

Abstract
Background
Mannose-binding lectin (MBL) acts in the innate immune response to Helicobacter pylori. Interleukin 8 (IL-8) is a potent cytokine produced by gastric epithelial cells in response to H. pylori. We aimed to investigate whether polymorphisms in MBL2 and IL-8 influence susceptibility to H. pylori infection, and the associations of these polymorphisms with the risk of gastroduodenal diseases in a Korean population.
Methods
We consecutively enrolled 176 H. pylori-negative control subjects, 221 subjects with H. pylori-positive non-atrophic gastritis, 52 mild atrophic gastritis (AG), 61 severe AG, 175 duodenal ulcer, and 283 gastric cancer (GC). Allele-specific PCR-RFLP was conducted for polymorphisms in MBL2 exon 1 (codon 52, 54, and 57) and IL-8 -251 T > A. IL-8 levels in gastric mucosal tissues and serum MBL levels were measured by enzyme-linked immunosorbent assay.
Results
MBL2 exon 1 polymorphic variants were found only in codon 54, and the allele frequencies did not differ significantly between the control and disease groups. Although serum MBL levels in codon 54 A/A mutants were markedly low, it did not influence susceptibility to H. pylori infection or the risk of gastroduodenal diseases. IL-8 levels were significantly different between T/T wild type, T/A heterozygote, and A/A mutant genotypes. IL-8 -251 A allele carriers (A/A + T/A) showed increased IL-8 levels, and were significantly associated with the risk of severe AG and GC.
Conclusions
We suggest that a combination of H. pylori infection and the IL-8 -251 T > A polymorphism might increase the risk of severe AG and GC in a Korean population.
Keywords: Mannose-binding lectin 2, Interleukin 8, Helicobacter pylori, Gastric cancer
Background
The Helicobacter pylori infection rate is about 50% among the worldwide adult population [1]. In Korea, the adult H. pylori infection rate was 66.9% in 1998, 59.6% in 2005, and dropped to 54.4% in 2011 [2]. The main cause of this decrease in infection rate is the improvement in unsanitary environmental conditions. Besides environmental factors, bacterial and host factors are involved in the pathogenesis of H. pylori infection. With regard to bacterial factors, H. pylori strains possessing the virulence factors cagA, vacA s1a/m1, and iceA1 are known to be particularly virulent, and are frequently associated with severe gastric epithelial damage [3]. In contrast to Western populations, the cagA protein is commonly found in Korean patients with gastric cancer (GC) and duodenal ulcer (DU) [4]. However, there have been no associations reported between different H. pylori genotypes and clinical outcome in Korean patients [5, 6].
With regard to host factors, host genetic variants may influence susceptibility to H. pylori and the pathogenesis of gastroduodenal diseases. Host factors are mainly related to two processes: recognition of H. pylori by the innate immune system, and the level of the cytokine response [7, 8]. Polymorphisms in pro- and anti-inflammatory cytokines are associated with the risk of atrophic gastritis (AG) and GC. Interleukin 1 beta (IL-1β), tumor necrosis factor-alpha, IL-6, and IL-8 are up-regulated in response to H. pylori infection [9]. Several anti-inflammatory cytokines such as IL-Rα and IL-10 are also related to H. pylori infection [10, 11].
IL-8 is a major neutrophil-activating cytokine and plays a central role in the immuno-pathogenesis of H. pylori-induced gastric mucosal injury. IL-8 levels are 10-fold higher in GC specimens than in normal gastric tissues [12]. The IL-8 -251 T > A polymorphism has been reported to be associated with increased production of IL-8 protein, and higher risks of AG, gastric ulcer, and GC [13–17]. However, many other reports are inconsistent with these findings [18–23], and a meta-analysis of epidemiological studies revealed no overall association [24].
The innate immune response to H. pylori infection is a further candidate host factor. Toll-like receptors (TLRs) recognize conserved pathogen-associated molecular patterns expressed by many pathogens, including H. pylori [25]. Mannose-binding lectin (MBL), a pattern recognition receptor encoded by the MBL2 gene, recognizes lipopolysaccharide in the cell wall of gram-negative bacteria such as H. pylori [26, 27]. H. pylori activates MBL in vitro, resulting in complement deposition [28, 29]. Some studies have found a possible association of MBL2 haplotype with susceptibility to H. pylori infection, as well as with risk of GC [30, 31]. However, other studies did not find any significant association between MBL genotype and H. pylori infection prevalence or GC risk [32, 33].
Serum MBL levels vary widely between healthy individuals, mainly due to genetic variation [34–36]. The variation in serum MBL levels is correlated with point mutations in the coding and promoter regions of MBL2. Three mutations within exon 1 (in codons 52, 54, and 57) interfere with MBL function and are associated with low serum levels of MBL. In African populations, point mutations at codons 52 and 57 occur frequently [36, 37]. In Caucasians, mutations at codons 52 and 54 are common [38]. In Chinese, Japanese, and Korean populations, mutations are predominantly common in codon 54, but not in codons 52 or 57 [39–41]. Polymorphisms within the promoter and 5′-untranslated regions of MBL2 also affect serum levels of MBL, but the effects were found to be lower than those of the exon 1 polymorphisms [41].
The aims of this study were: 1) to examine the influence of the polymorphisms in codons 52, 54, and 57 of MBL2 (related to innate immunity) on susceptibility to H. pylori infection; 2) to evaluate the association of the IL-8 -251 T > A polymorphism with the risk of gastroduodenal diseases in a Korean population; and 3) to analyze our and other investigators’ large-scale data regarding the IL-8 -251 T > A polymorphism and GC risk in Korean, Japanese, Chinese, and Caucasian populations.
Methods
From January 2012 to May 2015, H. pylori-negative healthy control subjects (control, n = 176), H. pylori-positive non-atrophic gastritis patients (NAG, n = 108), H. pylori-positive mild AG patients (n = 52), H. pylori-positive severe AG patients (n = 61), DU patients (n = 175), and GC patients (n = 283) were consecutively enrolled.
All participants (n = 855) underwent upper gastrointestinal endoscopy and routine laboratory tests. The controls were asymptomatic subjects who visited the Health Screening Center for a health status check-up, and their endoscopic findings were normal. Exclusion criteria were H. pylori eradication history; use of antibiotics, proton pump inhibitors, nonsteroidal anti-inflammatory drugs, or anticoagulant drugs; and severe systemic illnesses. Age, sex, alcohol consumption (current or never), smoking habits (current or never), salt intake (high, low-moderate), and family history of GC (first-degree relatives) were recorded. Informed consent was obtained from all included subjects. The Institutional Review Board of the Kyung Hee University Hospital approved the study protocol (KMC IRB 1523–04).
Diagnosis of H. pylori infection
The rapid urease test (or urea breath test) and serum anti-H. pylori immunoglobulin G antibody test were performed. A subject was defined as H. pylori infection-positive if both tests were positive. A subject was defined as H. pylori infection-negative if both tests were negative. Subjects with only one positive test were excluded from this study.
Histologic examination of chronic gastritis
One pathologist histologically evaluated chronic gastritis status in biopsy specimens. AG was graded based on the presence and proportion of glandular loss (mild, moderate, and severe) according to the updated Sydney system [42].
Genotyping of MBL2 exon 1 codons 52, 54 and 57, and of IL-8 -251
Genomic DNA was extracted from peripheral venous blood using a genomic DNA purification method. Polymerase chain reaction (PCR) amplification, restriction fragment length polymorphism (RFLP) analysis, and gel electrophoresis were performed for MBL2 (codons 52, 54, and 57 in exon 1) and IL-8 (−251 promoter region) as described previously [7, 34]. The PCR product involving codon 52 was digested by incubation with MluI at 37 °C for 3 h, resulting in two bands of 204 and 94 bp for the T/T genotype (mutant), three bands of 298, 204, and 94 bp for the A/T genotype (heterozygote), and one band of 298 bp for the A/A genotype (wild type). The PCR product involving codon 54 was digested by BanI at 50 °C for 3 h, resulting in two bands of 195 and 103 bp for the G/G genotype (wild type), three bands of 298, 195, and 103 bp for the G/A genotype (heterozygote), and one band of 298 bp for the A/A genotype (mutant). The PCR product involving codon 57 was digested with MboI at 37 °C for 3 h, resulting in two bands of 190 and 108 bp for the A/A genotype (mutant), three bands of 298, 190, and 108 bp for the G/A genotype (heterozygote), and one band of 298 bp for the G/G genotype (wild type). For genotyping of the IL-8 -251 T > A polymorphism, PCR products were digested with MfeI at 37 °C for 3 h, resulting in two bands of 449 and 92 bp for the A/A genotype (mutant), three bands of 541, 449, and 92 bp for the T/A genotype (heterozygote), and one band of 541 bp for the T/T genotype (wild type).
Measurement of serum MBL levels
MBL is a serum protein produced mainly by hepatocytes, and expressed in immune cells, but not in epithelial cells [43]. Circulatory MBL levels were taken as an indicator of the functional activity of MBL protein. Serum MBL levels were measured after overnight fasting by enzyme-linked immunosorbent assay (ELISA; MBL Oligomer ELISA kit; BioProto Diagnostics, Denmark).
Measurement of IL-8 levels in gastric mucosal tissues
Although measurement of serum IL-8 levels is straightforward, serum IL-8 levels do not reflect the severity of H. pylori-associated gastritis [44]. Therefore, we measured IL-8 levels in gastric mucosal tissues rather than serum IL-8 levels.
Three biopsy specimens were taken from the greater curvature side of the proximal antrum during endoscopic procedures. The specimens were put into a tube with 2.0 mL phosphate-buffered saline (pH 7.4), frozen on dry ice, and stored at −70 °C. Samples were homogenized and centrifuged, and the supernatants were aliquoted. Total protein was measured using the bicinchoninic acid assay (Thermo Scientific, Rockford, IL, USA). Gastric mucosal IL-8 levels were measured by ELISA (R&D Systems Inc., Minneapolis, MN, USA). The mucosal level of IL-8 was expressed as picograms per milligram of gastric biopsy protein.
Analysis of global raw data regarding IL-8 -251 T > A polymorphism and GC risk
The results obtained regarding the association of GC risk with IL-8 -251 T > A genotype was not consistent with previous epidemiological results [18–24]. Therefore, we collected large-scale raw data of GC patients (n = 3217) and controls (n = 3810) from Asian (Korea, Japan, and China), and Caucasian (Poland, Finland, and Portugal) populations [13–23], and analyzed GC risk according to IL-8 -251 T > A genotype.
Statistical analysis
Data are expressed as mean values ± standard deviations or as frequencies and percentages. Chi-squared and Kruskal–Wallis tests were performed to compare clinical parameters between the control and disease groups. Hardy–Weinberg equilibrium for polymorphisms in MBL2 and IL-8 was tested using R version 3.1.0 (R Development Core Team). Biases caused by differences in clinical parameters were adjusted using the chi-squared and Kruskal–Wallis tests. Multiple logistic regression analysis was performed to evaluate the associations of the genetic polymorphisms with susceptibility to H. pylori infection and the risk of gastroduodenal diseases using the SAS statistical software package version 9.4 (SAS Institute Inc.). All clinical parameters with a p value <0.20 in the univariate analysis were included in the full logistic regression model. The odds ratios (ORs) and their 95% confidence intervals (CIs) were used to compare the risks between the control and disease groups. P values <0.05 were considered statistically significant.
Results
Table 1 shows clinical features of the control and disease groups. Age, sex ratio, and alcohol consumption were similar among all groups. Risk factors for GC, such as smoking, high salt intake, and family history of GC, were more frequently observed in the H. pylori-positive GC group than in the control group. The differences were statistically significant.
Table 1.
Basic clinical features of the control and disease groups
| Hp (−) Control (n = 176) |
Hp (+) NAG (n = 108) |
Hp (+) Mild AG (n = 52) |
Hp (+) Severe AG (n = 61) |
DU (n = 175) |
GC (n = 283) |
p | |
|---|---|---|---|---|---|---|---|
| Sex (M:F) | 87:89 | 41:67 | 23:29 | 37:24 | 90:85 | 151:132 | 0.1832a |
| Mean age (years) | 52.5 ± 6.7 | 51.58 ± 6.65 | 59.52 ± 8.30 | 60.89 ± 8.55 | 53.2 ± 12.6 | 55.5 ± 8.8 | 0.1431b |
| Alcohol consumption (%) | 52.27% | 51.85% | 48.46% | 47.54% | 57.71% | 55.48% | 0.1751a |
| Smoking habits (%) | 19.89% | 18.52% | 30.77% | 26.23% | 58.86% | 40.99% | <0.0001a |
| High salt intake (%) | 25.00% | 26.85% | 16.92% | 49.18 | 39.43% | 48.41% | <0.0001a |
| FHx of GC (%) | 10.23% | 15.52% | 13.79% | 16.39% | 10.86% | 20.85% | 0.0043a |
| Hp positivity (%) | 0% | 100% | 100% | 100% | 96.00% | 73.14% | <0.0001a |
a: Chi square test for comparison of percentage (GC vs control)
b: Kruskal-Wallis test for comparison of mean ± SD (GC vs control)
The frequencies of the MBL2 codon 54 and IL-8 -251 polymorphisms in the control group did not deviate significantly from those expected under Hardy–Weinberg equilibrium (p = 1.000 and p = 0.184, respectively). In this study population, MBL exon 1 polymorphic variants were found only in codon 54. There were no variants at codons 52 or 57; only the wild type was observed.
The frequencies of MBL2 codon 54 and IL-8 -251 genotypes in the control and disease groups
The frequencies of MBL2 codon 54 genotypes were similar among the control and disease groups (Table 2). The frequency of IL-8 -251 A allele carriers was higher in the H. pylori-positive severe AG and H. pylori-positive GC groups than in the control group, but the differences did not reach statistical significance (Table 2).
Table 2.
The frequencies of MBL2 codon 54 and IL-8 -251 genotypes in the control and disease groups
| Genotype | Control n = 176 (%) |
Hp (+) NAG n = 108 (%) |
Hp (+) Mild AG n = 52 (%) |
Hp (+) Severe AG n = 61 (%) |
Hp (−) DU n = 7 (%) |
Hp (+) DU n = 168 (%) |
Hp (−) GC n = 76 (%) |
Hp (+) GC n = 207 (%) |
|
|---|---|---|---|---|---|---|---|---|---|
| MBL2 codon 54 | G/G (wild type) | 103 (58.52) | 65 (60.19) | 29 (55.77) | 32 (52.46) | 4 (57.14) | 95 (56.55) | 41 (53.95) | 129 (62.32) |
| G/A(heterozygote) | 63 (35.80) | 37 (34.26) | 22 (43.31) | 27 (44.26) | 3 (42.86) | 69 (41.07) | 29 (32.37) | 67 (32.37) | |
| A/A (mutant) | 10 (5.68) | 6 (5.56) | 1 (1.92) | 2 (3.28) | 0 (0.00) | 4 (2.38) | 6 (5.31) | 11 (5.31) | |
| G/A + A/A (A carrier) | 73 (41.48) | 43 (39.81) | 23 (44.23) | 29 (47.54) | 4 (57.14) | 73 (43.45) | 35 (46.05) | 78 (37.68) | |
| IL-8 -251 | T/T (wild type) | 70 (39.77) | 52 (48.15) | 20 (38.46) | 15 (24.59) | 3 (42.86) | 81 (48.21) | 23 (30.26) | 58 (28.02) |
| T/A (heterozygote) | 89 (50.57) | 49 (45.37) | 27 (51.92) | 37 (60.66) | 3 (42.86) | 73 (43.45) | 46 (60.53) | 122 (58.94) | |
| A/A (mutant) | 17 (9.66) | 7 (6,48) | 5 (9.62) | 9 (16.75) | 1 (14.29) | 14 (8.33) | 7 (9.21) | 27 (13.04) | |
| T/A + A/A (A carrier) | 106 (60.23) | 56 (51.85) | 32 (61.54) | 46 (75.41) | 4 (57.14) | 87 (51.79) | 53 (69.74) | 149 (71.99) | |
The frequencies of MBL2 codon 54 and IL-8 -251 genotypes were not significantly different between the control and disease groups
Association between MBL2 codon 54 G > A polymorphism and the risk of gastroduodenal diseases
We examined the association between the MBL2 codon 54 G > A polymorphism and the risk of gastroduodenal disease using univariate and multivariate logistic regression analysis. We regarded the control group as the reference subject group, and considered G/G (wild type) as the reference genotype. The MBL2 codon 54 G > A polymorphism did not increase susceptibility to H. pylori-positive NAG, mild AG, or severe AG, and also was not associated with the risk of DU and GC (Table 3).
Table 3.
MBL codon 54 G > A polymorphism and the risk of gastroduodenal diseases
| Genotype | Disease group | Unadjusted OR (95% CI) by univariate analysis | p | Adjusted OR (95% CI) by multivariate analysis | p |
|---|---|---|---|---|---|
| G/A (heterozygote) | Hp(+) NAG (n = 49) | 0.93 (0.56–1.55) | 0.7828 | 1.02 (0.59–1.75) | 0.9466 |
| Hp(+) mild AG (n = 27) | 1.24 (0.66–2.34) | 0.5074 | 1.16 (0.60–2.26) | 0.6527 | |
| Hp(+) severe AG (n = 37) | 1.38 (0.76–2.52) | 0.2937 | 1.16 (0.61–2.18) | 0.6498 | |
| Hp(−) DU(n = 3) | 1.23 (0.27–5.66) | 0.7938 | 1.21 (0.26–5.71) | 0.8082 | |
| Hp(+) DU (n = 73) | 1.19 (0.76–1.85) | 0.4450 | 1.24 (0.78–1.99) | 0.3605 | |
| Hp(−) GC(n = 46) | 1.16 (0.65–2.04) | 0.6170 | 1.11 (0.61–2.00) | 0.7356 | |
| Hp(+) GC (n = 122) | 0.85 (0.55–1.31) | 0.4566 | 0.85 (0.54–1.34) | 0.7356 | |
| A/A (mutant) | Hp(+) NAG (n = 7) | 0.95 (0.33–2.74) | 0.9255 | 1.19 (0.39–3.61) | 0.7561 |
| Hp(+) mild AG (n = 5) | 0.36 (0.04–2.89) | 0.3332 | 0.33 (0.04–2.81) | 0.3126 | |
| Hp(+) severe AG (n = 9) | 0.64 (0.13–3.09) | 0.5822 | 0.61 (0.12–3.12) | 0.5560 | |
| Hp(−) DU (n = 1) | 1.25(0.32–5.88) | 0.9521 | 1.24(0.31–5.77) | 0.9434 | |
| Hp(+) DU (n = 14) | 0.43 (0.13–1.43) | 0.1697 | 0.45 (0.13–1.55) | 0.2031 | |
| Hp(−) GC (n = 7) | 1.51 (0.52–4.42) | 0.4543 | 1.93 (0.63–5.89) | 0.2484 | |
| Hp(+) GC (n = 27) | 0.88 (0.36–2.15) | 0.7762 | 1.10 (0.43–2.79) | 0.8496 | |
| G/A + A/A (A carrier) | Hp(+) NAG (n = 56) | 0.93 (0.57–1.52) | 0.7820 | 1.04 (0.62–1.75) | 0.8774 |
| Hp(+) mild AG (n = 32) | 1.12 (0.60–2.09) | 0.7239 | 1.05 (0.55–2.00) | 0.8893 | |
| Hp(+) severe AG (n = 46) | 1.28 (0.71–2.29) | 0.4103 | 1.09 (0.59–2.01) | 0.7929 | |
| Hp(−) DU (n = 4) | 1.06 (0.23–4.87) | 0.9421 | 1.06 (0.23–4.98) | 0.9386 | |
| Hp(+) DU (n = 87) | 1.08 (0.71–1.66) | 0.7110 | 1.13 (0.72–1.78) | 0.5868 | |
| Hp(−) GC (n = 53) | 1.20 (0.70–2.07) | 0.5008 | 1.19 (0.68–2.09) | 0.5480 | |
| Hp(+) GC (n = 149) | 0.85 (0.57–1.29) | 0.4488 | 0.88 (0.57–1.35) | 0.5528 |
The control was regarded as the reference subject group, and the G/G wild type was considered as the reference genotype
Serum MBL levels
Serum levels of MBL were high in carriers of the G/G (wild type) genotype, intermediate in those with the G/A heterozygous genotype, and low in those with the A/A (mutant) genotype in all subjects (n = 855, Fig. 1). The differences between the three genotypes were highly statistically significant (p < 0.0001). However, there were no significant differences in serum MBL levels between the control (139.9 ± 83.2 ng/mL), H. pylori-positive NAG (149.3 ± 81.2 ng/mL), mild AG (146.9 ± 81.8 ng/mL), severe AG (140.2 ± 87.3 ng/mL), DU (143.8 ± 82.5 ng/mL), and GC (149.8 ± 82.6 ng/mL) groups.
Fig. 1.
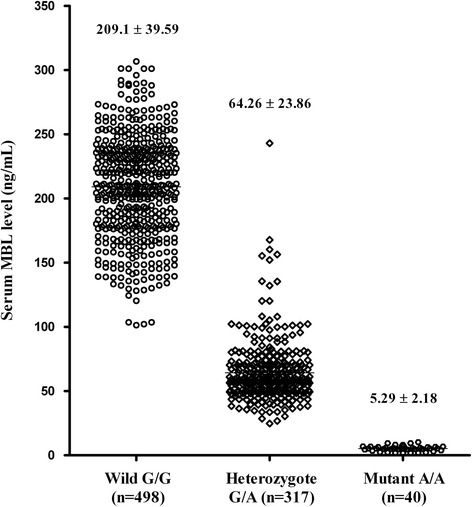
Serum MBL levels in all subjects according to MBL2 codon 54 genotype. Serum MBL levels differed significantly between the three genotypes, as determined by the Kruskal–Wallis test (p < 0.0001)
Association between IL-8 -251 T > A polymorphism and the risk of disease development
Because the IL-8 cytokine response is mainly dependent on the H. pylori-associated inflammatory severity, we sub-classified the H. pylori-positive chronic gastritis group (n = 221) into H. pylori-positive NAG, mild AG, and severe AG. We regarded the control group as the reference subject group, and considered T/T (wild type) as the reference genotype. The IL-8 -251 A allele significantly increased the risk of severe AG and GC, as determined by both univariate and multivariate logistic regression analysis (Table 4).
Table 4.
IL-8 -251 T > A polymorphism and the risk of gastroduodenal diseases
| Genotype | Disease group | Unadjusted OR (95% CI) by univariate analysis | p | Adjusted OR (95% CI) by multivariate analysis | p |
|---|---|---|---|---|---|
| T/A (heterozygote) | Hp(+) NAG (n = 49) | 0.74 (0.45–1.22) | 0.2406 | 0.76 (0.46–1.29) | 0.3018 |
| Hp(+) mild AG (n = 27) | 1.06 (0.55–2.05) | 0.8581 | 1.09 (0.55–2.16) | 0.8052 | |
| Hp(+) severe AG (n = 37) | 1.94 (0.99–3.82) | 0.0549 | 2.06 (1.01–4.21) | 0.0471 | |
| Hp(−) DU(n = 3) | 0.79 (0.15–4.02) | 0.7729 | 0.84 (0.16–4.36) | 0.8342 | |
| Hp(+) DU (n = 73) | 0.71 (0.45–1.11) | 0.1297 | 0.80 (0.50–1.29) | 0.3616 | |
| Hp(−) GC(n = 46) | 1.57 (0.87–2.84) | 0.1326 | 1.66 (0.89–3.07) | 0.1058 | |
| Hp(+) GC (n = 122) | 1.65 (1.06–2.58) | 0.0257 | 1.71 (1.07–2.72) | 0.0252 | |
| A/A (mutant) | Hp(+) NAG (n = 7) | 0.55 (0.21–1.43) | 0.2237 | 0.59 (0.21–1.59) | 0.2952 |
| Hp(+) mild AG (n = 5) | 1.03 (0.34–3.14) | 0.9593 | 1.00 (0.31–3.17) | 0.9941 | |
| Hp(+) severe AG (n = 9) | 2.47 (0.93–6.59) | 0.0710 | 2.42 (0.85–6.84) | 0.0966 | |
| Hp(−) DU (n = 1) | 1.37 (0.13–14.03) | 0.7895 | 1.66 (0.16–17.71) | 0.6743 | |
| Hp(+) DU (n = 14) | 0.71 (0.33–1.55) | 0.3905 | 0.72 (0.32–1.63) | 0.4289 | |
| Hp(−) GC (n = 7) | 1.25 (0.46–3.40) | 0.6577 | 1.19 (0.43–3.34) | 0.7403 | |
| Hp(+) GC (n = 27) | 1.92 (0.95–3.86) | 0.0683 | 1.94 (0.93–4.06) | 0.0744 | |
| T/A + A/A (A carrier) |
Hp(+) NAG (n = 56) | 0.71 (0.44–1.15) | 0.1669 | 0.73 (0.44–1.22) | 0.2282 |
| Hp(+) mild AG (n = 32) | 1.06 (0.56–1.99) | 0.8651 | 1.07 (0.55–2.08) | 0.8336 | |
| Hp(+) severe AG (n = 46) | 2.03 (1.05–3.90) | 0.0351 | 2.12 (1.06–4.25) | 0.0335 | |
| Hp(−) DU (n = 4) | 0.88 (0.19–4.05) | 0.8703 | 0.96 (0.20–4.48) | 0.3066 | |
| Hp(+) DU (n = 87) | 0.71 (0.46–1.09) | 0.1152 | 0.79 (0.50–1.24) | 0.3035 | |
| Hp(−) GC (n = 53) | 1.52 (0.86–2.70) | 0.1524 | 1.58 (0.87–2.87) | 0.1346 | |
| Hp(+) GC (n = 149) | 1.70 (1.11–2.60) | 0.0155 | 1.74 (1.11–2.74) | 0.0161 |
The control was regarded as the reference subject group, and the T/T wild type was considered as the reference genotype
Comparison of IL-8 levels according to disease phenotypes and IL-8 -251 each genotype
IL-8 levels were low in subjects with the T/T (wild type) genotype, intermediate in those with the T/A heterozygous genotype, and high in those with the A/A (mutant) genotype. The differences between the three genotypes were statistically significant (p = 0.0262, Fig. 2).
Fig. 2.
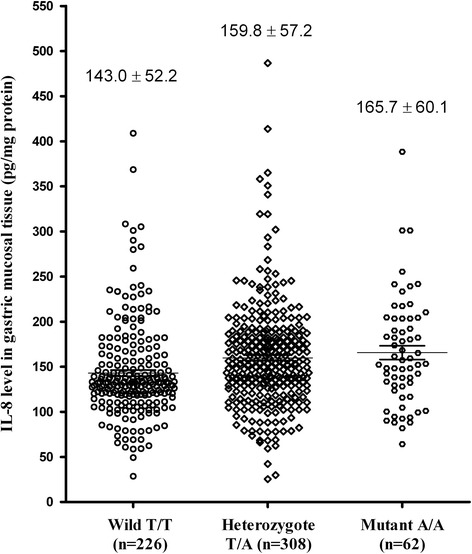
IL-8 levels in all H. pylori-positive subjects according to IL-8 -251 genotype. IL-8 levels differed significantly between the three genotypes, as determined by the Kruskal–Wallis test (p = 0.0262)
IL-8 levels were markedly low in H. pylori-negative control (n = 176, 4.43 ± 3.30 pg/mg protein) and H. pylori-negative subjects regardless of any disease phenotypes (n = 83, 5.02 ± 3.80 pg/mg protein) compared with H. pylori-positive subjects (n = 596, 154.05 ± 56.26 pg/mg protein; p < 0.0001). IL-8 levels were very low in the H. pylori-negative DU and H. pylori-negative GC groups as well as in the control group. Therefore, we regarded the H. pylori-positive NAG group as the reference subject group instead of the control group. We selected five H. pylori-positive disease groups to evaluate the gastric precancerous cascade: NAG, mild AG, severe AG, DU, and GC (Fig. 3). The five disease groups did not show any significant differences in IL-8 levels for the T/T (p = 0.7979), T/A (p = 0.2200), or A/A (p = 0.1000) genotypes, or A allele carriers (p = 0.0550), as analyzed by a multiple group comparison test. However, H. pylori-positive GC A allele carriers (n = 207, 172.3 ± 65.4 pg/mg protein) showed significantly higher IL-8 levels than NAG A allele carriers (n = 108, 148.3 ± 42.9 pg/mg protein, p = 0.0229), as determined by the two-group comparison test (Fig. 3).
Fig. 3.
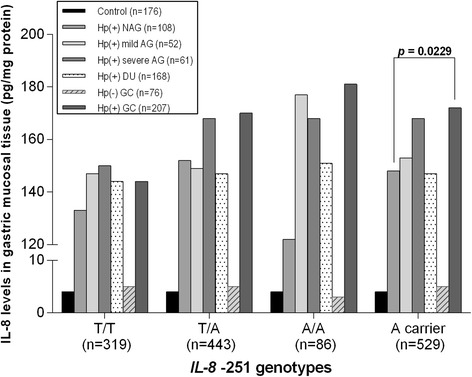
IL-8 levels according to disease phenotypes and IL-8 -251 genotype. The disease groups did not show any significant differences in IL-8 levels between the T/T, T/A, and A/A genotypes or for A allele carriers, as determined by multiple group comparison test. However, H. pylori-positive GC A allele carriers showed significantly higher IL-8 levels than NAG A allele carriers (p = 0.0229), as determined by two group comparison test
Analysis of global results of IL-8 -251 T > A polymorphism and GC risk
The Korean population, including the subjects of this study, showed a significant positive association between the IL-8 -251 T > A polymorphism and GC risk. On the contrary, the Chinese and Caucasian populations showed a negative association. The Japanese population was similar to the Korean population. The combined Korean and Japanese populations showed significantly increased GC risk for the IL-8 -251 T/A and A/A genotypes compared to the T/T genotype, and for A allele carriers compared to non-carriers (Table 5).
Table 5.
The association of IL-8 -251 T > A polymorphism with the risk of GC in different ethnicities
| Ethnicity | TT / total GC (%) TT / total controls (%) OR [95% CI] |
TA / total GC (%) TA / total controls (%) OR [95% CI] |
AA / total GC (%) AA / total controls (%) OR [95% CI] |
A* / total GC (%) A* / total controls (%) OR [95% CI] |
|---|---|---|---|---|
| This study (Korean) |
81/283 (28.62%) 70/176 (39.77%) 0.60 [0.40, 0.90] |
168/283(59.36%) 89/176 (50.57%) 1.43 [0.98, 2.09] |
34/283(12.01%) 17/176(9.66%) 1.27 [0.70, 2.31] |
202/283(71.38%) 106/176 (60.23%) 1.66 [1.11, 2.47] |
| Korean [16, 17] |
180/487(36.96%) 244/528(46.21%) 0.68 [0.53, 0.88] |
241/487(49.49%) 234/528(44.32%) 1.23 [0.96, 1.57] |
66/487(13.55%) 50/528(9.47%) 1.50 [1.02, 2.20] |
307/487(63.04%) 284/528 (53.79%) 1.46 [1.14, 1.88] |
| Japanese [13–15] |
337/789(42.71%) 485/964(50.31%) 0.74 [0.61, 0.89] |
375/789(47.53%) 397/964(41.18%) 1.29 [1.07, 1.56] |
77/789(9.76%) 82/964(8.51%) 1.16 [0.84, 1.62] |
452/789(57.29%) 479/964(49.69%) 1.36 [1.12, 1.64] |
| Chinese [18–20] |
329/926(35.53%) 270/814(33.17%) 1.11 [0.91, 1.35] |
425/926(45.90%) 406/814(49.88%) 0.85 [0.71, 1.03] |
172/926(18.57%) 138/814(16.95%) 1.12 [0.87, 1.43] |
597/926(64.47%) 544/814(66.83%) 0.90 [0.74, 1.10] |
| Caucasian [21–23] |
224/732(30.60%) 381/1328(28.69%) 1.10 [0.90, 1.34] |
365/732(49.86%) 669/1328(50.38%) 0.98 [0.82, 1.17] |
143/732(19.54%) 278/1328(20.93%) 0.92 [0.73, 1.15] |
508/732(69.40%) 947/1328(71.31%) 0.91 [0.75, 1.11] |
| Combined Korean and Japanese |
598/1559(38.36%) 799/1668(47.90%) 0.70 [0.61, 0.81] |
784/1559(50.29%) 720/1668(43.17%) 1.29 [1.12, 1.48] |
177/1559(11.35%) 149/1668(8.93%) 1.29 [1.02, 1.62] |
961/1559(61.64%) 869/1668(52.10%) 1.42 [1.24, 1.64] |
A*: A allele carriers (T/A + A/A)
The combined Korean and Japanese populations showed significant GC risk in IL-8 -251 T/A, A/A, and A allele carriers
Discussion
The innate immune response is the first line of defense against H. pylori infection in the human stomach. TLR and MBL are recognized as important proteins in innate immunity. Several studies have demonstrated that TLR4 and TLR2 polymorphisms are associated with the risk of GC [45–47]. However, some of the associations are controversial, and there are discrepancies between the results for Asian and Western populations [48]. A recent study in the Netherlands found that only the TLR1 polymorphism is associated with the prevalence of H. pylori seropositivity [49]. Further studies are needed in other populations worldwide to confirm these associations.
MBL binds to bacteria, yeasts, and viruses via specific repeated oligosaccharide moieties on the cell surface. MBL activates the complement-lectin pathway, facilitates opsonization and phagocytosis, and induces direct cellular lysis. MBL deficiency or a low serum MBL level has been associated with several infectious and autoimmune diseases, including meningococcal meningitis, pneumonia, arterial thrombosis, systemic lupus erythematosus, and celiac disease [50, 51].
At the time of its discovery, H. pylori was considered an extracellular bacterium that mainly colonized the gastric mucus layer or attached to gastric epithelial cells. However, it has since been demonstrated that H. pylori invades the lamina propria and gastric epithelial cells [52]. Therefore, H. pylori might be a target of phagocytosis by MBL activation. There have been few clinical studies regarding the role of MBL in H. pylori infection. Various microorganisms such as H. pylori, Neisseria meningitidis groups B and C, Nocardia farcinica, and Legionella pneumophila induce MBL activity in vitro [28]. Activated complements are found in the epithelium of patients with H. pylori-associated gastritis [29]. One pediatric study reported that MBL2 mRNA expression in gastric biopsy specimens was higher in H. pylori-positive chronic gastritis than in H. pylori-negative chronic gastritis patients [53]. However, the study had two weaknesses in terms of its ability to reach conclusions regarding the role of MBL2 expression in the development of H. pylori-infected chronic gastritis. The first weakness is the small number of biopsy specimens that were obtained, with only five H. pylori-positive children and four control children included. The second weakness is that they could not find any association between MBL2 genotype and the risk of H. pylori-infected chronic gastritis.
The association between the MBL2 haplotype and the risk of GC has been studied previously [30, 31]. A study conducted in Southern Italy found that the HYP + D haplotype (H/Y promoter region mutation + P untranslated region mutation + codon 52 mutation) may be a genetic marker for H. pylori-positive GC risk [30]. Another study performed in Warsaw, Poland found that the HY + D haplotype (H/Y promoter region mutation + codon 52 mutation) was related to an increased risk of GC compared with the HY+ A haplotype (H/Y mutation + codon 52 wild type) [31]. Therefore, the codon 52 D variant (cysteine > arginine) was specifically related to the risk of GC in two populations. In contrast to the above studies, which reported positive associations, Australian researchers evaluated healthy individuals for H. pylori infection, MBL2 genotype, mannan binding level, and complement 4 level in plasma, and found that MBL deficiency, defined by either genotype or plasma activity, was not associated with higher susceptibility to H. pylori infection [33]. In a Japanese study, they could no significant differences were found in MBL2 genotypes between GC patients and healthy controls [32]. Instead, the investigators found that the MBL2 codon 54 polymorphism was weakly associated with severe AG and advanced GC [32, 54]. In the present study, we first demonstrated that the codon 54 polymorphism did not increase susceptibility to H. pylori infection in a Korean population. Secondly, we did not find any evidence of a role for MBL2 in the development of gastroduodenal diseases. Thirdly, we did not find a higher risk of advanced GC or severe AG compared to early GC or mild AG, respectively, associated with MBL2 genotype.
With regard to interracial differences, the Korean population differs from the European (Italian and Polish) and African populations reported previously. However, the results for the Korean population are very similar to those reported for the Chinese and Japanese populations [35, 38–40]. The frequencies of point mutations in European populations are in between those of the East Asian and African populations.
In the present study, serum levels of MBL, an indicator of the functional activity of MBL, differed significantly according to the genotype. However, serum MBL levels were not significantly different between the control and disease groups, because the frequency of each genotype was similar in these groups.
H. pylori infection stimulates IL-8 gene expression and increases the IL-8 cytokine level in gastric epithelial cells. A significant correlation between a high level of IL-8 in the gastric mucosa and the risk of GC has been reported [13]. Our previous study found that the IL-8 level in gastric mucosal tissues was significantly higher in H. pylori-infected subjects compared with that in H. pylori non-infected subjects, irrespective of their gastroduodenal disease phenotype. After H. pylori eradication, the IL-8 level decreased dramatically, to the same level observed in non-infected subjects [55]. In this study, we confirmed once again that the IL-8 level in gastric mucosal tissues is mainly dependent on H. pylori-positive status.
It has been reported that the IL-8 -251 T > A polymorphism is related to higher levels of IL-8 and to an increased risk of AG, gastric ulcer, and GC [13, 14]. In this study, we also demonstrated that the IL-8 -251 T > A polymorphism increased IL-8 production, and was significantly associated with the risk of GC and severe AG. However, many other epidemiological studies have reported negative associations between the IL-8 -251 polymorphism and GC risk (18–23), and a meta-analysis revealed no overall association (24). In this study, we analyzed large-scale raw data from controls and GC patients from Korean, Japanese, Chinese, and Caucasian (Poland, Finland, and Portugal) populations (13–23). Korean results, including ours, were consistent with Japanese results, but not with Chinese or Caucasian results. The concordance between the Korean and Japanese results might be explained by genetic similarities. In a large study of single nucleotide polymorphism (SNP) maps covering the human genome performed in African Americans, Asians (Japanese-Chinese-Korean), and European Americans (Caucasians) [56], SNP differences in autosomes were only 5.86% between Korean and Japanese populations. Therefore, the Korean population is very similar to the Japanese population with respect to the pattern of SNPs [56].
Conclusions
The MBL2 codon 54 G > A polymorphism does not influence susceptibility to H. pylori infection and does not increase the risk of gastroduodenal diseases. We suggest that a combination of the IL-8 -251 T > A polymorphism and increased IL-8 production in response to H. pylori infection may be a risk factor for severe AG and GC development in a Korean population.
Acknowledgements
We thank Ms. Eunhee Lee for encouragement and scientific discussions.
Funding
The work was not granted.
Availability of data and materials
The datasets of the current study available from the corresponding author on reasonable request.
Authors’ contributions
YWC, CHO, and JWK participated in study design and the conception. YWC wrote the manuscript. JWL and MJP participated in the design of the study and performed the statistical analysis. JJS and CKL are responsible for acquisition of data. JYJ, SHD and HJK carried out quality control of data and algorithms. SSK, BHK and JWK are responsible for critical revision of the work. All authors reviewed and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The Institutional Review Board of the Kyung Hee University Hospital approved this study. Written informed consent was obtained from the all included subjects.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- AG
Atrophic gastritis
- CI
Confidence intervals
- DU
Duodenal ulcer
- ELISA
Enzyme-linked immunosorbent assay
- GC
Gastric cancer
- Helicobacter pylori
H. pylori
- Hp(−)
Helicobacter pylori-negative
- Hp(+)
Helicobacter pylori-positive
- IL-1β
Interleukin 1 beta
- IL-8
Interleukin 8
- MBL
Mannose-binding lectin
- NAG
Non-atrophic gastritis
- OR
Odds ratios
- PCR
Polymerase chain reaction
- RFLP
Restriction fragment length polymorphism
- SNP
Single nucleotide polymorphism
- TLR
Toll-like receptor
Contributor Information
Young Woon Chang, Email: cywgi@chollian.net.
Chi Hyuk Oh, Email: harrison@daum.net.
Jung-Wook Kim, Email: iloveact@hanmail.net.
Jae Won Lee, Email: jael@korea.ac.kr.
Mi Ju Park, Email: wedding0425@hanmail.net.
Jae-Jun Shim, Email: joyshim@khu.ac.kr.
Chang Kyun Lee, Email: cklee92@paran.com.
Jae-Young Jang, Email: jyjang@khu.ac.kr.
Seok Ho Dong, Email: gidrdong@hanmail.net.
Hyo Jong Kim, Email: hjkim@khmc.or.kr.
Sung Soo Kim, Email: sgskim@khu.ac.kr.
Byung-Ho Kim, Email: kimbh@khu.ac.kr.
References
- 1.McColl KE. Clinical practice. Helicobacter pylori infection. N Engl J Med. 2010;362(17):1597–1604. doi: 10.1056/NEJMcp1001110. [DOI] [PubMed] [Google Scholar]
- 2.Lim SH, Kwon JW, Kim N, Kim GH, Kang JM, Park MJ, et al. Prevalence and risk factors of Helicobacter pylori infection in Korea: nationwide multicenter study over 13 years. BMC Gastroenterol. 2013;13:104–13. [DOI] [PMC free article] [PubMed]
- 3.Atherton JC. The clinical relevance of strain types of Helicobacter pylori. Gut. 1997;40(6):701–703. doi: 10.1136/gut.40.6.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miehlke S, Kibler K, Kim JG, Figura N, Small SM, Graham DY, et al. Allelic variation in the cagA gene of Helicobacter pylori obtained from Korea compared to the United States. Am J Gastroenterol. 1996;91(7):1322–5. [PubMed]
- 5.Park SK, Kim GH, Jeong EJ, Bae YM, Heo J, Chu HJ, et al. Clinical relevance between the cagA, vacA, and iceA status of Helicobacter pylori and benign gastroduodenal diseases. Korean J Gastroenterol. 2002;40(1):23–31.
- 6.Chang YW, Han YS, Lee DK, Kim HJ, Lim HS, Moon JS, et al. Role of Helicobacter pylori infection among offspring or siblings of gastric cancer patients. Int J Cancer. 2002;101(5):469–74. [DOI] [PubMed]
- 7.Ferrero RL. Innate immune recognition of the extracellular mucosal pathogen, Helicobacter pylori. Mol Immunol. 2005;42(8):879–885. doi: 10.1016/j.molimm.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Rad R, Dossumbekova A, Neu B, Lang R, Bauer S, Saur D, et al. Cytokine gene polymorphisms influence mucosal cytokine expression, gastric inflammation, and host specific colonisation during Helicobacter pylori infection. Gut. 2004;53(8):1082–9. [DOI] [PMC free article] [PubMed]
- 9.Yamaoka Y, Kita M, Kodama T, Sawai N, Kashima K, Imanishi J. Induction of various cytokines and development of severe mucosal inflammation by cagA gene positive Helicobacter pylori strains. Gut. 1997;41(4):442–451. doi: 10.1136/gut.41.4.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hwang IR, Kodama T, Kikuchi S, Sakai K, Peterson LE, Graham DY, et al. Effect of interleukin 1 polymorphisms on gastric mucosal interleukin 1beta production in Helicobacter pylori infection. Gastroenterology. 2002;123(6):1793–803. [DOI] [PubMed]
- 11.Bodger K, Wyatt JI, Heatley RV. Gastric mucosal secretion of interleukin-10: relations to histopathology, Helicobacter pylori status, and tumour necrosis factor-alpha secretion. Gut. 1997;40(6):739–744. doi: 10.1136/gut.40.6.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamaoka Y, Kodama T, Kita M, Imanishi J, Kashima K, Graham DY. Relation between cytokines and Helicobacter pylori in gastric cancer. Helicobacter. 2001;6(2):116–124. doi: 10.1046/j.1523-5378.2001.00017.x. [DOI] [PubMed] [Google Scholar]
- 13.Taguchi A, Ohmiya N, Shirai K, Mabuchi N, Itoh A, Hirooka Y, et al. Interleukin-8 promoter polymorphism increases the risk of atrophic gastritis and gastric cancer in Japan. Cancer Epidemiol Biomark Prev. 2005;14(11):2487–93. [DOI] [PubMed]
- 14.Ohyauchi M, Imatani A, Yonechi M, Asano N, Miura A, Iijima K, et al. The polymorphism interleukin 8–251 A/T influences the susceptibility of Helicobacter pylori related gastric diseases in the Japanese population. Gut. 2005;54(3):330–5. [DOI] [PMC free article] [PubMed]
- 15.Shirai K, Ohmiya N, Taguchi A, Mabuchi N, Yatsuya H, Itoh A, et al. Interleukin-8 gene polymorphism associated with susceptibility to non-cardia gastric carcinoma with microsatellite instability. J Gastroenterol Hepatol. 2006;21:1129–1135. doi: 10.1111/j.1440-1746.2006.04443.x. [DOI] [PubMed] [Google Scholar]
- 16.Ye BD, Kim SG, Park JH, Kim JS, Jung HC, Song IS. The interleukin-8 -251 a allele is associated with increased risk of noncardia gastric adenocarcinoma in Helicobacter pylori-infected Koreans. J Clin Gastroenterol. 2009;43:233–239. doi: 10.1097/MCG.0b013e3181646701. [DOI] [PubMed] [Google Scholar]
- 17.Kang JM, Kim N, Lee DH, Park JH, Lee MK, Kim JS, et al. The effects of genetic polymorphisms of IL-6, IL-8, and IL-10 on Helicobacter pylori induced gastroduodenal diseases in Korea. J Clin Gastroenterol. 2009;43:420–428. doi: 10.1097/MCG.0b013e318178d1d3. [DOI] [PubMed] [Google Scholar]
- 18.Lee WP, Tai DI, Lan KH, Li AF, Hsu HC, Lin EJ, et al. The −251T allele of the interleukin-8 promoter is associated with increased risk of gastric carcinoma featuring diffuse-type histopathology in Chinese population. Clin Cancer Res. 2005;11:6431–6441. doi: 10.1158/1078-0432.CCR-05-0942. [DOI] [PubMed] [Google Scholar]
- 19.Lu W, Pan K, Zhang L, Lin D, Miao X, You W. Genetic polymorphisms of interleukin (IL)-1B, IL-1RN, IL-8, IL-10 and tumor necrosis factor a and risk of gastric cancer in a Chinese population. Carcinogenesis. 2005;26:631–636. doi: 10.1093/carcin/bgh349. [DOI] [PubMed] [Google Scholar]
- 20.Zeng ZR, Zhou SZ, Liao SY, Chen B, Li CJ, Chen MH, et al. Correlation of polymorphism of interleukin 8 gene -251 locus and gastric cancer in high and low prevalence regions in China. J Sun Yat-Sen Univ (Med Sci) 2005;26:537–540.
- 21.Savage SA, Hou L, Lissowska J, Chow WH, Zatonski W, Chanock SJ, et al. Interleukin-8 polymorphisms are not associated with gastric cancer risk in a polish population. Cancer Epidemiol Biomark Prev. 2006;15:589–591. doi: 10.1158/1055-9965.EPI-05-0887. [DOI] [PubMed] [Google Scholar]
- 22.Kamangar F, Abnet CC, Hutchinson AA, Newschaffer CJ, Helzlsouer K, Shugart YY, et al. Polymorphisms in inflammation-related genes and risk of gastric cancer (Finland) Cancer Causes Control. 2006;17:117–125. doi: 10.1007/s10552-005-0439-7. [DOI] [PubMed] [Google Scholar]
- 23.Canedo P, Castanheira-Vale AJ, Lunet N, Pereira F, Figueiredo C, Gioia-Patricola L, et al. The interleukin-8 -251 T/a polymorphism is not associated with risk for gastric carcinoma development in a Portuguese population. Eur J Cancer Prev. 2008;17:28–32. doi: 10.1097/CEJ.0b013e32809b4d0f. [DOI] [PubMed] [Google Scholar]
- 24.Liu L, Zhuang W, Wang C, Chen Z, Wu XT, Zhou Y. Interleukin-8 -251 a/T gene polymorphism and gastric cancer susceptibility: a meta-analysis of epidemiological studies. Cytokine. 2010;50(3):328–334. doi: 10.1016/j.cyto.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Kutikhin AG. Association of polymorphisms in TLR genes and in genes of the toll-like receptor signaling pathway with cancer risk. Hum Immunol. 2011;72(11):1095–1116. doi: 10.1016/j.humimm.2011.07.307. [DOI] [PubMed] [Google Scholar]
- 26.Jack DL, Klein NJ, Turner MW. Mannose-binding lectin: targeting the microbial world for complement attack and opsonophagocytosis. Immunol Rev. 2001;180:86–99. doi: 10.1034/j.1600-065X.2001.1800108.x. [DOI] [PubMed] [Google Scholar]
- 27.Kuhlman M, Joiner K, Ezekowitz RA. The human mannose-binding protein functions as an opsonin. J Exp Med. 1989;169(5):1733–1745. doi: 10.1084/jem.169.5.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuipers S, Aerts PC, van Dijk H. Differential microorganism-induced mannose-binding lectin activation. FEMS Immunol Med Microbiol. 2003;36(1–2):33–39. doi: 10.1016/S0928-8244(03)00032-4. [DOI] [PubMed] [Google Scholar]
- 29.Berstad AE, Brandtzaeg P, Stave R, Halstensen TS. Epithelium related deposition of activated complement in Helicobacter pylori associated gastritis. Gut. 1997;40(2):196–203. doi: 10.1136/gut.40.2.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scudiero O, Nardone G, Omodei D, Tatangelo F, Vitale DF, Salvatore F, et al. A mannose-binding lectin-defective haplotype is a risk factor for gastric cancer. Clin Chem. 2006;52(8):1625–7. [DOI] [PubMed]
- 31.Baccarelli A, Hou L, Chen J, Lissowska J, El-Omar EM, Grillo P, et al. Mannose-binding lectin-2 genetic variation and stomach cancer risk. Int J Cancer. 2006;119(8):1970–5. [DOI] [PubMed]
- 32.Wang FY, Tahara T, Arisawa T, Shibata T, Yamashita H, Nakamura M, et al. Mannan-binding lectin (MBL) polymorphism and gastric cancer risk in Japanese population. Dig Dis Sci. 2008;53(11):2904–8. [DOI] [PubMed]
- 33.Worthley DL, Mullighan CG, Dean MM, Gordon DL, Phillips P, Heatley S, et al. Mannose-binding lectin deficiency does not increase the prevalence of Helicobacter pylori seropositivity. Eur J Gastroenterol Hepatol. 2007;19(2):147–52. [DOI] [PubMed]
- 34.Madsen HO, Garred P, Thiel S, Kurtzhals JA, Lamm LU, Ryder LP, et al. Interplay between promoter and structural gene variants control basal serum level of mannan-binding protein. J Immunol. 1995;155(6):3013–20. [PubMed]
- 35.Madsen HO, Satz ML, Hogh B, Svejgaard A, Garred P. Different molecular events result in low protein levels of mannan-binding lectin in populations from southeast Africa and South America. J Immunol. 1998;161(6):3169–3175. [PubMed] [Google Scholar]
- 36.Madsen HO, Garred P, Kurtzhals JA, Lamm LU, Ryder LP, Thiel S, et al. A new frequent allele is the missing link in the structural polymorphism of the human mannan-binding protein. Immunogenetics. 1994;40(1):37–44. [DOI] [PubMed]
- 37.Sumiya M, Super M, Tabona P, Levinsky RJ, Arai T, Turner MW, et al. Molecular basis of opsonic defect in immunodeficient children. Lancet. 1991;337(8757):1569–70. [DOI] [PubMed]
- 38.Lipscombe RJ, Sumiya M, Hill AV, Lau YL, Levinsky RJ, Summerfield JA, et al. High frequencies in African and non-African populations of independent mutations in the mannose binding protein gene. Hum Mol Genet. 1992;1(9):709–15. [DOI] [PubMed]
- 39.Shi H, Wang F, Jin L, Liu M, Hong W, Du Q, et al. Genotype polymorphism and its implications of mannose-binding protein allele in 5 Chinese nationalities. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2001;18(3):202–5. [PubMed]
- 40.Gomi K, Tokue Y, Kobayashi T, Takahashi H, Watanabe A, Fujita T, et al. Mannose-binding lectin gene polymorphism is a modulating factor in repeated respiratory infections. Chest. 2004;126(1):95–9. [DOI] [PubMed]
- 41.Lee SG, Yum JS, Moon HM, Kim HJ, Yang YJ, Kim HL, et al. Analysis of mannose-binding lectin2 (MBL2) genotype and the serum protein levels in the Korean population. Mol Immunol. 2005;42(8):969–77. [DOI] [PubMed]
- 42.Rugge M, Genta RM. Staging and grading of chronic gastritis. Hum Pathol. 2005;36(3):228–233. doi: 10.1016/j.humpath.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 43.Boniotto M, Radillo O, Braida L, Pirulli D, Città A, Not T, et al. Detection of MBL-2 gene expression in intestinal biopsies of celiac patients by in situ reverse transcription polymerase chain reaction. Eur J Histochem. 2003;47:177–80. [DOI] [PubMed]
- 44.Bayraktaroğlu T, Aras AS, Aydemir S, Davutoğlu C, Ustündağ Y, Atmaca H, et al. Serum levels of tumor necrosis factor-alpha, interleukin-6 and interleukin-8 are not increased in dyspeptic patients with Helicobacter pylori-associated gastritis. Mediat Inflamm. 2004;13:25–8. [DOI] [PMC free article] [PubMed]
- 45.Hold GL, Rabkin CS, Chow WH, et al. A functional polymorphism of toll-like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology. 2007;132(3):905–912. doi: 10.1053/j.gastro.2006.12.026. [DOI] [PubMed] [Google Scholar]
- 46.Santini D, Angeletti S, Ruzzo A, et al. Toll-like receptor 4 Asp299Gly and Thr399Ile polymorphisms in gastric cancer of intestinal and diffuse histotypes. Clin Exp Immunol. 2008;154(3):360–364. doi: 10.1111/j.1365-2249.2008.03776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tahara T, Arisawa T, Wang F, et al. Toll-like receptor 2–196 to 174del polymorphism influences the susceptibility of Japanese people to gastric cancer. Cancer Sci. 2007;98(11):1790–1794. doi: 10.1111/j.1349-7006.2007.00590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castaño-Rodríguez N, Kaakoush NO, Goh KL, Fock KM, Mitchell HM. The role of TLR2, TLR4 and CD14 genetic polymorphisms in gastric carcinogenesis: a case-control study and meta-analysis. PLoS One. 2013;8(4) doi: 10.1371/journal.pone.0060327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mayerle J, den Hoed CM, Schurmann C, Stolk L, Homuth G, Peters MJ, et al. Identification of genetic loci associated with Helicobacter pylori serologic status. JAMA. 2013;309(18):1912–20. [DOI] [PubMed]
- 50.Turner MW. The role of mannose-binding lectin in health and disease. Mol Immunol. 2003;40(7):423–429. doi: 10.1016/S0161-5890(03)00155-X. [DOI] [PubMed] [Google Scholar]
- 51.Øhlenschlaeger T, Garred P, Madsen HO, Jacobsen S. Mannose-binding lectin variant alleles and the risk of arterial thrombosis in systemic lupus erythematosus. N Engl J Med. 2004;351(3):260–267. doi: 10.1056/NEJMoa033122. [DOI] [PubMed] [Google Scholar]
- 52.Ko GH, Kang SM, Kim YK, Lee JH, Park CK, Youn HS, et al. Invasiveness of Helicobacter pylori into human gastric mucosa. Helicobacter. 1999;4(2):77–81. [DOI] [PubMed]
- 53.Bak-Romaniszyn L, Cedzyński M, Szemraj J, St Swierzko A, Zeman K, Kałuzyński A, et al. Mannan-binding lectin in children with chronic gastritis. Scand J Immunol. 2006;63(2):131–5. [DOI] [PubMed]
- 54.Tahara T, Shibata T, Wang FY, Nakamura M, Yamashita H, Yoshioka D, et al. Mannan-binding lectin B allele is associated with a risk of developing more severe gastric mucosal atrophy in Helicobacter pylori-infected Japanese patients. Eur J Gastroenterol Hepatol. 2009;21(7):781–6. [DOI] [PubMed]
- 55.Chang YW, Oh HC, Jang JY, Hwangbo Y, Lee JW, Lee HJ, et al. IL-1beta and IL-8, matrix metalloproteinase 3, and pepsinogen secretion before and after H. pylori eradication in gastroduodenal phenotypes. Scand J Gastroenterol. 2008;43(10):1184–93. [DOI] [PubMed]
- 56.Miller RD, Phillips MS, Jo I, Donaldson MA, Studebaker JF, Addleman N, et al. The SNP consortium allele frequency project. High-density single-nucleotide polymorphism maps of the human genome. Genomics. 2005;86:117–126. doi: 10.1016/j.ygeno.2005.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets of the current study available from the corresponding author on reasonable request.