension. J. R Soc. Interface 11, pii: 20140734 10.1098/rsif.2014.0734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata H., Tatsumi H., Lim C. T. and Sokabe M. (2014b). Force-dependent vinculin binding to talin in live cells: a crucial step in anchoring the actin cytoskeleton to focal adhesions. Am. J. Physiol. Cell Physiol., 306, C607-C620. 10.1152/ajpcell.00122.2013 [DOI] [PubMed] [Google Scholar]
- Hoffman L. M., Jensen C. C., Kloeker S., Wang C.-L. A., Yoshigi M. and Beckerle M. C. (2006). Genetic ablation of zyxin causes Mena/VASP mislocalization, increased motility, and deficits in actin remodeling. J. Cell Biol. 172, 771-782. 10.1083/jcb.200512115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton E. R., Humphries J. D., Stutchbury B., Jacquemet G., Ballestrem C., Barry S. T. and Humphries M. J. (2016). Modulation of FAK and Src adhesion signaling occurs independently of adhesion complex composition. J. Cell Biol. 212, 349-364. 10.1083/jcb.201508080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K., Ji L., Applegate K. T., Danuser G. and Waterman-Storer C. M. (2007). Differential transmission of actin motion within focal adhesions. Science 315, 111-115. 10.1126/science.1135085 [DOI] [PubMed] [Google Scholar]
- Humphries J. D., Wang P., Streuli C., Geiger B., Humphries M. J. and Ballestrem C. (2007). Vinculin controls focal adhesion formation by direct interactions with talin and actin. J. Cell Biol. 179, 1043-1057. 10.1083/jcb.200703036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes R. O. (2002). Integrins: bidirectional, allosteric signaling machines. Cell 110, 673-687. 10.1016/S0092-8674(02)00971-6 [DOI] [PubMed] [Google Scholar]
- Ilić D., Furuta Y., Kanazawa S., Takeda N., Sobue K., Nakatsuji N., Nomura S., Fujimoto J., Okada M. and Yamamoto T. (1995). Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377, 539-544. 10.1038/377539a0 [DOI] [PubMed] [Google Scholar]
- Kanchanawong P., Shtengel G., Pasapera A. M., Ramko E. B., Davidson M. W., Hess H. F. and Waterman C. M. (2010). Nanoscale architecture of integrin-based cell adhesions. Nature 468, 580-584. 10.1038/nature09621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Ouyang M., Van den Dries K., McGhee E. J., Tanaka K., Anderson M. D., Groisman A., Goult B. T., Anderson K. I. and Schwartz M. A. (2016). Talin tension sensor reveals novel features of focal adhesion force transmission and mechanosensitivity. J. Cell Biol. 213, 371-383. 10.1083/jcb.201510012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lele T. P., Pendse J., Kumar S., Salanga M., Karavitis J. and Ingber D. E. (2006). Mechanical forces alter zyxin unbinding kinetics within focal adhesions of living cells. J. Cell. Physiol. 207, 187-194. 10.1002/jcp.20550 [DOI] [PubMed] [Google Scholar]
- Lele T. P., Thodeti C. K., Pendse J. and Ingber D. E. (2008). Investigating complexity of protein–protein interactions in focal adhesions. Biochem. Biophys. Res. Commun. 369, 929-934. 10.1016/j.bbrc.2008.02.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Fukuda K., Xu Z., Ma Y.-Q., Hirbawi J., Mao X., Wu C., Plow E. F. and Qin J. (2011). Structural basis of phosphoinositide binding to kindlin-2 protein pleckstrin homology domain in regulating integrin activation. J. Biol. Chem. 286, 43334-43342. 10.1074/jbc.M111.295352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Wang Y., Goh W. I., Goh H., Baird M. A., Ruehland S., Teo S., Bate N., Critchley D. R., Davidson M. W. et al. (2015). Talin determines the nanoscale architecture of focal adhesions. Proc. Natl. Acad. Sci. USA, 112, E4864-E4873. 10.1073/pnas.1512025112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo C.-M., Wang H.-B., Dembo M. and Wang Y.-L. (2000). Cell movement is guided by the rigidity of the substrate. Biophys. J. 79, 144-152. 10.1016/S0006-3495(00)76279-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleverty C. J., Lin D. C. and Liddington R. C. (2007). Structure of the PTB domain of tensin1 and a model for its recruitment to fibrillar adhesions. Protein Sci. 16, 1223-1229. 10.1110/ps.072798707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menkel A. R., Kroemker M., Bubeck P., Ronsiek M., Nikolai G. and Jockusch B. M. (1994). Characterization of an F-actin-binding domain in the cytoskeletal protein vinculin. J. Cell Biol. 126, 1231-1240. 10.1083/jcb.126.5.1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S. K., Hanson D. A. and Schlaepfer D. D. (2005). Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56-68. 10.1038/nrm1549 [DOI] [PubMed] [Google Scholar]
- Nilufar S., Morrow A. A., Lee J. M. and Perkins T. J. (2013). FiloDetect: automatic detection of filopodia from fluorescence microscopy images. BMC Syst. Biol. 7, 66 10.1186/1752-0509-7-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes P. W., Beckham Y., Stricker J. and Gardel M. L. (2012). Tension is required but not sufficient for focal adhesion maturation without a stress fiber template. J. Cell Biol. 196, 363-374. 10.1083/jcb.201107042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otey C. A. and Carpen O. (2004). Alpha-actinin revisited: a fresh look at an old player. Cell Motil. Cytoskeleton 58, 104-111. 10.1002/cm.20007 [DOI] [PubMed] [Google Scholar]
- Parsons J. T. (2003). Focal adhesion kinase: the first ten years. J. Cell Sci. 116, 1409-1416. 10.1242/jcs.00373 [DOI] [PubMed] [Google Scholar]
- Pasapera A. M., Schneider I. C., Rericha E., Schlaepfer D. D. and Waterman C. M. (2010). Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 188, 877-890. 10.1083/jcb.200906012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek M. J., Zahir N., Johnson K. R., Lakins J. N., Rozenberg G. I., Gefen A., Reinhart-King C. A., Margulies S. S., Dembo M., Boettiger D. et al. (2005). Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241-254. 10.1016/j.ccr.2005.08.010 [DOI] [PubMed] [Google Scholar]
- Pelham R. J. Jr and Wang Y. L. (1997). Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA 94, 13661-13665. 10.1073/pnas.94.25.13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham R. J. Jr and Wang Y. L. (1998). Cell locomotion and focal adhesions are regulated by the mechanical properties of the substrate. Biol. Bull. 194, 348-349; discussion 349-350 10.2307/1543109 [DOI] [PubMed] [Google Scholar]
- Petit V., Boyer B., Lentz D., Turner C. E., Thiery J. P. and Vallés A. M. (2000). Phosphorylation of tyrosine residues 31 and 118 on paxillin regulates cell migration through an association with CRK in NBT-II cells. J. Cell Biol. 148, 957-970. 10.1083/jcb.148.5.957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov S. V., Pasapera A. M., Sabass B. and Waterman C. M. (2012). Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell 151, 1513-1527. 10.1016/j.cell.2012.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price L. S., Leng J., Schwartz M. A. and Bokoch G. M. (1998). Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol. Biol. Cell 9, 1863-1871. 10.1091/mbc.9.7.1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A. J. (2011). Life at the leading edge. Cell 145, 1012-1022. 10.1016/j.cell.2011.06.010 [DOI] [PubMed] [Google Scholar]
- Roca-Cusachs P., del Rio A., Puklin-Faucher E., Gauthier N. C., Biais N. and Sheetz M. P. (2013). Integrin-dependent force transmission to the extracellular matrix by alpha-actinin triggers adhesion maturation. Proc. Natl. Acad. Sci. USA 110, E1361-E1370. 10.1073/pnas.1220723110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossier O., Octeau V., Sibarita J.-B., Leduc C., Tessier B., Nair D., Gatterdam V., Destaing O., Albigès-Rizo C., Tampé R. et al. (2012). Integrins β1 and β3 exhibit distinct dynamic nanoscale organizations inside focal adhesions. Nat. Cell Biol. 14, 1057-1067. 10.1038/ncb2588 [DOI] [PubMed] [Google Scholar]
- Sai X., Naruse K. and Sokabe M. (1999). Activation of pp60(src) is critical for stretch-induced orienting response in fibroblasts. J. Cell Sci. 112, 1365-1373. [DOI] [PubMed] [Google Scholar]
- Schaller M. D., Hildebrand J. D. and Parsons J. T. (1999). Complex formation with focal adhesion kinase: a mechanism to regulate activity and subcellular localization of Src kinases. Mol. Biol. Cell 10, 3489-3505. 10.1091/mbc.10.10.3489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subauste M. C., Pertz O., Adamson E. D., Turner C. E., Junger S. and Hahn K. M. (2004). Vinculin modulation of paxillin-FAK interactions regulates ERK to control survival and motility. J. Cell Biol. 165, 371-381. 10.1083/jcb.200308011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan V., Fischer R. S. and Waterman C. M. (2016). The FAK-Arp2/3 interaction promotes leading edge advance and haptosensing by coupling nascent adhesions to lamellipodia actin. Mol. Biol. Cell 27, 1085-1100. 10.1091/mbc.E15-08-0590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodosiou M., Widmaier M., Bottcher R. T., Rognoni E., Veelders M., Bharadwaj M., Lambacher A., Austen K., Muller D. J., Zent R., and , R.et al. (2016). Kindlin-2 cooperates with talin to activate integrins and induces cell spreading by directly binding paxillin. Elife 5, e10130 10.7554/elife.10130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thievessen I., Thompson P. M., Berlemont S., Plevock K. M., Plotnikov S. V., Zemljic-Harpf A., Ross R. S., Davidson M. W., Danuser G., Campbell S. L. et al. (2013). Vinculin-actin interaction couples actin retrograde flow to focal adhesions, but is dispensable for focal adhesion growth. J. Cell Biol. 202, 163-177. 10.1083/jcb.201303129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson R. A., Epstein D. B. A., Anderson K. I. and Bretschneider T. (2010). High resolution tracking of cell membrane dynamics in moving cells: an electrifying approach. Math. Model. Nat. Pheno. 5, 34-55. 10.1051/mmnp/20105102 [DOI] [Google Scholar]
- Wang J. G., Miyazu M., Matsushita E., Sokabe M. and Naruse K. (2001). Uniaxial cyclic stretch induces focal adhesion kinase (FAK) tyrosine phosphorylation followed by mitogen-activated protein kinase (MAPK) activation. Biochem. Biophys. Res. Commun. 288, 356-361. 10.1006/bbrc.2001.5775 [DOI] [PubMed] [Google Scholar]
- Webb D. J., Donais K., Whitmore L. A., Thomas S. M., Turner C. E., Parsons J. T. and Horwitz A. F. (2004). FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 6, 154-161. 10.1038/ncb1094 [DOI] [PubMed] [Google Scholar]
- Xu W., Baribault H. and Adamson E. D. (1998). Vinculin knockout results in heart and brain defects during embryonic development. Development 125, 327-337. [DOI] [PubMed] [Google Scholar]
- Yamada K. M. and Geiger B. (1997). Molecular interactions in cell adhesion complexes. Curr. Opin. Cell Biol. 9, 76-85. 10.1016/S0955-0674(97)80155-X [DOI] [PubMed] [Google Scholar]
- Yao M., Goult B. T., Chen H., Cong P., Sheetz M. P. and Yan J. (2014). Mechanical activation of vinculin binding to talin locks talin in an unfolded conformation. Sci. Rep. 4, 4610 10.1038/srep04610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R., Milo R., Kam Z. and Geiger B. (2007). A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J. Cell Sci. 120, 137-148. 10.1242/jcs.03314 [DOI] [PubMed] [Google Scholar]
- Zhang X., Jiang G., Cai Y., Monkley S. J., Critchley D. R. and Sheetz M. P. (2008). Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat. Cell Biol. 10, 1062-1068. 10.1038/ncb1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Guo W.-H., Rape A. and Wang Y.-L. (2013). Micropatterning cell adhesion on polyacrylamide hydrogels. Methods Mol. Biol. 1066, 147-156. 10.1007/978-1-62703-604-7_13 [DOI] [PubMed] [Google Scholar]