

Abstract
The regulatory field for digital pathology (DP) has advanced significantly. A major milestone was accomplished when the FDA allowed the first vendor to market their device for primary diagnostic use in the USA and published in the classification order that this device, and substantially equivalent devices of this generic type, should be classified into class II instead of class III as previously proposed. The Digital Pathology Association (DPA) regulatory task force had a major role in the accomplishment of getting the application request for Whole Slide Imaging (WSI) Systems recommended for a de novo. This article reviews the past and emerging regulatory environment of WSI for clinical use in the USA. A WSI system with integrated subsystems is defined in the context of medical device regulations. The FDA technical performance assessment guideline is also discussed as well as parameters involved in analytical testing and clinical studies to demonstrate that WSI devices are safe and effective for clinical use.
Keywords: De novo, digital, digital pathology, Digital Pathology Association, Food and Drug Administration, premarket approval, regulation, whole slide imaging
Introduction
Digital pathology (DP) has been employed for many decades. However, it is in this last decade that we have witnessed significant technology advances in the evolution of whole slide imaging (WSI) devices. Many applications unique to DP have emerged that can be revolutionary for medicine.[1] However, adoption of DP has been slow, especially in the USA. DP, specifically WSI, for primary diagnostic use in clinical practice was awaiting the entrance of devices to be cleared by authorities of the United States Food and Drug Administration (FDA). Many have perceived the regulatory field as being one of the key barriers for DP adoption.[2,3]
Recently, the regulatory field for DP has advanced significantly when it was recommended that manufacturers of WSI devices for primary diagnosis in surgical pathology submit their applications to the FDA through their de novo process.[4] This advance was the result of collegial discussions between the Digital Pathology Association (DPA), a nonprofit organization focusing on advancing the field of DP, and the FDA Center for devices and radiological health. A major milestone was accomplished when the FDA allowed Philips IntelliSite Pathology Solutions to market their device for primary diagnostic use of surgical pathology slides prepared from biopsied tissue as well as excised tissue in the USA, and published in their classification order that this device, and substantially equivalent devices of this generic type, should be classified as a class II device. The aim of this article is to review the emerging and future regulatory environment of WSI for clinical use in the United States.
Medical Device Regulations
If a product is labeled, promoted, and/or used in the USA in a manner that meets specific definitions according the Federal Food, Drug, and Cosmetic (FD and C) Act (Title 21 Code of Federal Regulations part 201,[h]) it will be regulated by the FDA as a medical device and is accordingly subject to premarketing and postmarketing regulatory controls. Such a medical device and its intended use will drive the regulatory pathway. When a device is not promoted or sold but rather designed, developed, and validated for its specific intended use, it is not regulated by the FDA. An example of a nonregulated medical device is a laboratory developed test (LDT) or a particular component (e.g., centrifuge) within an entire laboratory. As soon as an LDT or component device undergoes modification to be used for diagnostic purposes, promoted, and sold, it will become regulated.
Table 1 summarizes the risk-based medical device regulatory classification system established by federal law in the United States (FD and C Act, Section 513).[5] The regulatory control increases as device class increases. The conventional light microscope is classified as a Class I (low-risk) device. Class III devices, on the other hand, are subject to the most stringent level of control, where vendors are required to provide assurance of the device's safety and effectiveness. A medical device, such as a WSI scanner, that does not have a type marketed before the Medical Device Amendments of 1976 is automatically classified into Class III. The FDA also classifies medical devices into Class III if they are intended to be used to (a) support/sustain human life, (b) prevent impairment of human health, (c) that may present a potential unreasonable risk of illness/injury for which general and special controls are insufficient to provide reasonable assurance of the device's safety and effectiveness, or (d) for which there is insufficient information to make such a determination.
Table 1.
Food and Drug Administration medical device regulatory classification system
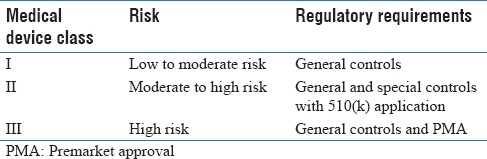
General controls, which apply to all medical devices unless exempted, include provisions that relate to device registration and listing, premarket notification, good manufacturing practices, and so on. Special controls, which are required for Class II devices, are typically device-specific and may include selected data sets used for testing, performance standards, clinical studies to ascertain intra-/inter-reader variable performance, human factors studies, user training, special labeling requirements, guidelines, and a plan to manage product design changes (postmarket control). Class III devices are also subject to approval of a premarket approval (PMA) application. PMA is the FDA process of scientific and regulatory review to evaluate the safety and effectiveness of Class III medical devices. PMA is the most stringent type of device marketing application required by the FDA. A PMA document requires valid clinical information, good scientific analysis, and well-organized writing. The technical section typically contains data derived from clinical investigations and if necessary nonclinical laboratory studies. Usually, it takes 180 days for the FDA to review the PMA and make a determination. An approved PMA grants the applicant (e.g., device manufacturer) permission to market their device for its intended use(s). A Class III device that fails to meet PMA requirements is considered to be adulterated and cannot be marketed.
De novo classification
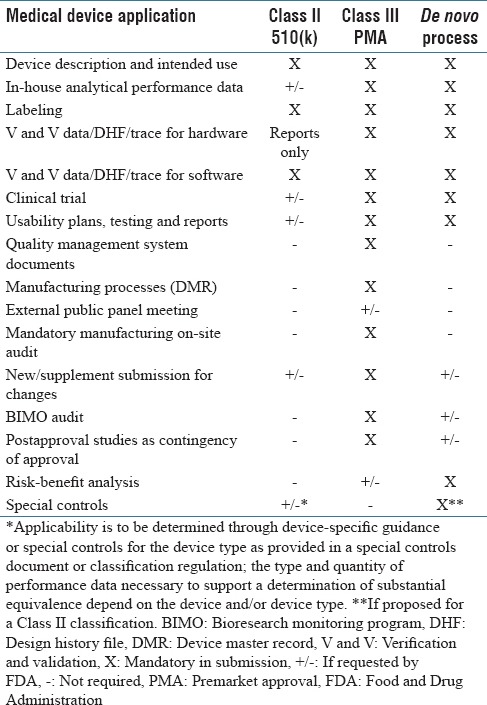
According to the FDA Modernization Act of 1997, certain medical devices that are found to be not substantially equivalent (NSE) to a cleared Class I, II, or III (not requiring PMA) device, may be eligible for the alternate de novo process as a Class I or Class II device. Under this new pathway, if there is no legally marketed predicate device manufacturers may submit a direct de novo without a preceding 510(k) and NSE. Typical content provided in a de novo submission includes the recommended Class (I or II) of the device, indications for use, description of functions, technological characteristics, components, proposed special controls, supporting performance and clinical testing data, summary of benefits, known and potential risks, and mitigation for each risk [Table 2].[6] If de novo is granted for a specific device it may then be “downgraded,” serve as a predicate device for future 510(k) submissions, and can be immediately marketed. If the de novo is declined, however, the medical device must remain in Class III and may thus not be marketed.
Table 2.
Overview of dossier contents for Food and Drug Administration medical device applications
Whole Slide Imaging Regulatory Background
Following a meeting (October 2009) with a public advisory panel that included lively discussion and presentations,[7] the FDA publicly announced that it considered WSI devices would fall in the highest risk classification, that is, a Class III device requiring a PMA regulatory path.[8] However, the FDA had never formally classified WSI devices. Until recently, the FDA had not yet cleared or approved WSI for routine surgical pathology diagnosis to replace conventional light microscopy, while several WSI systems had been cleared for limited use as Class II devices, for the quantification of immunohistochemical stains (e.g. ER, PR, HER2) using image algorithms,[9] and only after the primary diagnosis has been made using glass slides and a microscope. Indeed, the aforementioned news slowed efforts in the United States to employ WSI for primary diagnosis in pathology laboratories. During this time, a myriad of nonclinical applications for WSI including education and research flourished. In the interim, some vendors devoted their efforts to obtaining regulatory approval for their WSI devices by the Conformité Européenne (European Conformity; CE marking) as well as in Canada.[9] The successful use of WSI for routine diagnosis in general surgical pathology outside the United States provided evidence that this technology was safe and effective. In addition, several studies were published indicating that the risk of this technology was lower than originally perceived.[10,11,12] Since then, following constructive discussions with the FDA, the DPA suggested that DP manufacturers interested in marketing their WSI devices for primary diagnosis in the United States submit de novo applications rather than more burdensome PMAs to the FDA.[13]
Digital Pathology Association Regulatory Task Force
The DPA is a nonprofit organization focusing on advancing the field of DP. The DPA has a Regulatory Task Force that consists of pathologists and several industry members.[14] Their mission is to bring clarity to the DP regulatory process. In prior years, the main focus of this group centered on education and bringing awareness of regulatory issues to industry, intended end users, and regulatory authorities. This task force has had constant and collegial discussions with the FDA about issues such as the benefits and risks of WSI, industry concerns, and the regulatory path forward for DP systems. Communication also ensued regarding clinical study design and standardization for nonclinical/analytical performance and specifications. Over the years, with enhanced understanding of the characterization of WSI and its performance, the FDA was better positioned to offer the DPA clear guidance on classification and associated special controls. This has ultimately led to the FDA to recommend that industry follows the de novo regulatory pathway when pursuing approval of WSI for primary diagnosis.
Technical Performance Assessment Guideline
The FDA released (April 20, 2016) a guideline that contained recommendations regarding the technical performance assessment (TPA) data that should be submitted for regulatory evaluation of a digital WSI system.[15] This document lists the components of a WSI system and describes specifications, configurations and user interaction for each component including the slide feeder, light source, optics, mechanical scanner movement, sensor, image processing software, scanning methods, image file format, image review manipulation software, computer hardware, and display. It also incorporates quality control measures, system-level tests, and items that a usability validation test report should include in this study. The FDA has been commended for producing this document,[16,17] which not only recognizes WSI technology but also assures that all manufacturers now follow the same standards.[18]
Regulatory Device Description
A whole slide image refers to the digitized slide that represents a high-resolution replica of the original scanned glass slide. This image can be manipulated with software to mimic microscopic review (i.e., virtual microscopy). WSI refers largely to the acquisition process of creating a whole slide image on a scanner. There are several different manufacturers with varying designs of WSI devices. These devices have various magnifications, scanning methodologies, hardware, and software employed to convert a glass slide into a digital whole slide image. Given that each system has its own design inputs and output, there have been concerns related to color reproducibility, whole slide tissue coverage, stitching, and image features.[19,20]
For regulatory purposes, a WSI system is defined as consisting of two integrated subsystems,[15] the (a) image acquisition subsystem (whole slide scanner) that converts the content of a glass slide into a digital image file, and (b) a workstation environment, including the display, for viewing digital images [Figure 1]. Vendors are required to submit their manufactured device to the FDA as one system that encompasses the entire pixel pathway. The technical and clinical performance of these combined subsystems in the imaging chain needs to be validated as a whole. Decoupling the two subsystems and coupling them to any other subsystem does not ensure the safety and effectiveness for their intended use. This combined setup is further referred to as a “closed” system. Once such a system is released and cleared, this particular version of the system including all of its components and software configurations for its particular intended use, become a “locked down” device.
Figure 1.
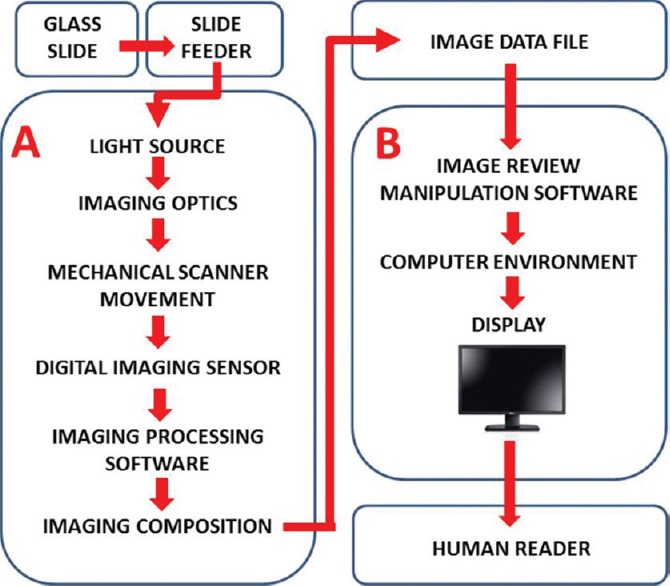
Overview of a digital pathology system. A digital pathology system is composed of two subsystems: (A) Image acquisition and (B) workstation environment. The arrows depict the pixel data pathway[15]
Clinical Component
When submitting an application to the FDA manufacturers of a WSI system need to perform testing that includes both analytical testing and a clinical study to demonstrate that their device is safe and effective for its intended clinical use. Requirements for validation studies and performance measures were outlined in talks from members of the FDA's Office of In vitro Diagnostics and Radiological Health at prior DPA Pathology Visions meetings.
Analytical testing
The objective of an analytical study conducted by the manufacturer is to show that despite multiple sources of variability a WSI system produces digital images accurately and reliably for interpretation in the hands of intended users. Studies accordingly need to demonstrate precision, instrument-to-instrument reproducibility and reader-to-reader reproducibility using morphological features critical to diagnosis or a differential diagnosis. For system precision and reproducibility, intra- and inter-system and intersite evaluation by multiple pathologist readers are required. For diagnostic features, these should be selected from ≥3 different organ systems and at a scanning magnification that is consistent with their identification.
Clinical study
One of the concerns when using WSI devices is that it may create suboptimal images that could cause a misdiagnosis. The intent of a clinical study is to, therefore, show that making a primary diagnosis with a WSI system by its intended users is no less accurate and reliable (i.e., noninferior) to optical microscopy using glass slides and a conventional light microscope. Manufacturers typically use a presubmission to solicit feedback from the FDA about their device and clinical trial design. Presubmissions often become an iterative process between the FDA and the manufacturer with several rounds of communication on a given topic. This process was central to the development of the current proposed clinical study design. The efforts of the first DP system manufacturers who engaged the FDA in this regard are commended for their pioneering effort. For a clinical study to validate the use of WSI for primary diagnosis different pathology sites (n = 4), each with its own WSI device and at least four readers should be recruited. The pathology readers for such a study should represent the intended use population, representing both general pathologists and subspecialties with varying years of experience. The study population should also be sufficiently empowered (n = ~2000 cases) to show that WSI is noninferior to the microscope for different organs and diseases and possibly different stains (e.g., H&E, special stains, immunohistochemistry). There should be a mix of easy and difficult cases that contain both single and multiple slides. Ideally, the reading method should be randomized where after a feasible washout period pathologists have to make a diagnosis for each case using digital and glass slide modalities. The primary end point is the error rate of comparing the readings to the “ground truth” diagnosis, which could be determined either by an expert panel or original sign-out report. Vendors have found such clinical studies to be challenging particularly with respect to case selection, defining major discordances, and utilizing unified diagnostic terminology. The use of standardized synoptic checklists and predetermined discordance tables has been recommended by some authors to help gather consistent, comprehensive diagnostic data for WSI validation studies.[21] So far, one vendor (Philips) completed a multicenter clinical validation study of their DP solution that was submitted to the FDA for clinical use in the United States.[22,23] This large multicenter, retrospective clinical study demonstrated that diagnosing 2000 surgical pathology cases (15,925 readings) with the Philips IntelliSite Pathology Solution was noninferior to optical microscopy.[24] The clinical trial yielded a digital to optical interpretation rate difference of 0.4% with a 95% confidence interval (−0.3–1.0) indicating noninferiority for digital versus optical reads. Central adjudication was carried out by three independent pathologists.
Conclusions
The regulatory path for DP in the United States has crystalized in recent years. The framework for manufacturers to follow to demonstrate technical, and to a lesser extent analytical and clinical performance, has been established. A similar document to the TPA guideline issued by the FDA that assists manufacturers when performing clinical studies would be beneficial. Now that the first DP manufacturer has obtained FDA clearance the FDA has consequently provided more clarity and guidance on the classification order for these devices and special controls required. A major step forward toward adoption of DP occurred with the FDA's classification of WSI for primary diagnosis of surgical pathology slides into a class II device. Following this first de novo classification, subsequent manufacturers can apply for a 510(k) clearance. This first authorized de novo WSI predicate device will pave the path to show substantial equivalence for other devices [Figure 2] and has provided clarity on the type of special controls required. WSI devices will likely change as technology evolves. This will undoubtedly encourage more manufacturers to enter the DP field. The challenge from a regulatory perspective will be how best to evaluate the safety and effectiveness of these newer devices. One suggestion is to develop a medical device development tool such as a phantom, validated test, or registration tool that can aid with device development and regulatory evaluation instead of executing expensive assessments over and over again. The sale of legally available WSI devices for clinical use will open doors for these DP platforms to be deployed within larger enterprise image ecosystems that facilitate next generation applications such as image analysis, streaming analytics, and computational pathology. The next challenge awaiting the DP community will be to resolve regulatory issues surrounding the use of such open systems and how they facilitate image algorithms that employ deep learning.
Figure 2.
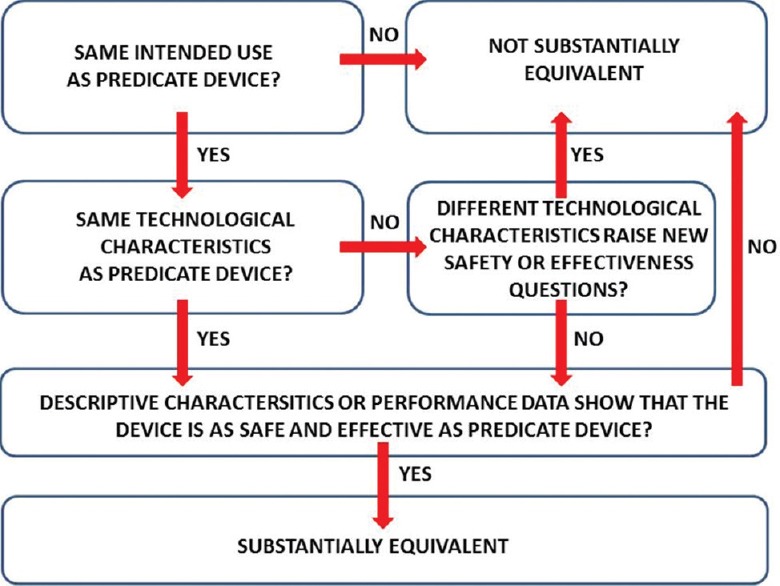
Algorithm for evaluating substantial equivalence in 510(k) premarket notifications
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
Esther Abels and Liron Pantanowitz are members of the DPA Regulatory Task Force. Esther Abels is also the Director of Regulatory, Clinical and Medical Affairs for Philips Digital Pathology Solutions. Liron Pantanowitz is a consultant for Hamamatsu.
Footnotes
Available FREE in open access from: http://www.jpathinformatics.org/text.asp?2017/8/1/23/206226
REFERENCES
- 1.Pantanowitz L, Parwani AV. Digital Pathology. Chicago, USA: ASCP Press; 2017. [Google Scholar]
- 2.Titus K. Regulators scanning the digital scanners. CAP Today. 2012;26:56–62. [Google Scholar]
- 3.Parwani AV, Hassell L, Glassy E, Pantanowitz L. Regulatory barriers surrounding the use of whole slide imaging in the United States of America. J Pathol Inform. 2014;5:38. doi: 10.4103/2153-3539.143325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. [Last accessed 2017 Feb 01];DPA Recommends whole Slide Imaging Manufacturers Submit De novo Applications to the FDA for Primary Diagnosis in the United States. Available from: http://www.prweb.com/releases/2016/01/prweb13163307.htm . [Google Scholar]
- 5. [Last accessed on 2017 Feb 01];FDA Overview of Device Regulation. Available from: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Overview/default.htm . [Google Scholar]
- 6. [Last accessed 2017 Feb 05];The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)] 2014 Available from: http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM284443.pdf#page=25 . [Google Scholar]
- 7.Faison TA. [Last accessed on 2017 Feb 01];Historical Overview of FDA Regulation of Digital Pathology Imaging Applications: The Safety and Effectiveness Issues. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/.../MedicalDevices/.../UCM201450.ppt . [Google Scholar]
- 8.Lange H. Digital pathology: A regulatory overview. Lab Med. 2011;42:587–91. [Google Scholar]
- 9.Têtu B, Evans A. Canadian licensure for the use of digital pathology for routine diagnoses: One more step toward a new era of pathology practice without borders. Arch Pathol Lab Med. 2014;138:302–4. doi: 10.5858/arpa.2013-0289-ED. [DOI] [PubMed] [Google Scholar]
- 10.Al-Janabi S, Huisman A, Vink A, Leguit RJ, Offerhaus GJ, Ten Kate FJ, et al. Whole slide images for primary diagnostics in dermatopathology: A feasibility study. J Clin Pathol. 2012;65:152–8. doi: 10.1136/jclinpath-2011-200277. [DOI] [PubMed] [Google Scholar]
- 11.Stathonikos N, Veta M, Huisman A, van Diest PJ. Going fully digital: Perspective of a Dutch academic pathology lab. J Pathol Inform. 2013;4:15. doi: 10.4103/2153-3539.114206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thorstenson S, Molin J, Lundström C. Implementation of large-scale routine diagnostics using whole slide imaging in Sweden: Digital pathology experiences 2006-2013. J Pathol Inform. 2014;5:14. doi: 10.4103/2153-3539.129452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. [Last accessed on 2017 Mar 07];FDA Open to Whole-slide Imaging as Class II Device. CAP Today. 2016 Available from: http://www.captodayonline.com/fda-open-whole-slide-imaging-class-ii-device/ [Google Scholar]
- 14. [Last accessed on 2017 Feb 01];DPA Regulatory Task Force. Available from: https://www.digitalpathologyassociation.org/digital-pathologyassociation-regulatory-committee . [Google Scholar]
- 15. [Last accessed on 2017 Feb 01];FDA Technical Performance Assessment of Digital Pathology Whole Slide Imaging Devices. Available from: http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM435355.pdf . [Google Scholar]
- 16.Allen TC. Comes digital pathology. Arch Pathol Lab Med. 2015;139:972. doi: 10.5858/arpa.2015-0083-ED. [DOI] [PubMed] [Google Scholar]
- 17. [Last accessed on 2017 Feb 05];FDA Draft Guidance for WSI, on Behalf of the Digital Pathology Association, Public Statement. Available from: https://www.digitalpathologyassociation.org/fda-draft-guidance-for-whole-slide-imaging . [Google Scholar]
- 18. [Last accessed 2017 Feb 05];Put it on the Board. Guidance Seen as Sign of FDA Openness to Digital Pathology. Available from: http://www.captodayonline.com/put-board-616/ [Google Scholar]
- 19.Badano A, Revie C, Casertano A, Cheng WC, Green P, Kimpe T, et al. Consistency and standardization of color in medical imaging: A consensus report. J Digit Imaging. 2015;28:41–52. doi: 10.1007/s10278-014-9721-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leo P, Lee G, Shih NN, Elliott R, Feldman MD, Madabhushi A. Evaluating stability of histomorphometric features across scanner and staining variations: Prostate cancer diagnosis from whole slide images. J Med Imaging (Bellingham) 2016;3:047502. doi: 10.1117/1.JMI.3.4.047502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wack K, Drogowski L, Treloar M, Evans A, Ho J, Parwani A, et al. A multisite validation of whole slide imaging for primary diagnosis using standardized data collection and analysis. J Pathol Inform. 2016;7:49. doi: 10.4103/2153-3539.194841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. [Last accessed on 2017 May 05];FDA Letter to Philips. Available from: https://www.accessdata.fda.gov/cdrh_docs/pdf16/DEN160056.pdf . [Google Scholar]
- 23. [Last accessed on 2017 May 05];FDA News Release. Availavle from: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm552742.htm . [Google Scholar]
- 24.Feldman M, Rubin BP, Moskaluk CA, Cacciabeve N, Lindberg G, Nelis M, et al. A large multicenter, retrospective non-inferiority study to evaluate diagnostic concordance between optical vs. digital microscopic diagnoses in 2000 surgical pathology cases. Mod Pathol. 2017;30:395A. [Google Scholar]